
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional polymeric dialdehyde dextrin network capped mesoporous silica nanoparticles for pH/GSH dual-controlled drug release†
Chao Chen‡
 a,
Wen Sun‡a,
Wenji Yaoa,
Yibing Wang*a,
Hanjie Ying*b and
Ping Wang*ac
a,
Wen Sun‡a,
Wenji Yaoa,
Yibing Wang*a,
Hanjie Ying*b and
Ping Wang*ac
aState Key Laboratory of Bioreactor Engineering, Shanghai Collaborative Innovation Center for Biomanufacturing, Biomedical Nanotechnology Center, School of Biotechnology, East China University of Science and Technology, Shanghai 200237, People's Republic of China. E-mail: ybwang@ecust.edu.cn; pwang11@ecust.edu.cn
bState Key Laboratory of Materials-Oriented Chemical Engineering, College of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, Puzhu South Road, Nanjing 211816, People's Republic of China. E-mail: yinghanjie@njtech.edu.cn
cDepartment of Bioproducts and Biosystems Engineering, University of Minnesota, St Paul, MN 55108, USA. E-mail: ping@umn.edu
First published on 7th June 2018
Abstract
Multi-stimulation responsive nanomaterial-based drug delivery systems promise enhanced therapeutic efficacy in cancer therapy. This work examines a smart pH/GSH dual-responsive drug delivery system by using dialdehyde dextrin (DAD) end-capped mesoporous silica nanoparticles (MSNs). Specifically, DAD was applied as a “gatekeeper polymer” agent to seal drug loads inside the mesoporous of MSNs via a pH-sensitive Schiff bond, whereas the formed DAD polymer shells were further cross-linked by GSH-sensitive disulfide bonds. Results revealed that the DAD gatekeeper polymer could tightly close the mesopores of MSNs to control premature drug release under physiological conditions and respond to acidic and GSH conditions to release the trapped drugs. Significantly, fluorescent microscopy observation and cytotoxicity studies indicated that drug-loaded nanoparticles could be rapidly internalized through a passive targeting effect to inhibit cancer growth. Taken together, these polymer-modified pH/GSH dual-responsive MSNs could be used as promising candidates for “on-demand” anticancer drug delivery applications.
Introduction
In recent decades, medical applications of nanocarriers for cancer therapeutics have fully transformed the area of drug delivery.1–3 Meanwhile, various nanostructured materials have been designed and synthesized to address some of the most obvious restrictions of traditional medicine, including inadequate pharmacokinetics, poor drug solubility, and multiple side effects.4–8 Furthermore, nanoscale-based drug delivery systems exhibited enhanced permeability and retention effects (EPR),9,10 which mainly reduced drug distribution in nonspecific tissues and improved local drug concentration without provoking adverse reactions in tumor therapy.11–13 Up to now, numerous types of nanoparticles have been developed as drug delivery systems for tumor therapy11 such as inorganic nanoparticles,14 high-molecular polymers,15,16 nanogels,17 micelles,18 and liposomes.19 Among these nanoparticles, biocompatible mesoporous silica nanoparticles (MSNs) were used as ideal nanocarriers to deliver drugs due to their unique advantages, including well-defined surfaces, large surface areas, tunable pore size, and a stable structure.20–24 Unfortunately, conventional bare MSNs-based nanocarriers still has some limitations because of premature leakage of the cytotoxic drugs during systemic circulation.8,25,26 Therefore, although a large number of studies have shown successful drug encapsulation by MSNs-based materials, delivering therapeutic drugs for tumor tissues in a controlled manner remains a research hotspot for the development of a smart drug delivery system.27Recently, polymer-based controlled drug delivery systems have been extensively explored for different biomedical applications due to their specific advantages9,25,28,29 where the polymer could be removed with different biological stimuli (temperature, pH, enzymes, glucose, and GSH). Therefore, modifying a stimuli-sensitive polymer on the surface of MSNs to cover the mesopores is a promising strategy for designing desired stimuli-responsive nanocarriers.30–33 During the past decade, various types of polymer-coated MSN carriers have been constructed and fabricated, and different stimuli have served as triggers for regulating drug release including redox conditions, pH, temperature, enzymes, and light.23,28,31,34–38 Among these stimuli, a pH-induced activation release system represents an effective strategy for tumor therapies since the human body exhibits variations in pH.39,40 Moreover, most tumor microenvironments have lower extracellular pH values (pH 7.0–6.0) than extracellular bloodstreams and normal tissues (pH 7.4), and so pH value will drop further inside cancer cell such as lysosomes (pH 5.0–4.5) and endosomes (pH 6.0–5.5).39–41 Therefore, we can combine the advantages of MSNs and the unusual pH gradients to construct pH-sensitive nanocarriers which can release entrapped guest drug molecules in a controlled manner under certain pH conditions.
Herein, in order to realize a pH/GSH dual-sensitive gatekeeper polymer for capping drug load and regulated release with MSNs, a biodegradable dextrin was selected as the “gatekeeper polymer” and modified on the surface of MSNs to fabricate a stimuli-responsive drug delivery system for controlled intercellular drug release. To achieve this, dextrin was oxidized by NaIO4 to obtain dialdehyde dextrin (DAD), and the novel DAD was first served as a “gatekeeper polymer” grafted on the surface of MSNs via pH-sensitive Schiff base reactions. In order to further realize optimally controlled drug release and delivery performance, the modified DAD polymer shell was further cross-linked with cystamine dihydrochloride to construct intermolecular redox-sensitive disulfide bonds. Furthermore, doxorubicin hydrochloride (DOX) was selected as a model drug to evaluate the drug loading and pH/GSH dual-sensitive controlled release behavior of the fabricated multifunctional MSNs. We hoped the designed and constructed multifunctional drug delivery system could respond to both acidic pH and elevated GSH concentrations; however, accelerated and complete release was only secured when both stimuli were presented. Moreover, the in vitro cellular uptake efficacy and anticancer activity of DOX-loaded nanoparticles were systemically indicated.
Material and methods
Materials
Tetraethylorthosilicate (TEOS), sodium periodate, sodium hydroxide (NaOH), 3-aminopropyl trimethoxysilane (APTES), cetyltrimethylammonium bromide (CTAB), dextrin (from maize starch) and ethylene glycol (EG) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Cystamine dihydrochloride (cystamine·2HCl, >98%) was purchased from Macklin Reagent Company. 2,5-Diphenyl-3-(4,5-dimethyl-2-thiazolyl) tetrazolium bromide (MTT) was purchased from Sigma-Aldrich. Doxorubicin hydrochlorides (DOX) were purchased from Sangon Biotech (Shanghai, China). All other chemicals were commercially available and used without further purification.Synthesis and ammonization of mesoporous silica nanoparticles (MSNs)
For the preparation of MSNs, a slightly modified method was employed as previously reported.31,42 Briefly, 250 mg of CTAB was dissolved in 100 mL of purified water, followed by 20 mL of EG and 0.875 mL of NaOH (2 M) added to the above solution. The mixture was heated to 80 °C and violently stirred for 1 h. Subsequently, 1.25 mL of TEOS was rapidly added to the above mixture and stirred for an additional 2 h. Then, the products were collected by centrifugation and purified three times with water and ethanol. Finally, the obtained sample was vacuum-dried to yield the as-synthesized MSNs.The amino-modified MSNs were synthesized according to previous procedures.32 To 500 mg of MSNs ultrasonically dispersed in 100 mL ethanol was added 2 mL of 3-aminopropyl trimethoxysilane (APTES). After stirring at 80 °C for 24 h, centrifugation was performed to collect the product which was then dried for further use. Finally, the amino-functionalized MSNs were calcined at 550 °C for 6 h to remove CTAB and MSNs-NH2 was synthesized.
Synthesis of dialdehyde dextrin (DAD)
The preparation of dialdehyde dextrin was similar to that of the dialdehyde starch.43 In short, 5 g of dextrin was dissolved in sodium periodate solution (50 mL, 0.3 M) with magnetic stirring for 24 h at room temperature in the dark. After that, 20 mL of ethylene glycol was added to the above mixture. The mixture was stirred for an additional 15 min to neutralize the solution. Then, the obtained product was transferred into a dialysis bag (MWCO = 1000 Da) against ultrapure water for 72 h with several changes of water. Finally, the collected dialysate was further centrifuged and rinsed with ultrapure water for three times and then dried by lyophilization.Drug loading into MSNs
For detailing, 100 mg of MSNs-NH2 was ultrasonically dispersed into PBS buffer (100 mL, 0.1 M, pH 7.4) containing 100 mg DOX. The mixture was stirred for 24 h at room temperature in the dark. Then, the DOX-loaded nanoparticles were obtained by centrifugation and rinsed thoroughly with deionized water to remove the adsorbed and unloaded drugs. The obtained sample was donated as DOX-MSNs-NH2.Preparation of DAD coated MSNs (MSNs-N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C-DAD)
C-DAD)
Typically, MSNs-NH2 (100 mg) was ultrasonically dispersed in MES buffer (100 mL, 0.01 M, pH 6.0) containing 100 mg dialdehyde dextrin and stirred for 24 h at room temperature. After that, cystamine dihydrochloride (1 g) was added to the mixture and it was stirred for 6 h at room temperature. The synthesized product was collected by centrifugation and washed several times with ethanol and water, respectively. The product was denoted as MSNs-N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-DAD. The DOX loaded sample was named DOX-MSNs-NC-DAD.
C-DAD. The DOX loaded sample was named DOX-MSNs-NC-DAD.
In vitro dual-responsive drug release
To investigate the pH and GSH dual-responsive release character, the obtained DOX-MSNs-NC-DAD was tested under different pH and GSH conditions. In brief, 3 mg of DOX-loaded nanoparticles were dispersed in PBS buffer (10 mL, 0.1 M, pH 7.4) and then transferred to a dialysis bag (cellulose membrane; MWCO is 7000 Da). For analysis, the stimuli-responsive release behavior of the DOX-loaded nanoparticles was dialyzed against PBS with different pH values of 5.0, 6.5, and 7.4 and different concentrations of GSH (0, 5, and 10 mM) and stirred at 37 °C. At predetermined time intervals, 2 mL of dialysate was taken out and analyzed with a U-5100 UV-Vis spectrophotometer (Hitachi). Meanwhile, fresh medium with equal volume was added to above solution.
Characterization
Transmission electron microscope (TEM) images of prepared nanoparticles were carried out on a JEM-1400 (JEOL, Japan) at 200 kV. The Brunauer–Emmett–Teller (BET) approach (ASAP 2010, Micromeritics, USA) and Barrett–Joyner–Halenda (BJH) method were performed to calculate pore size distributions and surface area, respectively. Fourier transform infrared (FTIR) spectra were measured by a Bruker IFS 55 spectrometer (Switzerland) using KBr pellets. The zeta potential and hydrodynamic size were determined using a Zetasizer Nano ZS90 (Malvern, UK). Small-angle powder X-ray diffraction patterns of the obtained materials were collected on a RINT2000 vertical goniometer (Rigaku, Japan) using Cu Kα irradiation. Thermal gravimetric analysis (TGA) was determined on a TGA-50 instrument (Shimadzu, Japan) with a heating rate of 10 °C min−1 under a nitrogen flow. The number-averaged molecular weight and the polydispersity index of DAD were determined on a WATERS 1515 equated with a series of PS gel columns, using THF as an eluent at 40 °C with a PS calibration. The concentration of DOX was evaluated with a U-5100 UV-Vis spectrophotometer at 480 nm.Cell culture
Hela cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum and antibiotics (100 U mL−1 penicillin and 100 μg mL−1 streptomycin) in 5% CO2 at 37 °C. Media in all samples were changed every two days and the cells were separated by trypsin before cell density reached 80%.Intracellular uptake experiment
Confocal laser scanning microscopy (CLSM) and flow cytometry (FCM) were performed to evaluate the cellular uptake of free DOX and drug-loaded nanoparticles. In brief, Hela cells were seeded in 20 mm glass bottom culture dishes overnight. After that, free DOX and DOX-loaded nanoparticles (equivalent DOX concentration: 1 μg mL−1) were dispersed in RPMI 1640 culture medium (1 mL) and replaced the above medium. After incubating at 37 °C for another 2 h and 6 h, the original medium was removed, and the cells were washed with PBS three times and then fixed with 4% paraformaldehyde for 10 min. Then, the nuclei were stained with DAPI for 20 min. Finally, the treated cells were observed by CLSM. For flow cytometry analysis, Hela cells were seeded in 6-well plates at a density of 2 × 105 cells per well and then treated as above. Non-treated cells were used as a control. After incubating for 2 h and 6 h, the treated cells were washed with PBS for several times, then trypsinized and resuspended in PBS buffer for FCM analysis.In order to determine whether the DOX release from MSNs-NC-DAD was triggered by intracellular low pH values and high GSH level, Hela cells were incubated with 20 mM NH4Cl (pH inhibitor)44 and 5 μM N-ethyl maleimide (NEM, GSH inhibitor)1 for 30 min, and then incubated with DOX-MSNs-NC-DAD nanoparticles for another 6 h. Finally, the treated cells were then washed with PBS three times and observed under CLSM. To quantitatively measure intracellular release of DOX from nanocarriers; FCM was performed to analyze the intracellular DOX fluorescence intensity.
Cytotoxicity assay
MTT assay was performed to determine the cytotoxicity of free DOX or drug-loaded nanoparticles. Hela cells were cultured in a 96-well plate with a density of 1 × 104 cells per well with 5% CO2 at 37 °C overnight. Afterward, fresh culture medium containing different concentrations of free DOX and drug-loaded nanoparticles was added to replace the medium, respectively. After being cultured for another 24 h and 48 h, 10 μL of MTT (5 mg mL−1) stock solution was added to each well and incubated for another 4 h. Then, MTT containing medium was removed and 150 μL of DMSO was added to each well and shaken for 10 min. Finally, the 96-well plates were measured on an ELx 808 micro-plate reader (Biotek, VT) at 570 nm. The cell viability was calculated as ODtest/ODcontrol × 100%.Statistical analysis
All experiments were conducted three times, and all statistical data were formed as means ± standard errors. The difference of each group in this study was analyzed by one-way ANOVA. The statistically significant values in all cases were at p-value < 0.05.Results and discussion
Synthesis and characterization of MSNs-NC-DAD
The detailed procedure to design the pH/GSH dual-responsive nanocarriers based on dialdehyde dextrin (DAD) end-capped mesoporous silica nanoparticles (MSNs) was presented in Scheme 1. In brief, MSNs were synthesized by a previously reported base-catalyzed sol–gel method and modified with amino groups.31,32 Subsequently, the obtained DAD polymer was conjugated on the surface of MSNs as caps by reaction between the amino groups of MSNs and aldehyde groups of DAD through pH-sensitive Schiff base reactions. Afterward, the formed DAD polymer shell was further cross-linked with cystamine dihydrochloride to construct intermolecular redox-sensitive disulfide bonds. Thereafter, along with the disruption of DAD polymer shells by hydrolysis of Schiff bonds and breakage of the disulfide bonds under low pH and high GSH conditions, the loaded drugs would be released to specific cells.
 | ||
| Scheme 1 Schematic structure of DOX-MSNs-NC-DAD and pH/GSH dual-stimuli responsive drug delivery. | ||
The structure and morphology of the as-prepared multi-functionalized MSNs were characterized by TEM and FE-SEM. Fig. 1A and S9† showed that the synthesized MSNs were uniformly spherical with a mean diameter around 106 nm according to the DLS analysis (Fig. S1†). Moreover, an array of ordered mesoporous networks could be clearly observed by a high magnification TEM image (Fig. S10†), and the mesoporous structure of MSNs was further confirmed by a low-angle XRD pattern. Characteristic diffraction peaks at 200, 110, and 100 in Fig. 1B revealed that the synthesized MSNs belong to a MCM-41 type with highly ordered mesoporous structure.31,44,45 After the DAD polymer was modified on the surface of the MSNs, the obtained MSNs-NC-DAD exhibited a blurry mesoporous structure with the legible polymer layer capping on the MSNs (Fig. 1C and S9†). Meanwhile, the measured hydrodynamic diameters increased from 112 to 154 nm (Fig. S1 and Table S1†), further indicating that the DAD polymer was successfully conjugated onto MSNs. In addition, the average surface potentials of MSNs were significantly changed during the preparation process (Fig. 1D and Table S2†). After being modified with APTES and conjugated with DAD, the zeta potential of obtained MSNs were changed from −25 to 20 and −6 mV due to the amino groups of MSNs-NH2 and aldehyde groups of DAD, indicating the DAD polymer's existence in the nanosystem. The number-averaged molecular weight of DAD was 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 041 Da (PDI 1.27).
041 Da (PDI 1.27).
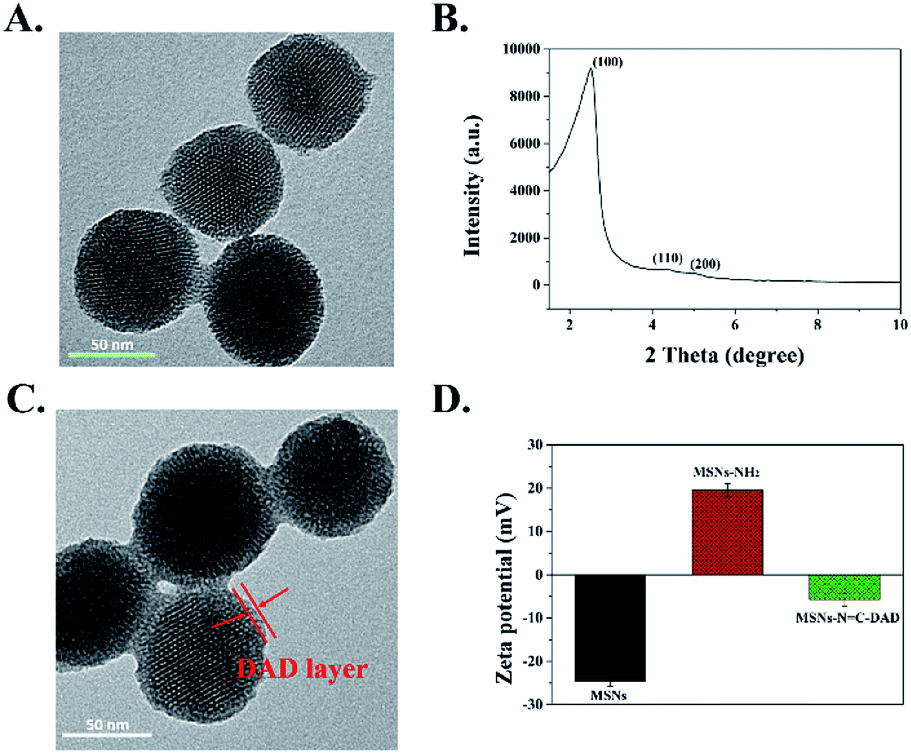 | ||
| Fig. 1 Morphology and structural characterization of different MSNs. (A) TEM image of MSNs. (B) Small-angle XRD patterns of MSNs. (C) TEM image of MSNs-NC-DAD, and (D) zeta potential of MSNs, MSNs-NH2 and MSNs-NC-DAD. | ||
FTIR spectra were performed to direct the progress of the surface functionalization of MSNs. Fig. S2† demonstrated that the surfactant template agent CTAB was successfully removed from MSNs after being calcined at 550 °C for 6 h. The FTIR spectrum of dialdehyde dextrin (DAD) exhibited a new peak at 1720 cm−1 compared with that of dextrin before oxidation,46 which could be attributed to the CO vibration in aldehyde groups (Fig. 2A). As a result, the dextrin has been successfully oxidized into dialdehyde dextrin (DAD). In addition, MSNs-NH2 displayed two new strong absorption signals at 1560 cm−1 and 1636 cm−1 compared to blank MSNs (Fig. 2B), which were assigned to the stretching vibration of –NH2 bending and amide I, indicating the successful modification of amino groups on MSNs.32,47 Following grafting with DAD polymer, the new distinctive absorption peak at 1720 cm−1 (CO) was observed when compared with that of MSNs-NH2. Most importantly, the distinctive absorption peak at 1640 cm−1 was attributed to the CN of the Schiff base reaction between the amino groups of MSNs and aldehyde groups of DAD.48 Furthermore, because the stretching vibration band of S–S on MSNs-NC-DAD was too weak to be measured through FTIR spectroscopy, the Raman spectrum at 502 cm−1 exhibited a strong stretch band, which contributed to the introduction of disulfide bonds (S–S) derived from cystamine dihydrochloride (Fig. 2C).49 These results suggested that the pH-sensitive Schiff bond and GSH-sensitive disulfide bonds were successfully constructed and fabricated.
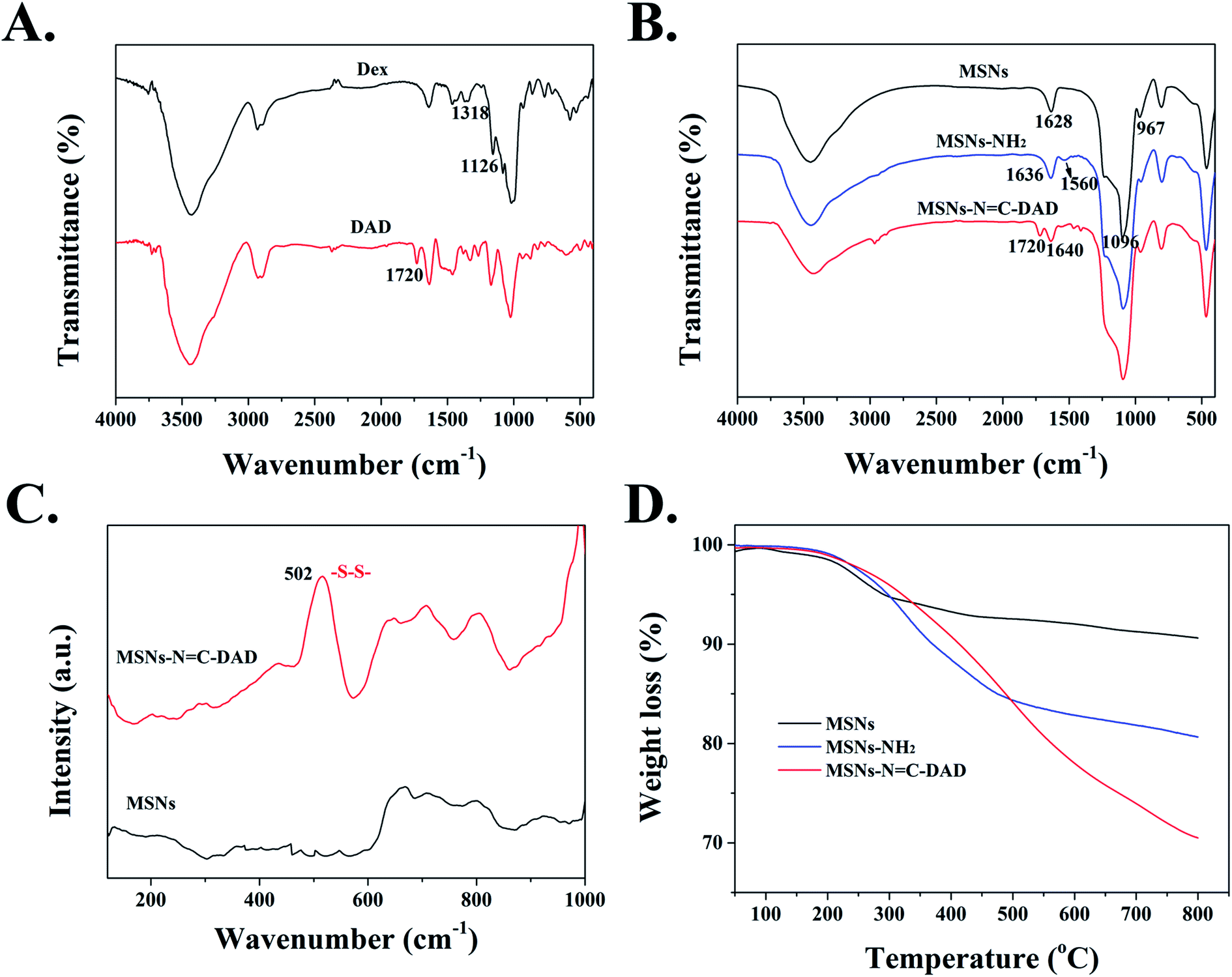 | ||
| Fig. 2 FTIR spectra of (A) Dex and DAD, (B) MSNs, MSNs-NH2 and MSNs-NC-DAD, (C) Raman spectra of MSNs and MSNs-NC-DAD, and (D) TGA curves of MSNs, MSNs-NH2, and MSNs-NC-DAD. | ||
To further demonstrate the successful surface modification process of MSNs, a series of characteristic techniques were performed. The surface modification extent of MSNs was measured by TGA (Fig. 2D) and the final weight losses of all materials is shown in Table S3.† After the temperature rose to 800 °C, the weight loss of MSNs, MSNs-NH2, and MSNs-NC-DAD was 9.38, 19.43, and 29.51 wt%, respectively. The increased weight losses further indicated that the MSNs-NC-DAD was successfully fabricated step by step. In addition, the N2 adsorption and desorption isotherm of blank MSNs exhibited a typical type IV isotherm (Fig. 3A and B), which further indicated the highly ordered mesoporous structure of the prepared MSNs.31,45 After a series of functionalizations, the surface areas of nanoparticles were reduced from 967 to 137 m2 g−1 after each modification (Fig. 3 and Table S4†). Meanwhile, the pores were decreased from 2.9 nm to undetectable, which also suggested the successful fabrication of multifunctional MSNs-NC-DAD. Taken together, all of these results suggested that MSNs-NC-DAD nanoparticles were successfully prepared.
 | ||
| Fig. 3 (A) N2 adsorption/desorption isotherms and (B) pore size distributions of MSNs, MSNs-NH2, DOX-MSNs-NH2, MSNs-NC-DAD, and DOX-MSNs-NC-DAD. | ||
Dual-responsive drug release performance
In order to evaluate the pH/GSH dual-responsive release behavior of the MSNs-NC-DAD nanoparticles, DOX was selected as a model drug due to its water solubility and fluorescence properties.50 The loading process of DOX mainly depends on the physical adsorption mechanism of the mesopores. Fig. S3† showed that a significant ultraviolet absorption spectrum of DOX molecules was observed after DOX was loaded into MSNs-NC-DAD nanoparticles, indicating that the DOX was successfully loaded on MSNs-NC-DAD nanoparticles.51 The loading capacity of DOX on MSNs-NC-DAD was about 20.1%, much higher than that of conventional MSNs (less than 10%) (according to the standard curve of DOX absorbance value at 480 nm Fig. S4†). The high DOX loading capacity might be due to the modification of the biocompatible polymer DAD, which could effectively prevent drug leakage in the process of drug loading. In addition, Fig. S5† showed that the existing state of DOX in MSNs-NC-DAD was in a non-crystalline state due to the confined effect of the mesoporous of MSNs.27,52
Next, in order to verify the blocking efficiency of DAD polymer, we evaluated the release behavior of MSNs-NC-DAD at physiological conditions. Fig. S6† showed that only 13% of DOX was released from DOX-MSNs-NC-DAD after 30 h, while about 81% of DOX was leaked out from DOX-MSNs, suggesting that the DAD polymer could effectively block DOX within the mesoporous area of MSNs under neutral conditions. Following, to investigate the pH-responsive release behavior of DAD polymer covered nanoparticles, the DOX release curves of DOX-MSNs-NC-DAD were examined in the presence of different pH values (pH 5.0, 6.5, and 7.4, respectively). Fig. 4A showed that a slow release of below 13% of incorporated DOX within 30 h was observed at pH 7.4, whereas the accumulative release of DOX at pH 6.5 and 5.0 increased to 41% and 55% in a total release period of 30 h, indicating that the DAD polymer covering the surface of MSNs could be cleaved under a mild acid environment. This pH-trigger release mechanism can be explained by the fact that the pH-sensitive Schiff based group is stable under physiological conditions but is easily cleaved under mild acid conditions.53
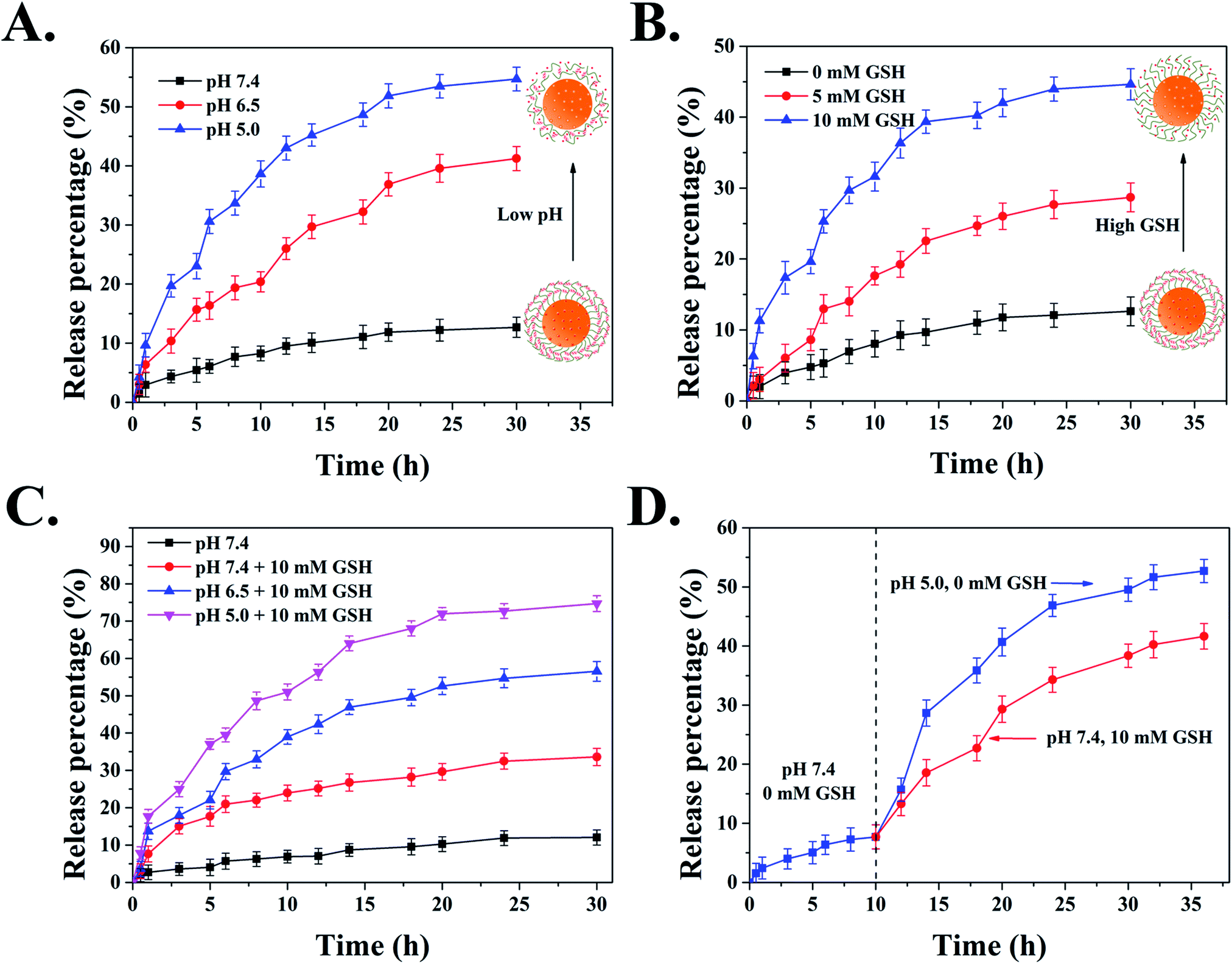 | ||
| Fig. 4 In vitro pH/GSH dually responsive release of DOX from MSNs-NC-DAD. (A) The release profiles of DOX from MSNs-NC-DAD at different pH conditions (5.0, 6.5, and 7.4) at 37 °C for 30 h. (B) The release profiles of DOX from MSNs-NC-DAD in pH 7.4 PBS in the presence of different concentrations of GSH (0, 5, and 10 mM) at 37 °C for 30 h. (C) The release profiles of DOX from MSNs-NC-DAD in pH 7.4, 6.5, and 5.0 PBS in the presence/absence of 10 mM GSH. (D) pH/GSH-responsive release of DOX from MSNs-NC-DAD. Data are represented as mean ± SD (n = 3). | ||
In addition to pH stimulus-responsive ability, the cross-linked DAD shell can be also activated by GSH due to its redox ability toward disulfide bonds in polymer chains. The rapid release of DOX from MSNs-NC-DAD was significantly observed in Fig. 4B with an increase of GSH concentration. Briefly, the accumulative release of DOX from DOX-MSNs-NC-DAD was 13% for 0 mM GSH, 29% for 5 mM GSH, and 44% for 10 mM GSH, respectively. These results evidenced that the intramolecular disulfide bonds cross-linked in the DAD polymer could be cleaved under reductive conditions, resulting in the cross-linked DAD polymer network becoming loosened and triggering the release of DOX from nanoparticles.
It is well known that the pH value and GSH concentration in cancer intracellular microenvironments are 5.0–5.5 and 2–10 mM, respectively, which are deviated from the normal level (pH 7.4, CGSH = 2–20 mM).45 Therefore, the constructed and fabricated pH/GSH dual-responsive MSNs-based nanocarrier is an ideal candidate as an anticancer drug delivery system. To evaluate the sensitivity of the DAD polymer shell covered system with different pH values and concentrations of GSH, the drug release curves of MSNs-NC-DAD in different conditions were traced by UV-Vis spectra. Fig. 4C and D clearly exhibited that the release efficiency of DOX significantly increased when the carriers were delivered under mild acid conditions (pH 6.5 or 5.0) in the presence of GSH (10 mM). Moreover, the introduction of GSH could remarkably increase the release rate of MSNs-NC-DAD under acidic environments. The rapid release of DOX from carriers could be explained by the fact that the combined disruption of DAD polymer shells by hydrolysis of Schiff bonds and breakage of the disulfide bonds under low pH and high GSH conditions. Based on the above analysis, it is reasonable that the designed MSNs-NC-DAD have promising applications in site-specific cancer chemotherapy thanks to the acidic microenvironments and high GSH concentration in cancer cells.
Cellular uptake and intracellular release of DOX
The internalization and intracellular distribution of free DOX and DOX-loaded nanoparticles were investigated by CLSM in real time. As shown in Fig. 5, both free DOX and DOX-loaded MSNs-NC-DAD were significantly observed in nucleus or cytoplasm after incubation for 2 h to 6 h. The number of intracellular uptakes of free DOX and DOX-MSNs-NC-DAD was obviously increased with prolonged incubation time. Moreover, the amount of DOX-MSNs-NC-DAD taken up by Hela cells was significantly increased in the cytoplasm after 2 h and large numbers of DOX were diffused into the surrounding nuclear membrane or the nuclear regions. These results demonstrated that the intracellular uptake of DOX was increased in a time-dependent manner and the DOX-MSNs-NC-DAD nanoparticles could effectively deliver DOX to cancer cells because of the pH-triggered release. Furthermore, flow cytometry (FCM) was performed to quantitatively monitor the amount of cellular uptake of the DOX-loaded nanoparticles. Fig. S7† shows that the mean fluorescence intensity of DOX-MSNs-NC-DAD entered into Hela cells was more than 1.36-fold higher than that of free DOX treatment, potentially because of the enhanced permeability and retention (EPR) effects induced by MSNs-NC-DAD.
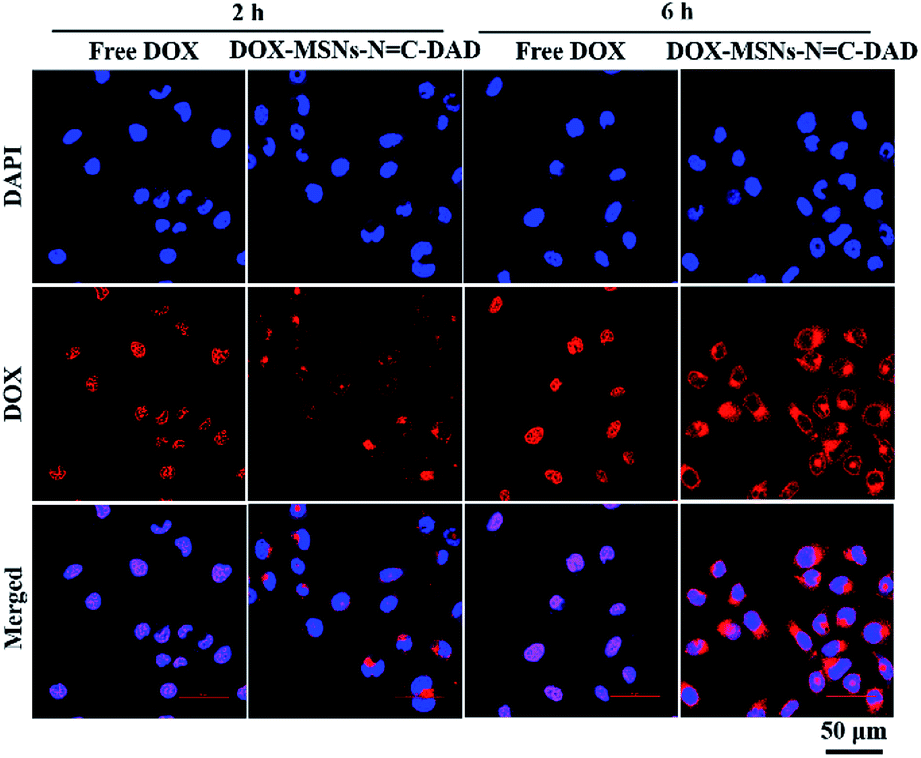 | ||
| Fig. 5 CLSM images of Hela cells incubated with free DOX and DOX-MSNs-NC-DAD for 2 and 6 h, respectively (equivalent DOX concentration: 1 μg mL−1). Scale bar: 50 μm. | ||
Furthermore, in order to detect whether the DOX release from MSNs-NC-DAD was triggered by tumor intracellular low pH and high concentrations of GSH, intracellular pH inhibitor NH4Cl and GSH inhibitor NEM were used to treat Hela cells for 30 min before being incubated with MSNs-NC-DAD. As shown in Fig. 6A and B, compared with the control group (DOX-MSNs-NC-DAD only), weaker DOX fluorescence was observed in Hela cells pretreated with NH4Cl and NEM, suggesting that most DOX molecules remained encapsulated in the MSNs. These results demonstrated that DOX release from DOX-MSNs-NC-DAD was triggered by intracellular low pH and a high concentration GSH level. In summary, the above results from qualitative and quantitative analysis suggested that the cellular uptake of DOX-MSNs-NC-DAD was time dependent and the DOX release was triggered by an intracellular low pH and high GSH level.
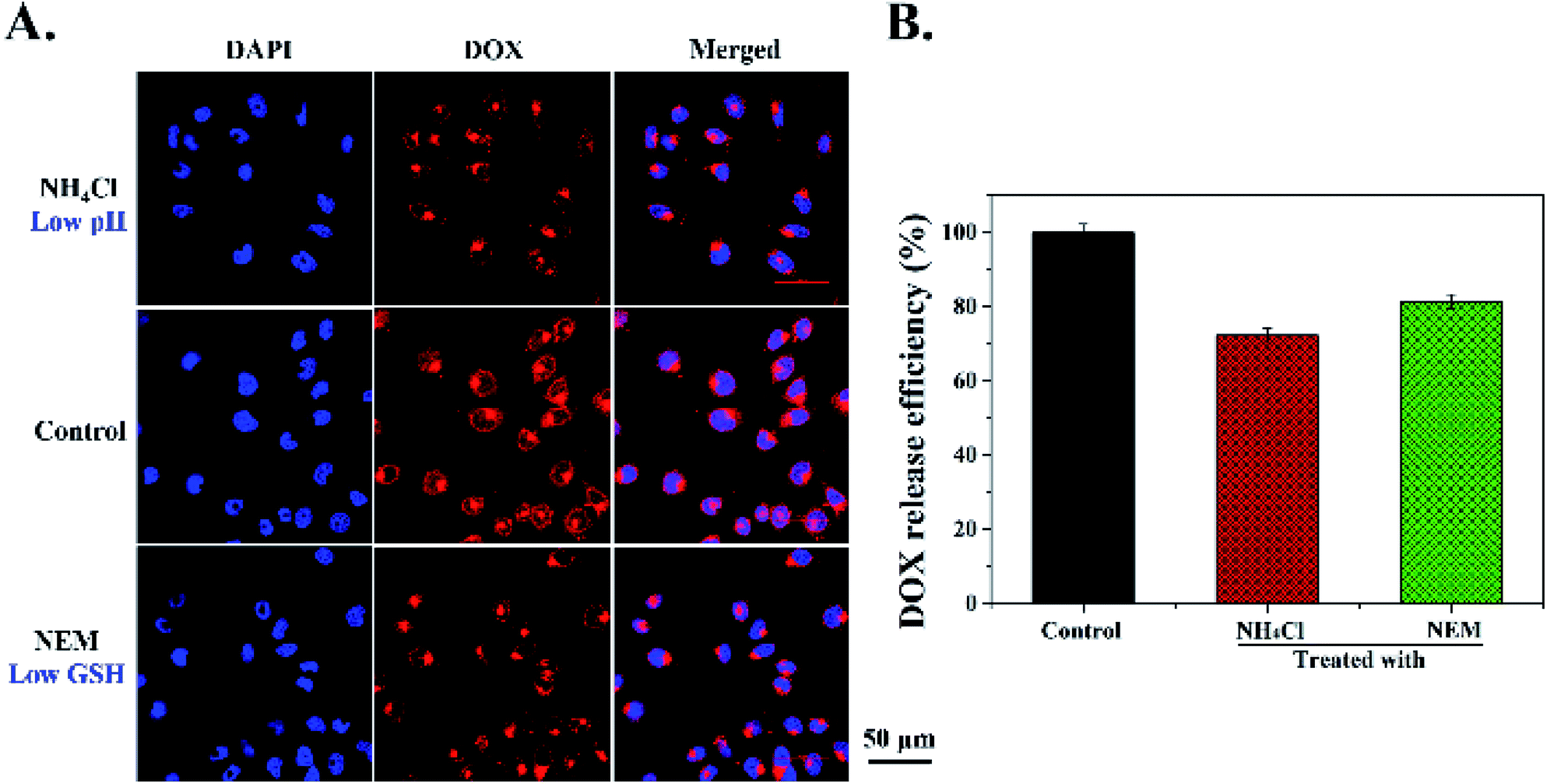 | ||
| Fig. 6 (A) CLSM images of intracellular DOX release from DOX-MSNs-NC-DAD in Hela cells. Cells first treated with NH4Cl, nothing, and NEM, then incubated with DOX-MSNs-NC-DAD for 6 h, respectively. Scale bar: 50 μm. (B) The DOX release efficiency of DOX-MSNs-NC-DAD at different treatments. | ||
In vitro cytotoxicity assay
In cancer therapy, biocompatibility and low toxicity of the fabricated MSNs as nanocarriers are required to avoid side effects. Therefore, the in vitro cytotoxicity of MSNs and MSNs-NC-DAD against Hela and L-02 cells was determined by a MTT assay. Fig. S8† shows that more than 80% cell viability was observed at test particle concentrations against both Hela and L-02 cells, demonstrating excellent biocompatibility. However, the cell viability of Hela cells was significantly reduced with dose and time after being incubated with free DOX or DOX-loaded MSNs-NC-DAD (Fig. 7A and B). Meanwhile, DOX-loaded MSNs-NC-DAD exhibited more cytotoxicity compared with free DOX at the same equivalent concentration both at 24 h and 48 h. A possible reason for this could be assigned as different cellular uptake mechanisms that the nanocarriers use to enter into cells through “passive targeting” which is higher than that of free DOX through diffusion. Furthermore, the anticancer activity of DOX was quantified by half maximal inhibitory concentration (IC50). As shown in Table 1, the IC50 values of DOX-loaded MSNs-NC-DAD at 24 h and 48 h were 1.01 and 0.35 μg mL−1, respectively, versus those of free DOX (1.48 and 0.51 μg mL−1, respectively), indicating that the former were 1.47- and 1.46-fold more effective than the latter. Therefore, these results revealed that the constructed DOX-MSNs-NC-DAD could enhance the cytotoxicity effects of DOX in cancer therapy.
 | ||
| Fig. 7 Effect of free DOX and DOX-MSNs-NC-DAD on cell viability of Hela cells by MTT assay after (A) 24 h and (B) 48 h incubation at 37 °C. | ||
| Formulations | IC50 (μg ml−1) | |
|---|---|---|
| 24 h | 48 h | |
| a IC50, half maximal inhibitory concentration. | ||
| Free DOX | 1.48 | 0.51 |
| DOX-MSNs-NC-DAD |
1.01 | 0.35 |
Conclusions
In summary, we have synthesized multifunctional MSNs that can be employed as pH/GSH dual-responsive drug delivery systems for intracellular controlled drug release. The obtained DAD polymer as “gatekeeper polymer” agents were coated onto the surfaces of MSNs via pH-sensitive Schiff bonds and the formed DAD polymer shells were further cross-linked by GSH-sensitive disulfide bonds. Such smart novel systems not only offer high drug loading capacity (20.1%), but also exhibited excellent biocompatibility. Furthermore, the drug-loaded functional nanoparticles could effectively prevent drug release under physiological conditions, and rapidly release the drug in the presence of acidic environments and high levels of GSH concentrations, which mimic the pH and reduction conditions in cancer cells. Moreover, in vitro experiments confirmed that these functional nanocarriers could greatly increase drug accumulation at cancer cells and significantly enhance cytotoxicity to cancer cells than free drugs. Therefore, the designed pH/GSH dual-responsive nanoparticles are expected to be an effective chemotherapeutic platform in oncotherapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (31471659 and 21636003). Authors thank the support from Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM).Notes and references
- S. Zhao, Q. Yu, J. Pan, Y. Zhou, C. Cao, J.-M. Ouyang and J. Liu, Acta Biomater., 2017, 54, 294–306 CrossRef PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef PubMed.
- N. Ahmed, H. Fessi and A. Elaissari, Drug Discovery Today, 2012, 17, 928–934 CrossRef PubMed.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2013, 26, 435–451 CrossRef.
- H. Chen, C. Khemtong, X. Yang, X. Chang and J. Gao, Drug Discovery Today, 2011, 16, 354–360 CrossRef PubMed.
- M. C. L. Giudice, F. Meder, E. Polo, S. S. Thomas, K. Alnahdi, S. Lara and K. A. Dawson, Nanoscale, 2016, 8, 16969–16975 RSC.
- T. Führmann, M. Ghosh, A. Otero, B. Goss, T. R. Dargaville, D. D. Pearse and P. D. Dalton, Neurosci. Lett., 2015, 602, 126–132 CrossRef PubMed.
- J. L. Paris, M. V. Cabañas, M. Manzano and M. Vallet-Regí, ACS Nano, 2015, 9, 11023–11033 CrossRef PubMed.
- X. Zhang, L. Han, M. Liu, K. Wang, L. Tao, Q. Wan and Y. Wei, Mater. Chem. Front., 2017, 1, 807–822 RSC.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef PubMed.
- K.-N. Yang, C.-Q. Zhang, W. Wang, P. C. Wang, J.-P. Zhou and X.-J. Liang, Cancer Biol. Med., 2014, 11, 34 Search PubMed.
- K. Cho, X. Wang, S. Nie and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef PubMed.
- B. Chang, X. Sha, J. Guo, Y. Jiao, C. Wang and W. Yang, J. Mater. Chem., 2011, 21, 9239–9247 RSC.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef PubMed.
- X. Wang, Y. Du, J. Luo, B. Lin and J. F. Kennedy, Carbohydr. Polym., 2007, 69, 41–49 CrossRef.
- Q. Wang, X. Hu, Y. Du and J. F. Kennedy, Carbohydr. Polym., 2010, 82, 842–847 CrossRef.
- M. Zan, J. Li, S. Luo and Z. Ge, Chem. Commun., 2014, 50, 7824–7827 RSC.
- E. Blanco, C. W. Kessinger, B. D. Sumer and J. Gao, Exp. Biol. Med., 2009, 234, 123–131 CrossRef PubMed.
- Y. Malam, M. Loizidou and A. M. Seifalian, Trends Pharmacol. Sci., 2009, 30, 592–599 CrossRef PubMed.
- Q. Zhao, H. Geng, Y. Wang, Y. Gao, J. Huang, Y. Wang, J. Zhang and S. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 20290–20299 Search PubMed.
- Q. Zhao, J. Liu, W. Zhu, C. Sun, D. Di, Y. Zhang, P. Wang, Z. Wang and S. Wang, Acta Biomater., 2015, 23, 147–156 CrossRef PubMed.
- N. Han, Q. Zhao, L. Wan, Y. Wang, Y. Gao, P. Wang, Z. Wang, J. Zhang, T. Jiang and S. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 3342–3351 Search PubMed.
- D. Xiao, H. Z. Jia, J. Zhang, C. W. Liu, R. X. Zhuo and X. Z. Zhang, Small, 2014, 10, 591–598 CrossRef PubMed.
- J. G. Croissant, Y. Fatieiev, A. Almalik and N. M. Khashab, Adv. Healthcare Mater., 2018, 7, 1700831 CrossRef PubMed.
- Y. Zhang, C. Y. Ang, M. Li, S. Y. Tan, Q. Qu, Z. Luo and Y. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 18179–18187 Search PubMed.
- J. G. Croissant, Y. Fatieiev and N. M. Khashab, Adv. Mater., 2017, 29, 1604634 CrossRef PubMed.
- J. Jiao, X. Li, S. Zhang, J. Liu, D. Di, Y. Zhang, Q. Zhao and S. Wang, Mater. Sci. Eng., C, 2016, 67, 26–33 CrossRef PubMed.
- Y. Yang, X. Yan, Y. Cui, Q. He, D. Li, A. Wang, J. Fei and J. Li, J. Mater. Chem., 2008, 18, 5731–5737 RSC.
- Q. Wang, Z. Dong, Y. Du and J. F. Kennedy, Carbohydr. Polym., 2007, 69, 336–343 CrossRef.
- P. Zhang, T. Wu and J.-L. Kong, ACS Appl. Mater. Interfaces, 2014, 6, 17446–17453 Search PubMed.
- L. Tan, M.-Y. Yang, H.-X. Wu, Z.-W. Tang, J.-Y. Xiao, C.-J. Liu and R.-X. Zhuo, ACS Appl. Mater. Interfaces, 2015, 7, 6310–6316 Search PubMed.
- Z. Luo, K. Cai, Y. Hu, L. Zhao, P. Liu, L. Duan and W. Yang, Angew. Chem., Int. Ed., 2011, 50, 640–643 CrossRef PubMed.
- X. L. Chen, H. Sun, J. Hu, X. Han, H. L. Liu and Y. Hu, Colloids Surf., B, 2017, 152, 77–84 CrossRef PubMed.
- J. Liu, B. Zhang, Z. Luo, X. Ding, J. Li, L. Dai, J. Zhou, X. Zhao, J. Ye and K. Cai, Nanoscale, 2015, 7, 3614–3626 RSC.
- N. Ž. Knežević, B. G. Trewyn and V. S. Y. Lin, Chem. Commun., 2011, 47, 2817–2819 RSC.
- H. A. Meng, M. Xue, T. A. Xia, Y. L. Zhao, F. Tamanoi, J. F. Stoddart, J. I. Zink and A. E. Nel, J. Am. Chem. Soc., 2010, 132, 12690–12697 CrossRef PubMed.
- W. Cheng, J. P. Nie, L. Xu, C. Y. Liang, Y. Peng, G. Liu, T. Wang, L. Mei, L. Q. Huang and X. W. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 18462–18473 Search PubMed.
- J. G. Croissant, C. Qi, O. Mongin, V. Hugues, M. Blanchard-Desce, L. Raehm, X. Cattoen, M. W. C. Man, M. Maynadier, M. Gary-Bobo, M. Garcia, J. I. Zink and J. O. Durand, J. Mater. Chem. B, 2015, 3, 6456–6461 RSC.
- Y. Chen, H. Zhang, X. Cai, J. Ji, S. He and G. Zhai, RSC Adv., 2016, 6, 92073–92091 RSC.
- L. Xing, H. Zheng, Y. Cao and S. Che, Adv. Mater., 2012, 24, 6433–6437 CrossRef PubMed.
- Z. Li, D. L. Clemens, B.-Y. Lee, B. J. Dillon, M. A. Horwitz and J. I. Zink, ACS Nano, 2015, 9, 10778–10789 CrossRef PubMed.
- C. Chen, W. Sun, X. Wang, Y. Wang and P. Wang, Mater. Sci. Eng., C, 2018, 85, 88–96 CrossRef PubMed.
- J. Wang, H. Liu, F. Leng, L. Zheng, J. Yang, W. Wang and C. Z. Huang, Microporous Mesoporous Mater., 2014, 186, 187–193 CrossRef.
- M. Zhang, J. Liu, Y. Kuang, Q. Li, D.-W. Zheng, Q. Song, H. Chen, X. Chen, Y. Xu and C. Li, Int. J. Biol. Macromol., 2017, 98, 691–700 CrossRef PubMed.
- Q.-L. Li, S.-H. Xu, H. Zhou, X. Wang, B. Dong, H. Gao, J. Tang and Y.-W. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 28656–28664 Search PubMed.
- H. Chen, D. Zheng, J. Liu, Y. Kuang, Q. Li, M. Zhang, H. Ye, H. Qin, Y. Xu and C. Li, Int. J. Biol. Macromol., 2016, 85, 596–603 CrossRef PubMed.
- L. Dai, Q. Zhang, J. Li, X. Shen, C. Mu and K. Cai, ACS Appl. Mater. Interfaces, 2015, 7, 7357–7372 Search PubMed.
- C. Hou, Z. Qi and H. Zhu, Colloids Surf., B, 2015, 128, 544–551 CrossRef PubMed.
- Y. Tian, R. Guo, Y. Jiao, Y. Sun, S. Shen, Y. Wang, D. Lu, X. Jiang and W. Yang, Nanoscale Horiz., 2016, 1, 480–487 RSC.
- Z. Zou, D. He, X. He, K. Wang, X. Yang, Z. Qing and Q. Zhou, Langmuir, 2013, 29, 12804–12810 CrossRef PubMed.
- T. Li, X. Shen, Y. Geng, Z. Chen, L. Li, S. Li, H. Yang, C. Wu, H. Zeng and Y. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 13748–13758 Search PubMed.
- B. Tian, S. Liu, S. Wu, W. Lu, D. Wang, L. Jin, B. Hu, K. Li, Z. Wang and Z. Quan, Colloids Surf., B, 2017, 154, 287–296 CrossRef PubMed.
- M. Massaro, R. Amorati, G. Cavallaro, S. Guernelli, G. Lazzara, S. Milioto, R. Noto, P. Poma and S. Riela, Colloids Surf., B, 2016, 140, 505–513 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03163k |
| ‡ These authors contribute equally in this work. |
| This journal is © The Royal Society of Chemistry 2018 |