
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,3-Phenylene-bridged naphthalene wheels synthesized by one-pot Suzuki–Miyaura coupling and the complex of the hexamer with C60†
Peifeng Mei,
Akinobu Matsumoto,
Hironobu Hayashi ,
Mitsuharu Suzuki,
Naoki Aratani* and
Hiroko Yamada*
,
Mitsuharu Suzuki,
Naoki Aratani* and
Hiroko Yamada*
Graduate School of Science and Technology, Nara Institute of Science and Technology (NAIST), Takayama-cho, Ikoma, Nara 630-0192, Japan. E-mail: aratani@ms.naist.jp; hyamada@ms.naist.jp
First published on 7th June 2018
Abstract
A large 1,3-phenylene-bridged hexameric naphthalene wheel N6 and a heptameric wheel N7 were synthesized simply by Suzuki–Miyaura coupling via one-pot reaction from monomers. We could control the distribution of N6 and N7 via the reaction conditions. The hexameric wheel structure was revealed by X-ray diffraction analysis. The wheel N6 exhibited C60 encapsulation ability in the solid state, which was also confirmed by single crystal X-ray analysis.
Introduction
Bottom-up “designed organic synthesis” of benzene-based nano-carbon materials has been extensively attempted, since such synthesis will allow for tailored fine-tuning of their structures, properties, and functions.1 These attempts are also important for cyclic aromatic molecules.2–4 Such cyclic systems are expected to act as a host molecule and to display multiple electronic interactions with guest molecules that are not shared with normal linear compounds. Pd-catalyzed cross coupling reactions have been demonstrated to be quite powerful in the synthesis of a variety of oligomers of polycyclic aromatic hydrocarbons (PAHs). Despite this progress, 1,3-phenylene bridged cyclic PAH hexamers that can be regarded as benchmark wheels in terms of their simple hexagonal structure have been rarely synthesized.5–8 Cyclic heptamers and higher analogues are scarcer. Schlüter et al. made a cyclotetraicosaphenylene by using a repetitive Suzuki–Miyaura cross coupling protocol.5 The first [6]cyclo-m-phenylene was prepared by Staab et al.6 and recently a series of [n]cyclo-m-phenylenes were synthesized by a one-pot Ni-mediated Yamamoto coupling.7 The cyclic porphyrin hexamer is interesting not only as an artificial light-harvesting photosynthetic antenna but also as a shape-persistent organic molecule.8 The conformationally rather restricted cyclic structure is amenable for studies on the structure–optical property relationship but likely poses a synthetic challenge. In this paper, we report the quick synthesis of the first 1,3-phenylene bridged hexameric and heptameric naphthalene wheels via one-step cross-coupling at multiple sites starting from simple monomers, and the complexization of hexamer N6 with C60. Encapsulation of C60 in the wheel N6 was examined and the structure was confirmed by single-crystal X-ray analysis.Results and discussion
For construction of cyclic frameworks, our synthetic strategy lies in using Suzuki–Miyaura cross-coupling reaction. Namely, 1,3-diborylbenzene and 1,4-dibromonaphthalene in DMF/toluene were treated with 10 mol% of Pd catalyst at 80 °C under an inert atmosphere, giving a 1,3-phenylene-bridged cyclic naphthalene hexamer N6 in 6% yield without any detection of N7 after repeated separations over a preparative GPC column and a silica gel column (Scheme 1). | ||
| Scheme 1 Synthesis of naphthalene wheels N6 and N7. Reaction conditions: (a) Pd cat., Cs2CO3, toluene/DMF, 80 °C, 6% (N6) and 0% (N7). (b) Pd cat., CsF, 18-crown-6, THF/H2O, r.t. 4% (N6) and 13% (N7). | ||
High-resolution matrix assisted laser desorption/ionization time-of-flight (HR-MALDI-TOF) mass spectrum of N6 displays the parent ion peaks at m/z 1212.4686 (calcd for C96H60 = 1212.4690 [M]+). Although the 1H NMR spectrum of N6 in CDCl3 at room temperature was very broad, that in C2D2Cl4 at 60 °C became sharper and simple, exhibiting only a single set of signals that consists of two singlet peaks at 7.67 and 7.56 ppm due to the Hc and Hd, respectively, and signals due to Ha and Hb in the range of 7.66 ppm and He and Hf at 8.10 and 7.47 ppm, respectively. These data indicate that the wheel N6 takes a C6 symmetric structure in solution at 60 °C.
Definitive structural assignment of N6 was accomplished through a single crystal X-ray diffraction analysis, which unveiled a distinct hexagonal conformation (Fig. 1a).‡ The phenylene-bridges are on the co-plane, suggesting less structural strain. The dihedral angles between the naphthalene mean-planes and phenylene groups are in the range of 58–64°. Interestingly, the hexagons are interconnected through phenylene C–H and naphthalene π-plane interactions in the crystal, forming an infinite one-dimensional tubular packing structure along the c-axis (Fig. 1b).
 | ||
| Fig. 1 (a) Single crystal X-ray structure of N6 and (b) packing structure of N6. Thermal ellipsoids are scaled at 50% probability. Solvent molecules are omitted for clarity. | ||
Fig. 2 shows the UV-vis absorption and fluorescence spectra of N6 in CH2Cl2. Cyclic hexamer N6 shows a single absorption band at 306 nm and a blue emission at 383 nm. This broad single band can be qualitatively understood in terms of the weak π-conjugation and the exciton coupling,9 as similarly to previously reported 1,3-phenylene naphthalene dimer.10 Given the rigid hexagonal conformation for the wheel, J-type exciton coupling of transition dipoles is effective. The interacting components lead to red-shifted absorption band compared with naphthalene monomer (275 nm in CH2Cl2). The steady-state fluorescence spectrum in toluene is also displayed in Fig. 2.
 | ||
| Fig. 2 UV-vis absorption (solid line) and fluorescence (dotted line) spectra of N6 and N7 in CH2Cl2. Fluorescence spectra were taken for excitation (λex = 306 nm for N6 and 308 nm for N7) with the absorbance adjusted to 0.1. | ||
To further understand the electronic features of N6, the density functional theory (DFT) and the time-dependent (TD)-DFT calculations both at the B3LYP/6-31G(d) level using the Gaussian 09 software package were carried out (Fig. 3).11 It is revealed that the frontier orbitals are degenerated. The coefficients of HOMO and LUMO of N6 localize on the six naphthalene units. The main absorption band of N6 at 306 nm predominantly comprises the S2 and S3 transitions (oscillator strength, f = 1.03 and f = 1.02), whereas the long wavelength S1 absorption is forbidden (f = 0.00). The transition energies and oscillator strengths simulated by TD-DFT calculations showed a good agreement with the observed absorption spectrum of N6.
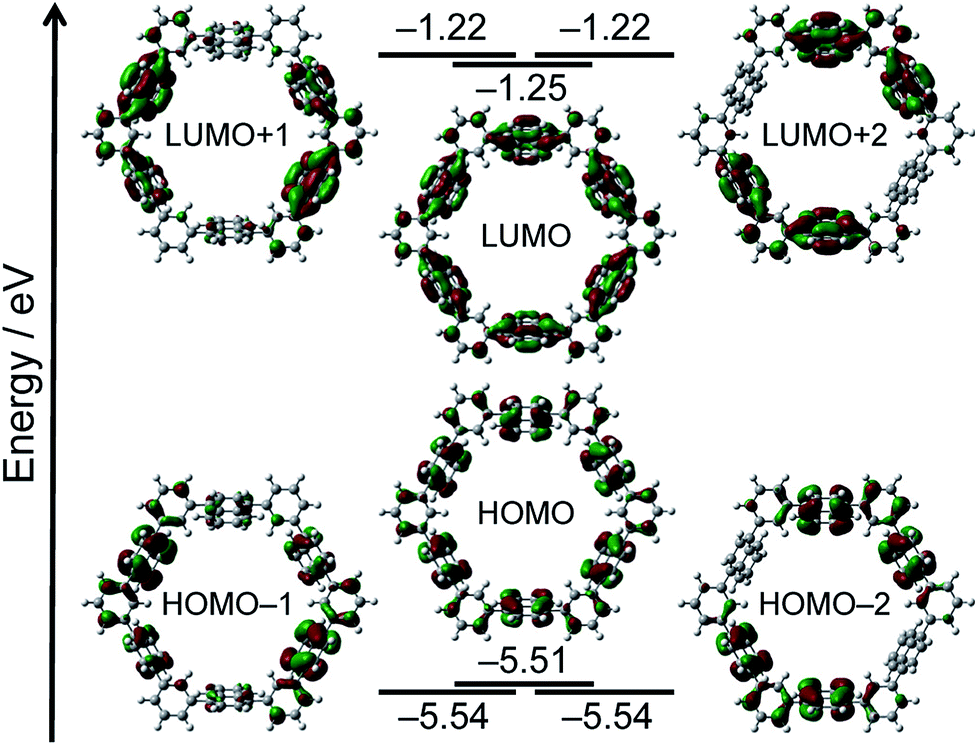 | ||
| Fig. 3 MO diagrams of N6 calculated at the B3LYP/6-31G(d) level. | ||
In the next step, the encapsulation of C60 into N6 was examined, since the diameter of the interior cavity of N6 is ca. 15 Å, being possibly fit to the diameter of C60.12 Unfortunately, however, the addition of C60 into a toluene solution of N6 did not change the absorption spectrum probably because of weak interactions between two components. The encapsulation was also not confirmed by NMR spectroscopy. Thus we attempted to make co-crystals of N6 with C60. The host–guest binding structure was unambiguously confirmed by the single-crystal X-ray diffraction analysis (Fig. 4).‡ Higher concentration on the crystallization process could give the encapsulation complex. In the solid-state, the naphthalene units of C60@N6 take a similar structure to those of N6 with respects to dihedral angles of phenylene toward naphthalene (51–72°), and an inside space (15 Å diameter). The positions of C60 are disordered at two parts (66:34). As shown in Fig. 4, a C60 molecule is nicely captured within the cavity. Closer inspection of the crystal structure reveals that the naphthalene planes are protruding their planar face toward the interior space, which interacts with C60. Interestingly, the C60 molecules in the crystal are aligned with the aid of N6 agent to form a 1D structure along the a-axis (Fig. 4b).
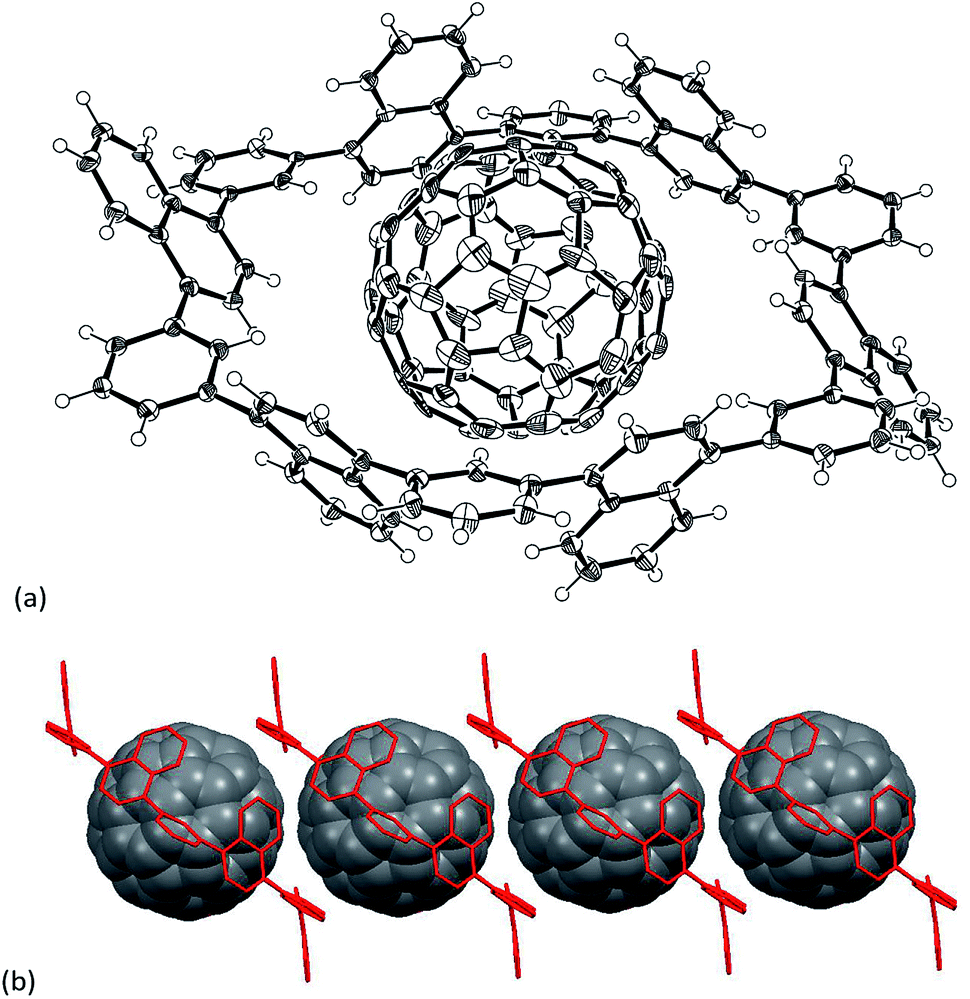 | ||
| Fig. 4 (a) Single crystal X-ray structure of C60@N6 and (b) packing structure of C60@N6. Thermal ellipsoids are scaled at 50% probability. Solvent molecules are omitted for clarity. | ||
During this research, Yokozawa and co-workers reported an efficient cyclization reaction of o- and m-alternate polyphenylenes.13 In order to check the effect of the reaction conditions, we applied Yokozawa's conditions on our cyclic naphthalene synthesis. Interestingly, the distribution of the wheel size was shifted to the larger size, and we successfully isolated N6 and N7 in 4% and 13% yields, respectively (Scheme 1). HR-MALDI-TOF mass spectrum of N7 displayed the parent ion peaks at m/z 1414.5469 (calcd for C112H70 = 1414.5472 [M]+). To observe a relatively clear 1H NMR spectrum in C2D2Cl4, it was needed to measure the spectrum at higher temperature than that for N6. The 1H NMR spectrum of N7 in C2D2Cl4 at 120 °C was simple, exhibiting only a single set of signals that consists of two singlet peaks at 7.66 and 7.59 ppm due to the Hd and Hc, respectively, at 7.75 ppm due to Ha and signals due to Hb in the range of 7.61–7.66 ppm and He and Hf at 8.13 and 7.40 ppm, respectively.
UV-vis absorption and fluorescence spectra of N7 in CH2Cl2 are also shown in Fig. 2. Cyclic heptamer N7 exhibits a slightly red-shifted absorption band at 308 nm and a blue-shifted emission at 382 nm. These are presumably because the conformational deformation from N6 to N7 makes the forbidden S1 transition to be just a little allowed.
Conclusions
In summary, 1,3-phenylene-linked cyclic naphthalene hexamer and heptamer were simply constructed by Suzuki–Miyaura cross-coupling reaction via a one-pot route and the hexagonal structure of N6 was confirmed by X-ray structural analysis. The hexameric wheel N6 formed co-crystal with C60 and acted as an alignment agent in the solid state. The host–guest chemistry of N7 with higher fullerenes, template synthesis of N6 and N7 and the measurement of their conductivities are currently being explored in our laboratory.Experimental
Materials and methods
1H NMR (400 MHz and 600 MHz) and 13C NMR (151 MHz) spectra were recorded with a JEOL JNM-ECX 400, a JEOL JNM-ECP 400 and a JEOL JNM-ECA 600 spectrometers by using tetramethylsilane as an internal standard. The HR-MALDI-TOF mass spectra were measured by a Bruker Autoflex II spectrometer using positive ion mode.UV/Vis absorption spectra were measured with a JASCO UV/Vis/NIR spectrophotometer V-570.
TLC and gravity column chromatography were performed on Art. 5554 (Merck KGaA) plates and silica gel 60N (Kanto Chemical), respectively. All other solvents and chemicals were reagent-grade quality, obtained commercially, and used without further purification. For spectral measurements, spectral-grade solvents were purchased from Nacalai Tesque.
All DFT calculations were performed with a Gaussian 09 program package. The geometries were fully optimized at the Becke's three-parameter hybrid functional combined with the Lee–Yang–Parr correlation functional abbreviated as the B3LYP level of density functional theory. The 6-31G(d) bases set implemented was used for structure optimizations and frequency analyses.
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 = 3:1). After being purified by a preparative GPC, 4.0 mg of N6 was gained in 6% yield as a white solid. 1H NMR (C2D2Cl4, 600 MHz, ppm, 60 °C) δ 7.47 (q, J = 9.6 Hz, 12H), 7.56 (s, 12H), 7.66–7.67 (m, 24H) and 8.10 (q, J = 9.6 Hz, 12H). 13C NMR (C2D2Cl4, 151 MHz, ppm, 60 °C) δ 126.14, 126.60, 126.71, 128.31, 129.09, 132.05, 132.22, 139.83 and 141.01. UV-vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 306(8.7 × 104) nm. Fluorescence (CH2Cl2, λex = 306 nm): λmax = 383 nm.:CH2Cl2 = 3:1). After being purified by GPC, 7.0 mg of N7 was gained in 13% yield and 2.0 mg of N6 in 4% yield. 1H NMR (C2D2Cl4, 400 MHz, ppm, 120 °C) δ 7.49 (m, 14H), 7.59 (s, 7H), 7.61 (d, 7H), 7.66 (d, 21H), 7.75 (d, 7H) and 8.13 (m, 14H). 13C NMR (C2D2Cl4, 151 MHz, ppm, 120 °C) δ 120.46, 125.92, 125.93, 126.50, 126.66, 128.24, 129.28, 131.60, 132.30, 139.85 and 141.08. UV-vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 308(1.1 × 105) nm. Fluorescence (CH2Cl2, λex = 308 nm): λmax = 382 nm.
:CH2Cl2 = 3:1). After being purified by a preparative GPC, 4.0 mg of N6 was gained in 6% yield as a white solid. 1H NMR (C2D2Cl4, 600 MHz, ppm, 60 °C) δ 7.47 (q, J = 9.6 Hz, 12H), 7.56 (s, 12H), 7.66–7.67 (m, 24H) and 8.10 (q, J = 9.6 Hz, 12H). 13C NMR (C2D2Cl4, 151 MHz, ppm, 60 °C) δ 126.14, 126.60, 126.71, 128.31, 129.09, 132.05, 132.22, 139.83 and 141.01. UV-vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 306(8.7 × 104) nm. Fluorescence (CH2Cl2, λex = 306 nm): λmax = 383 nm.:CH2Cl2 = 3:1). After being purified by GPC, 7.0 mg of N7 was gained in 13% yield and 2.0 mg of N6 in 4% yield. 1H NMR (C2D2Cl4, 400 MHz, ppm, 120 °C) δ 7.49 (m, 14H), 7.59 (s, 7H), 7.61 (d, 7H), 7.66 (d, 21H), 7.75 (d, 7H) and 8.13 (m, 14H). 13C NMR (C2D2Cl4, 151 MHz, ppm, 120 °C) δ 120.46, 125.92, 125.93, 126.50, 126.66, 128.24, 129.28, 131.60, 132.30, 139.85 and 141.08. UV-vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 308(1.1 × 105) nm. Fluorescence (CH2Cl2, λex = 308 nm): λmax = 382 nm.Crystallography
X-ray crystallographic data were recorded at 90 K on a Bruker APEX II X-ray diffractometer equipped with a large area CCD detector by using graphite monochromated Mo-Kα radiation.The structure was solved by using direct methods (SHELXT program).14 Structure refinements were carried out by using SHELXL-2014/7 program.15
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by CREST JST (No. JPMJCR15F1) and by JSPS KAKENHI Grant Numbers JP16H02286 and JP26105004 to H.·Y., JP16K17949 to M. S. and JP17H03042 to N. A. A. M. appreciates JSPS for Research Fellowships for Young Scientists. We also thank Ms Y. Nishikawa and Mr F. Asanoma, NAIST for the mass and NMR spectroscopy measurements, respectively.Notes and references
- Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef.
- K. Yazaki, L. Catti and M. Yoshizawa, Chem. Commun., 2018, 54, 3195–3206 RSC.
- D. Lorbach, A. Keerthi, T. M. Figueira-Duarte, M. Baumgarten, M. Wagner and K. Müllen, Angew. Chem., Int. Ed., 2016, 55, 418–421 CrossRef PubMed.
- Y. Yamamoto, K. Wakamatsu, T. Iwanaga, H. Sato and S. Toyota, Chem.–Asian J., 2016, 11, 1370–1375 CrossRef PubMed.
- V. Hensel, K. Lützow, J. Jakob, K. Gessler, W. Saenger and A. D. Schlüter, Angew. Chem., Int. Ed. Engl., 1997, 36, 2654–2656 CrossRef; V. Hensel and A. D. Schlüter, Chem.–Eur. J., 1999, 5, 421–429 CrossRef.
- H. A. Staab and F. Binnig, Tetrahedron Lett., 1964, 5, 319–321 CrossRef.
- J. Y. Xue, K. Ikemoto, N. Takahashi, T. Izumi, H. Taka, H. Kita, S. Sato and H. Isobe, J. Org. Chem., 2014, 79, 9735–9739 CrossRef PubMed and the references are therein..
- H.-W. Jiang, S. Ham, N. Aratani, D. Kim and A. Osuka, Chem.–Eur. J., 2013, 19, 13328–13336 CrossRef PubMed; Y. Nakamura, N. Aratani and A. Osuka, Chem. Soc. Rev., 2007, 36, 831–845 RSC.
- M. Kasha, Rad. Res., 1963, 20, 55–70 CrossRef PubMed; A. Osuka and K. Maruyama, J. Am. Chem. Soc., 1988, 110, 4454–4456 CrossRef.
- G. F. Woods, F. T. Reed, T. E. Arthur and H. Ezekiel, J. Am. Chem. Soc., 1951, 73, 3854–3856 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- B. W. Smith, M. Monthioux and D. E. Luzzi, Nature, 1998, 396, 323 CrossRef; S. Bandow, M. Takizawa, H. Kato, T. Okazaki, H. Shinohara and S. Iijima, Chem. Phys. Lett., 2001, 347, 23 CrossRef.
- H. Sugita, M. Nojima, Y. Ohta and T. Yokozawa, Chem. Commun., 2017, 53, 396–399 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 CrossRef PubMed.
- Squeeze-Platon and A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht, The Netherlands, 2005 CrossRef; P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray crystal analysis and NMR and mass spectra. CCDC 1838834 (N6) and 1838835 (C60@N6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra03601b |
‡ Crystallographic data for N6: C96H60·O, Mw = 1229.44, monoclinic, space group P21/c (#14), a = 11.962(6), b = 32.625(16), c = 15.124(8) Å, β = 93.619(8)°, V = 5890(5) Å3, T = 90(2) K, Z = 2, reflections measured 29202, 9464 unique. The final R1 was 0.1193 (>2σ(I)), and the final wR on F2 was 0.3390 (all data), GOF = 1.109. Crystallographic data for C60@N6: C96H60·C60·4(C6H5Cl), Mw = 2384.24, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2), a = 11.110(5), b = 14.775(6), c = 17.361(7) Å, α = 94.214(7)°, β = 94.286(7)°, γ = 101.721(7)°, V = 2771(2) Å3, T = 90(2) K, Z = 1, reflections measured 15703, 10790 unique. The final R1 was 0.0691 (>2σ(I)), and the final wR on F2 was 0.2025 (all data), GOF = 1.036. The contributions to the scattering arising from the presence of disordered solvents in the crystals of N6 were removed by use of the utility SQUEEZE in the PLATON software package.16 (#2), a = 11.110(5), b = 14.775(6), c = 17.361(7) Å, α = 94.214(7)°, β = 94.286(7)°, γ = 101.721(7)°, V = 2771(2) Å3, T = 90(2) K, Z = 1, reflections measured 15703, 10790 unique. The final R1 was 0.0691 (>2σ(I)), and the final wR on F2 was 0.2025 (all data), GOF = 1.036. The contributions to the scattering arising from the presence of disordered solvents in the crystals of N6 were removed by use of the utility SQUEEZE in the PLATON software package.16 |
| This journal is © The Royal Society of Chemistry 2018 |