
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogen bonding for the design of inhibitors by targeting the S1 pocket of serine proteases†
Longguang
Jiang‡
 a,
Xu
Zhang‡
b,
Yang
Zhou
a,
Yayu
Chen
a,
Zhipu
Luo
c,
Jinyu
Li
a,
Cai
Yuan
d and
Mingdong
Huang
*a
a,
Xu
Zhang‡
b,
Yang
Zhou
a,
Yayu
Chen
a,
Zhipu
Luo
c,
Jinyu
Li
a,
Cai
Yuan
d and
Mingdong
Huang
*a
aCollege of Chemistry, Fuzhou University, Fuzhou 350116, China. E-mail: jianglg@fzu.edu.cn; HMD_lab@fzu.edu.cn
bCenter for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China
cSynchrotron Radiation Research Section, NCI, Argonne National Laboratory, Argonne, Illinois 60439, USA
dCollege of Biological Science and Engineering, Fuzhou University, Fuzhou 350116, China
First published on 6th August 2018
Abstract
Halogen bonding (or X bonding) has attracted increasing interest due to its significant role in molecular recognition in biological systems. Trypsin-like serine proteases have many physiological and pathophysiological functions. There is therefore extensive interest in generating specific inhibitors for pharmacological intervention in their enzymatic activity. We study here if it is possible to use halogenated compounds as the P1 group to bind to the S1 specificity pocket of trypsin-like serine proteases to avoid the low bioavailability of the amidine or guanidine P1 group that is typically used in many inhibitors. We used 4-chlorobenzylamine (ClBA), 4-bromobenzylamine (BrBA) and 4-iodobenzylamine (IBA) as probes to test their binding modes to a trypsin-like serine protease, urokinase-type plasminogen activator (uPA), which has been recognized as a marker for breast cancer and an important target for inhibitor development. The results showed that these compounds inhibited uPA with stronger efficacies compared with their non-halogenated analogues. We also determined the high-resolution crystal structures of uPA in complex with BrBA and IBA, respectively. The structures revealed that BrBA bound to the S1 pocket of uPA via halogen bonds, but IBA did not make halogen bonds with uPA, demonstrating that the iodine may not be the best choice as a target moiety for serine proteases. These results advocate halogen bonding, especially bromine bonding, as an efficient strategy for the future design of novel inhibitors against trypsin-like serine proteases to provide strong potency and promote bioavailability.
1. Introduction
Halogen bonding is the non-covalent interaction between an electron-rich donor (D) and a halogen atom (X) that is linked to an electron-withdrawing acceptor (A).1,2 Halogen bonds are strong, specific, and directional interactions. All four halogens are found to support halogen bonding and generally follow the trend: F ≪ Cl < Br < I, with iodine normally forming the strongest interaction.3–5 A typical halogen bond should have at least these features: (1) the interatomic distance between X and the appropriate nucleophilic atom of A needs to be less than the sum of the van der Waals radii; (2) the length of the D–X covalent bond should be longer than the unbonded D–X; (3) the angle of D⋯X-A should be close to 180°.1 In general, halogen bonds are energetically and geometrically comparable to hydrogen bonding.6 Halogen bond strengths depend on structural and chemical environments and can be quite strong (up to 43 kcal mol−1).7Halogen bonding has attracted increasing interest in the molecular recognition in biological system. Halogen bonding has been shown to be a stabilizing factor and a conformational determinant in protein-ligand and DNA structures.8 About the half of the small weight molecules used in high-throughput drug screening contain halogen atoms. Halogen atom has basicity weaker than arginine/amine groups that typically used in serine protease inhibitors, and likely to have better bioavailability. The prevalence of halogenated compounds has made them widely used as inhibitors against biomedical important targets.9–12 In this work, we consider the possibility to include halogen bonds in the design of inhibitors to inhibit proteolytic activity of serine proteases. Serine proteases have many physiological and pathophysiological functions.13–15 There is extensive interest in generating specific inhibitors for pharmacological intervention with their enzymatic activity. Moreover, serine proteases are classical subjects for studies of catalytic and inhibitory mechanisms of enzymes. Serine proteases share a common mechanism of catalysis that relies upon the coordinate action of three catalytic residues (Ser195, His57 and Asp102, in chymotrypsinogen numbering).16–18 Trypsin-like serine proteases cleave peptide bonds following a positively charged amino acid (lysine or arginine). This specificity is driven by the presence of a primary specificity pocket (S1 pocket) of the enzyme that accommodates the lysine or arginine residue of the substrates. The S1 pocket is deep and has a negatively charged residue (Asp189) at its base. The majority of trypsin-like proteases inhibitors contain argininal or its mimetics (guanidine or amidine groups) and utilize this binding site for potencies.19
One interesting member of the trypsin family of serine proteases is urokinase-type plasminogen activator (uPA, EC.3.4.21.73), which catalyses the conversion of the zymogen plasminogen into the active protease plasmin through cleavage of plasminogen's Arg15-Val16 bond.20 Abnormal expression of uPA is responsible for tissue damage in several pathological conditions, including rheumatoid arthritis, allergic vasculitis, and in particular, is a key factor for the invasive capacity of malignant tumors.21 Therefore, uPA is a potential therapeutic target. Many small molecular organic inhibitors of uPA use a guanidine or benzamidine-like moieties to interact with the S1 pocket (thus named P1 group). A major problem in these amidine- or guanidine-based small molecular inhibitors for uPA or serine proteases is the low bioavailability and poor pharmacokinetic profile due to the presence of positive charges on these amidine or guanidine groups at physiological pH.22–24 Thus, intensive research efforts have been dedicated to find non-amidine/guanidine inhibitors that maintain interaction to the S1 specificity pocket of uPA.25–27
Fragment-based drug discovery is a method used for finding lead compounds as part of the drug discovery process. It is based on identifying small chemical fragments, which may have weak potency (>100 μM) but are efficient binders relative to their size and may therefore represent suitable starting points for lead compounds.28 Is it possible to use small molecular compounds with the halogen atom to replace the amidine/guanidine group as the P1 group to target to the S1 pocket of trypsin-like serine proteases by halogen bonding? In this study, we select 4-chlorobenzylamine (ClBA), 4-bromobenzylamine (BrBA) and 4-iodobenzylamine (IBA) to test the feasibility of this idea. These compounds contain one halogen atom (chlorine, bromine or iodine) at one end of benzene ring, which has higher polarization of the electrostatic potential at its iodine atom surfaces compared to Cl and Br atom (Fig. 1), and two lighter atoms (–CH2NH2) at another end, so that the orientation of the inhibitor can be easily distinguished by X-ray crystallography. We determined the crystal structures of BrBA and IBA in complex with uPA, respectively. These structures showed that BrBA bound to the S1 pocket of uPA with Br atom pointing to Asp189, forming the halogen bonds between the halogen atom and the Asp189 of uPA. Unexpectedly, IBA did not make the halogen bond with uPA but formed the hydrogen bonds by the amino group. These results advocate that halogen bonding, especially bromine bonding, as an efficient strategy for the future design of novel inhibitors against trypsin-like serine proteases to provide strong potency and promote bioavailability.
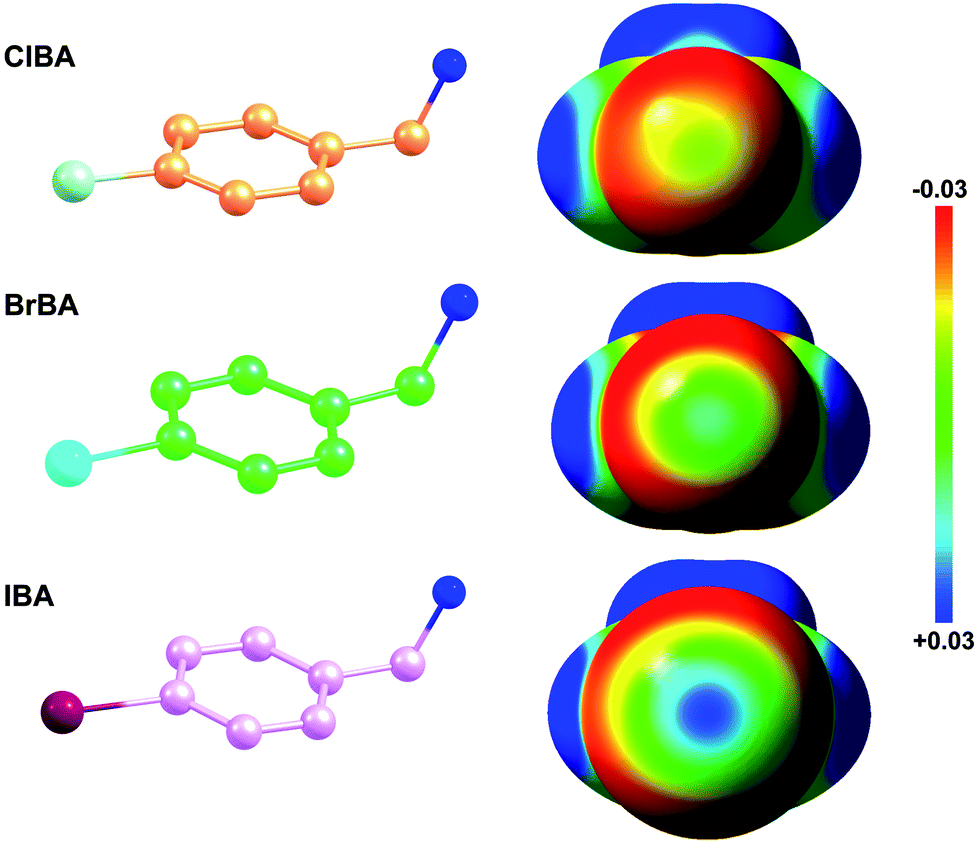 | ||
| Fig. 1 Electrostatic potential mapped on the surface of molecular electron density of ClBA, BrBA and IBA. The configurations of ClBA, BrBA and IBA were optimized at B3LYP/6-31G* level using GAMESS package. The electrostatic potential maps were visualized by Avogadro (0.03 (red) to 0.03 (blue) au). | ||
2. Materials and methods
2.1 Materials
4-Chlorobenzylamine (ClBA, CAS number: 104-86-9), 4-bromobenzylamine (BrBA, CAS number: 3959-07-7) 4-iodobenzylamine (IBA, CAS number: 39959-59-6) and 4-hydroxybenzylamine (CAS number: 696-60-6) were purchased from Sigma-Aldrich (St. Louis, MO, USA), the chromogenic substrate S-2444 (BIOPHEN CS-61(44), pyroGlu-Gly-Arg-pNA·HCl) was purchased from DiaPharma Group, Inc (Ohio, USA).2.2 Expression and purification of recombinant uPA
Expression and purification of recombinant uPA serine protease domain was described previously by Zhao et al.29 Briefly, uPA16-244 was secreted from a stably transfected pichia pastoris strain (X-33) after induction with methanol. A purified product was obtained after cation exchange chromatography (SP sepharose Fast Flow from, GE Healthcare) followed by gel filtration chromatography using a Superdex 75 HR 10/300 column (GE Healthcare) equilibrated with 20 mM phosphate buffer (pH 6.5) containing 150 mM NaCl. The protein was eluted as a single peak under these conditions with a retention volume of approximately 13.6 ml, corresponding to its molecular weight. The protein was dialyzed in 20 mM potassium phosphate (pH 6.5) overnight and concentrated to 10 mg ml−1 using stirred ultra-filtration cells (Millipore and Amicon Bioseparations, Model-5124).2.3 Determination of the inhibition constant (Ki) for the inhibition of uPA by various compounds
For routine determinations of Ki values for the inhibition of uPA under steady-state inhibition conditions, a fixed concentration of uPA (2 nM) was pre-incubated in 200 μl of HEPES-buffered saline with 0.1% bovine serum albumin at 37 °C with various concentrations of compounds for 15 min before the addition of the chromogenic substrate S-2444 at concentrations approximately equal to the KM. The initial reaction velocities were monitored at an absorbance of 405 nm. The inhibition constants (Ki) were subsequently determined from the nonlinear regression analyses of plots for Vi/V0versus [I]0 using eqn (1) and derived under an assumption of competitive inhibition:| Vi/Vo = (Ki × (KM + [S]0))/((Ki × [S]0) + (KM × (Ki + [I]0))) | (1) |
2.4 Crystallization and data collection of uPA:halogenated compound complexes
The crystallization trials were carried out using the sitting drop vapor diffusion method. The crystals were obtained by equilibrating against a reservoir solution containing 50 mM sodium citrate (pH 4.6) and 2.0 M ammonium sulfate supplemented with 5% PEG400 at room temperature. The crystals appeared after approximately three days. For uPA:BrBA complex at pH4.6, the crystals of uPA were soaked for 72 hours in crystallization mother liquor containing 1 mM the halogenated compounds. For uPA:halogenated compound complexes at pH7.4, the crystals were soaked for one week in new soaking buffer (40% PEG4000, 100 mM Tris–HCl pH 7.4) containing 1 mM halogenated compounds. Prior to X-ray data collection, the crystals were soaked briefly in a cryoprotectant solution containing 20% glycerol. X-ray diffraction data of the crystals were collected at the BL17U beamline, Shanghai Synchrotron Radiation Facility (SSRF). The diffraction data was indexed and integrated by HKL2000 program package.30 The data set of uPA:IBA was processed both with and without merging the Bijvoet pairs. The anomalous data were used only to confirm the iodine atom site by the anomalous difference map and all diffraction data and refinement statistics are based on the merged “native” data. The data collection statistics are presented in Table S1.†2.5 Crystal structure determination and refinement
The crystal structure of the different complexes were solved by molecular replacement31 using the uPA structure (PDB code: 4DVA)32 as the search model. The electron density for the compound was clearly visible in the uPA active sites and was modelled based on the Fo − Fc difference map. The orientation of IBA was determined based on the anomalous scattering of iodine atom at 0.979 Å. The structures was refined by ccp4 program package31 and manually adjusted by the molecular graphics program COOT33 iteratively until convergence of the refinement. Solvent molecules were added using a Fo − Fc Fourier difference map at 2.5σ in the final refinement step. Statistics of data collection and final model refinement are summarized in Table S1.† The final structures were analysed by software Pymol.342.6 Binding free energy calculations
The free energy of binding between each ligands and uPA was derived using the molecular mechanics/Poisson–Boltzmann solvent accessible surface area (MM/PBSA) approach.35,36 The binding free energies (ΔGbind) were computed with following equations:| ΔGbind = ΔHbind − TΔSbind | (2) |
| ΔHbind = Egas + ΔGbind | (3) |
| Egas = Eele + Evdw | (4) |
| ΔGsol = ΔGpb + ΔGnonpolar | (5) |
| ΔGnonpolar = γ(SASA) + β | (6) |
The sum of the molecular mechanical gas-phase energies, Egas is a standard force field energy, including the electrostatic potential (Eele) and the van der Waals potential (EvdW). The solvation free energy (ΔGsol) was computed as the sum of a polar solvation free energy (ΔGpb) and a nonpolar solvation free energy (ΔGnonpolar). ΔGnonpolar was calculated from eqn (6), where SASA is the solvent-accessible surface area estimated using gsas implemented in GROMACS 4.5 code,37 with a solvent probe radius of 1.4 Å. γ and β are empirical constants set to 0.00542 kcal mol−1 Å−2 and 0.92 kcal mol−1, respectively. The conformational entropy (ΔSbind) was not considered in this work due to high computational cost and low prediction accuracy. The ΔGbind and each energy components for ligands bound to uPA, determined by MM/PBSA calculations were given in Table S2.† The configurations of halogenated compounds were optimized at B3LYP/6-31G* level using GAMESS package.38 The electrostatic potential maps were visualized by Avogadro.39
3. Results
3.1 Inhibition of the enzyme activity of human uPA by the halogenated compounds
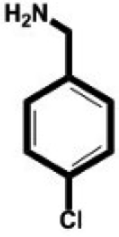
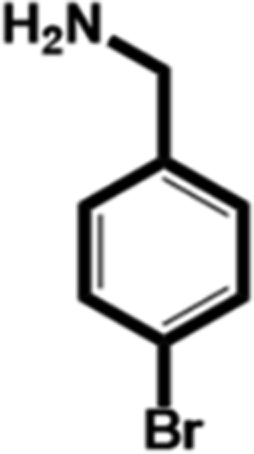
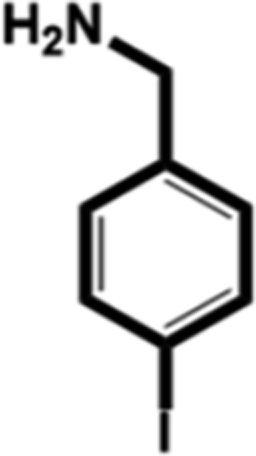
In this study, we select 4-chlorobenzylamine (ClBA), 4-bromobenzylamine (BrBA) and 4-iodobenzylamine (IBA) to test whether the use of small molecular compounds containing halogen atom can function as the P1 group to target to the S1 pocket of trypsin-like serine proteases by halogen bonding. We measured their Ki values for competitive inhibition of human uPA at 37 °C by chromogenic assays. The results indicated that these halogenated compounds indeed inhibited the enzyme activity of human uPA to an extent comparable to small molecules targeting at the S1 pocket,40 and the inhibition of BrBA (1.28 mM) is slightly stronger than that of IBA (1.38 mM), the inhibition of ClBA (9.15 mM) is weakest among them (Table 1). It is understandable that the inhibitory capabilities were not very strong because these compounds only targeted to the S1 pocket of the protease.| Compounds | Chemical structures | K i for inhibition (mM) |
|---|---|---|
| 4-Chlorobenzylamine (ClBA) |
|
9.15 ± 0.02 (3) |
| 4-Bromobenzylamine (BrBA) |
|
1.28 ± 0.06 (3) |
| 4-Iodobenzylamine (IBA) |
|
1.38 ± 0.09 (3) |
| 4-Hydroxybenzylamine |
|
>5 mM |
| 4-(Aminomethyl-phenyl)-methanol |
|
13.9 mM (ref. 40) |
3.2 BrBA binds to Asp189 of uPA at the specific pocket through halogen bonds
We determined the crystal structure of BrBA in complex with uPA at low pH (pH 4.6) to high resolution (1.17 Å), which helps us to identify the orientation of BrBA. The binding of the small molecule BrBA did not perturb the structural conformation of uPA, as shown by the low RMSD (0.13 Å) when compared to the unbound uPA. The position of BrBA was located at the major specificity pocket of uPA (S1 pocket) with its position well supported by the electron density maps (Fig. 2A). The bromine atom of BrBA buried deeply inside the S1 pocket and formed two halogen bonds (2.9 Å and 2.9 Å) with the carboxyl side chain of Asp189 at the bottom of the S1 pocket, respectively (Fig. 2B, Table S3, ESI†). Here we measured only the distances between Asp189 oxygen atom and the bromine atom because our crystal structures did not have enough resolution to observe the positions of protons in between. The bromine atom of BrBA has also short distances (3.0 Å) to the carbonyl group and hydroxyl group of Ser190 (Fig. 2B). Furthermore, the phenyl ring of BrBA formed van der Waals interactions with the backbone atoms of residues 190–192 and 215–217.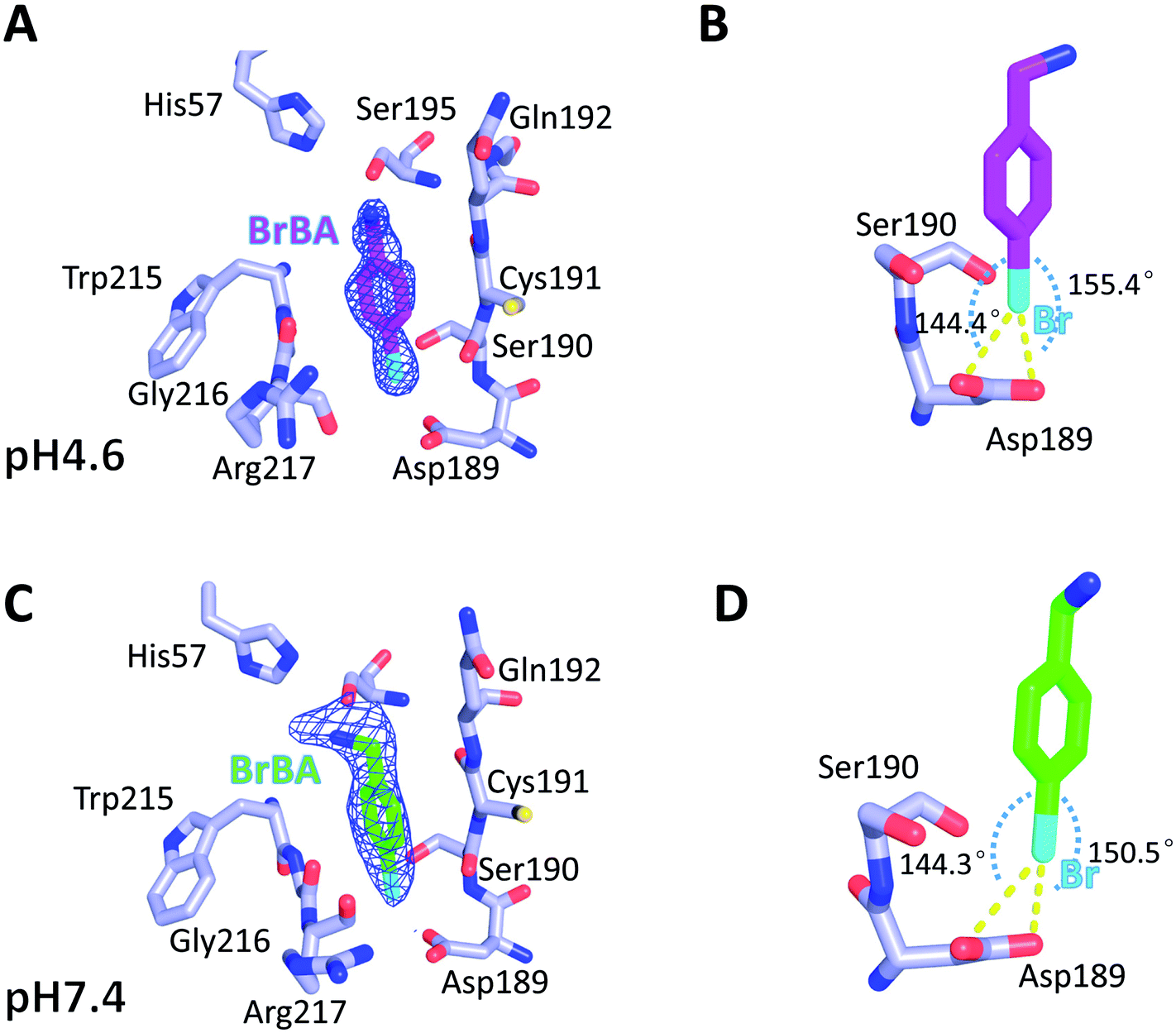 | ||
| Fig. 2 4-Bromobenzylamine (BrBA, shown as magenta stick) bound to the S1 pocket of uPA at pH 4.6 and pH 7.4. (A) At pH 4.6, BrBA located at the major specificity pocket of uPA (S1 pocket) with its position well supported by the electron density maps. (B) At pH 4.6, the bromine atom (cyan) of BrBA interacts to the Asp189 of the S1 pocket and formed two halogen bonds with the carboxyl side chain of Asp189. The angles of D⋯Br–A are 144.4° and 155.4°, respectively. (C) The higher pH (7.4) structure supports the results of low pH. (D) At pH 7.4, the bromine atom interacts to the Asp189 of the S1 pocket and formed two halogen bonds with the carboxyl side chain of Asp189. The angles of D⋯Br–A are 144.3° and 150.4°, respectively. Electron density maps (σ-weighted 2Fo − Fc omit map contoured at 1.0σ, in blue) corresponding to the peptide residues are represented as cages. Halogen bonds are indicated as yellow dashed lines. Active site residues of uPA are presented by lightblue sticks. | ||
To determine the molecular binding mode of BrBA with uPA at physiological pH, we soaked the uPA crystals with BrBA at pH 7.4 and solved the crystal structure of the uPA:BrBA complex at 2.0 Å. We found that BrBA is still inserted in the S1 pocket of uPA (Fig. 2C). Similar to the uPA:BrBA structure at low pH, the bromine group of BrBA bound to the Asp189 of the bottom of the S1 pocket. The bromine was 3.0 Å and 3.3 Å away from the carboxyl oxygen atoms of Asp189; in addition, the angles of D⋯Br–A were 150.5° and 144.3°, respectively. Such distances and angles demonstrate the presence of halogen bonding between BrBA and Asp189 of uPA (Fig. 2D, Table S3, ESI†). The bromine atom is also quite close to the main chain oxygen atom of uPA Ser190 with a distance of 3.3 Å. The phenyl ring maintains van der Waals interaction with the backbone atoms of residues 190–192 and 215–217 of uPA as presented at the low pH.
Although the binding model of BrBA at low pH is similar to that at neutral pH, there are some obvious differences of BrBA between these two structures. BrBA at low pH inserts to the S1 pocket deeper (0.4 Å) than it at neutral pH. The amine group of BrBA points toward the solvent environment at the physiological pH, and has a hydrogen bond with Ser195 of uPA, which is different from the complex structure in the low pH. In addition, BrBA at low pH is slanted (11.7°) and near Ser195 of uPA comparing to BrBA at pH7.4, but the amino group of BrBA at low pH did not make any hydrogen bond with uPA, in contrast, the amino group of BrBA at pH 7.4 has one hydrogen bond with Ser195 of uPA (Fig. S1 and Table S4, ESI†).
3.3 IBA uses amine moiety, but not halogen, to bind to Asp189 of uPA at neutral pH
To further evaluate the role of halogen binding in the design of the serine protease inhibitors, we study the iodo-analogue of BrBA, 4-iodobenzylamine (IBA), which has higher polarization of the electrostatic potential at its iodine atom surfaces compared to two other halogen atoms.41 We determined the crystal structure of IBA in complex with uPA at neutral pH (pH 7.4) to a high resolution of 1.45 Å (Table S1, ESI†). Interestingly, the 2Fo − Fc density map of IBA (Fig. 3A) and the anomalous difference map of iodine atom (Fig. 3B) both clearly demonstrated that IBA adopted a completely different conformation in the S1 specific pocket of uPA. The iodine atom exposed to the solvent but was remained in the active pocket, mediated by two water molecules (Fig. S2, ESI†). At the other end, the amino group of IBA binds to Asp189 by two hydrogen bonds and binds to Ser190 by hydrogen bonds (Fig. 3C, Table S4, ESI†). The anomalous map of iodine atom (Fig. 3B) showed that IBA used the amino group rather than the iodine atom to make interactions with uPA, which is consistent with above-mentioned result that the inhibition of IBA is weaker than that of BrBA. Thus, our results demonstrated that the iodine might not be the best choice for the inhibitor of serine proteases investigated here.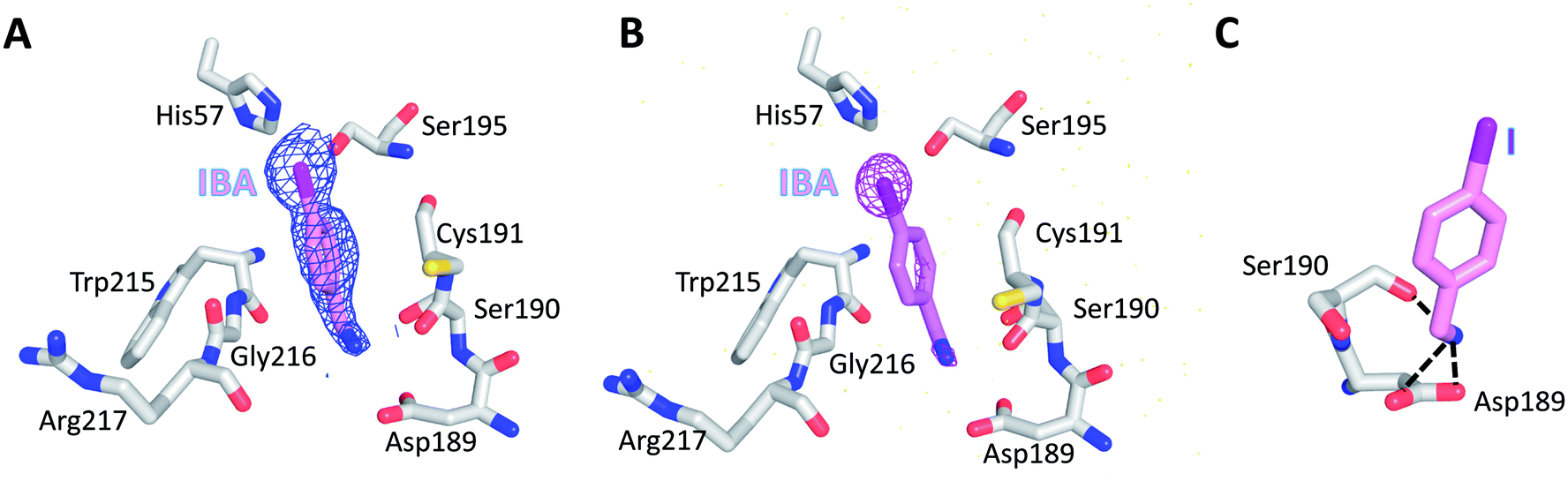 | ||
| Fig. 3 IBA did not make the halogen bonding with Asp189 of uPA at neutral pH. Both the 2Fo − Fc density map of IBA (A) and the anomalous difference map of iodine atom (B) clearly demonstrated that the iodine atom exposes to the solvent and did not form the halogen bonding with uPA. (C) The amine group of IBA binds to Asp189 of uPA by making two hydrogen bonds with Asp189 and one hydrogen bond (black dashed lines) with Ser190 of uPA. | ||
In addition, we also tried to determine the structure of 4-chlorobenzylamine (ClBA) bound to uPA by soaking CIBA into uPA crystals, but we did not observe electron density of ClBA in the crystal structure. The reason may be that the affinity of ClBA is too weak to give visible electron density.
4. Discussion
Many newly developed drugs contain one or more halogen atoms. The presence of halogens improves oral absorption. In addition, halogens fill hydrophobic cavities in the binding site, and facilitate the crossing of blood–brain barrier and prolong the drug lifetime.2,12 The number of biological halogen bonds identified in Protein Data Bank (PDB) has increased significantly from 116 in 2004 to well over 600 in 2013.41,42 There are a number of reviews on the role of halogen bonds in the recognition of inhibitors against several classes of proteins.11,12,42–45 Serine protease is one of the significant target to develop halogenated inhibitors.46,47 Indeed, halogen-based inhibitor for blood coagulation factor Xa has been developed.48–50 Matter et al. reported two series of halogenated inhibitors of serine protease factor Xa and showed that organic chlorines/bromines in both series are favorably in close contact with the phenyl ring of Tyr228 at the back wall of the protease S1 pocket.51,52 Rivaroxaban, a factor Xa inhibitor, contains an aromatic carbon–chlorine moiety and is buried inside the S1 pocket of factor Xa (Fig. 4). However, the chlorine of rivaroxaban does not form halogen bond to the Asp189. The chlorine atom is quite far away from the Asp189 located at the bottom of the S1 pocket. Instead, the chlorine atom interacts with the phenyl ring of the Tyr228 residue located at one side of the S1 pocket through the halogen–π interaction. Rivaroxaban shows favourable pharmacokinetic profile and oral bioavailability and has been approved for clinical use as an anticoagulant.53,54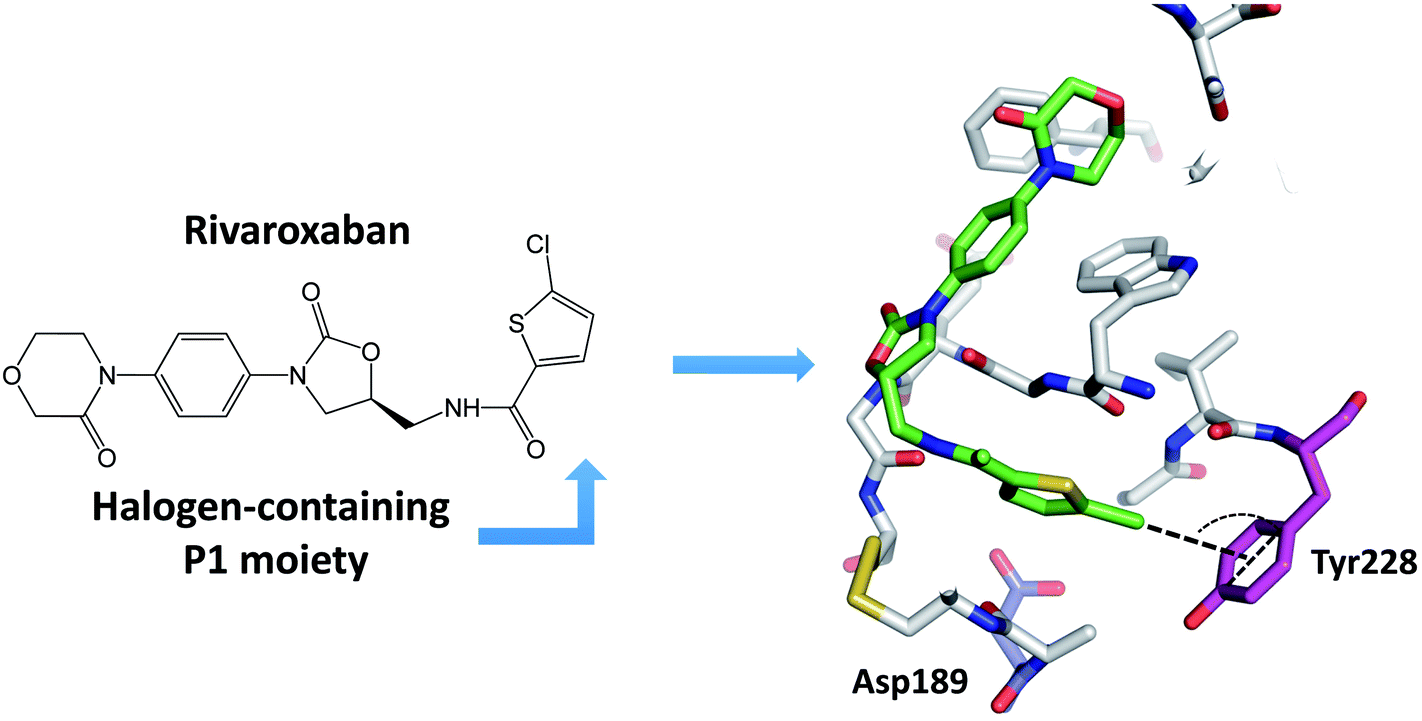 | ||
| Fig. 4 A halogen-based inhibitor (rivaroxaban) for blood coagulation factor Xa. The chlorine interacts with the aromatic ring of Tyr228 located at the bottom of the S1 pocket. As a result of this chlorine–Tyr228 interaction in the S1 pocket, highly basic groups such as amidine or other positively charged groups are not required for high factor Xa affinity. The inhibitor is colored green, and protein residue Asp189 is colored blue, the residue Tyr228 is colored magenta. Dash lines indicate the chlorine–π interaction. | ||
The major specificity pocket of trypsin-like serine protease (S1 pocket) typically accommodates substrates or inhibitors with an arginine group (P1 group) through the charged hydrogen bonds between the P1 group and the Asp189 of the S1 pocket. Hydrogen bonds between charged groups are much stronger (3.5–4.5 kcal mol−1) than the regular hydrogen bonds (0.5–1.8 kcal mol−1).55 Thus, many trypsin-like serine protease inhibitors contain arginine mimetics, e.g., amidine or guanidine, as P1 group to target serine proteases. Because such P1 group usually has high pKa and is quite basic at physiological pH, the molecules containing such moiety tend to have lower bioavailability.22–24
Can halogen atoms replace amidine or guanidine as a part of P1 group to target the S1 pocket of serine proteases by forming the halogen bonding with Asp189? Our present study aims to answer this question by using uPA as a target, and 4-chlorobenzylamine (ClBA), 4-bromobenzylamine (BrBA) and 4-iodobenzylamine (IBA) as probes. We found that ClBA, BrBA and IBA indeed inhibited the proteolytic activity of uPA with Ki values of 9.15 mM, 1.28 mM and 1.38 mM, respectively. Although these halogenated compounds are still weak binder to uPA and likely to have low specificity toward uPA compared to other serine proteases, they are better than the analogues lacking the halogen atom, such as 4-hydroxybenzylamine with no obvious inhibition on uPA at 5 mM. A non-halogenated analogue, 4-(aminomethyl-phenyl)-methanol (APM), inhibited uPA of a Ki of 13.9 mM, by binding to the S1 pocket of uPA (Fig. 5),40 which is about 10-fold weaker than the Br analogue, demonstrating the feasibility of halogen compound as P1 group. The weaker potency of APM was also supported by theoretical calculation (Table S2, ESI†).
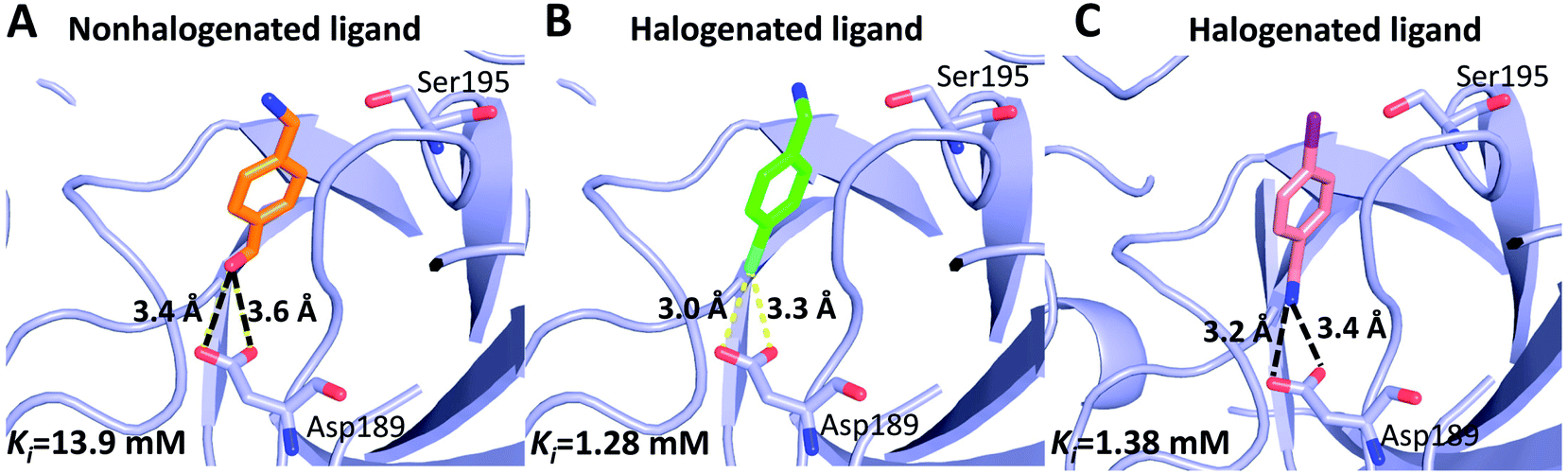 | ||
| Fig. 5 Comparison of halogenated and nonhalogenated inhibitors to the protein target (uPA). uPA in complex with the nonhalogenated ligand 4-(aminomethyl-phenyl)-methanol ((A) PDB code 3KHV), its brominated analog 4-bromobenzylamine (B) and iodinated analog 4-iodobenzylamine (C). The polypeptides backbone are traced as a lightblue ribbon, with amino acids and ligands involved in halogen bonding interactions and hydrogen bonds (black dashed lines) shown as ball stick models with carbons (orange, green and pink, respectively), oxygen (red), nitrogen (blue), bromine (cyan), and iodine (purple). | ||
Our results demonstrate that iodine is not the best choice for binding to S1 pocket of uPA, in contrast to typical halogen bonding strength: Cl < Br < I. Similar observation of inversed halogen bond strength was also reported and rationalized recently through computational chemistry method.56 In addition, there are some reports showing the inhibition of bromine-based inhibitors is better than the other halogen-based inhibitors. A Br-containing compound was found to be better than I analogue to inhibit aldose reductase. The reason was proposed that the favorable energetics of halogen bonding may be compromised by factors, such as solvation/desolvation changes, which plays an important role in halogen bonds of protein–ligand complexes.57 However, we should point out that iodine is a better inhibitor in some cases. One example is PhiKan5196, which is the inhibitor of the p53 tumour suppressor and displays halogen bonding between the iodine atom of the drug and the target protein. The substitution of iodine by bromine and chlorine led to reduced binding affinity of the inhibitor.58
Our current halogen compound have low inhibitory capabilities (Ki > 1 mM). Previous study demonstrated the possibility of small molecule compounds with weak inhibition to uPA (IC50 > 1 mM) can be developed into high potent inhibitor.59 Further derivatization of BrBA at the amine moiety will be needed to improve it binding to uPA. Another advantage of halogen-based inhibitor development is the relative low chemical reactivity of carbon–halogen bond, compared to amidine or guanidine moieties, which facilitates chemical synthesis of these novel halogen-based inhibitors for serine proteases.
5. Conclusions
In summary, we studied the binding of halogen-containing compounds (ClBA, BrBA and IBA) to uPA. The proteolytic and structural studies showed that these compounds' inhibitory activities to uPA were enhanced compared to the non-halogen indeed found for BrBA, but not IBA, through structural analysis. These results advocate that halogen bonding, especially bromine bonding, as an efficient strategy for the future design of novel inhibitors against trypsin-like serine proteases to provide strong potency and promote bioavailability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Shanghai Synchrotron Radiation Facility and the scientists at beam line BL17U for assistance with X-ray data collection. This work was supported by grants from National Key R&D Program of China (2017YFE0103200), Natural Science Foundation of China (31370737, 31400637, 31570745 and 31670739) and Fujian province (2018J01729), Fuzhou University Testing Fund of Precious Apparatus (2017T010).References
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS.
- A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef.
- Y. Lu, T. Shi, Y. Wang, H. Yang, X. Yan, X. Luo, H. Jiang and W. Zhu, J. Med. Chem., 2009, 52, 2854–2862 CrossRef CAS PubMed.
- D. A. Kraut, M. J. Churchill, P. E. Dawson and D. Herschlag, ACS Chem. Biol., 2009, 4, 269–273 CrossRef CAS.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS.
- A. R. Voth, F. A. Hays and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6188–6193 CrossRef CAS.
- N. F. Valadares, L. B. Salum, I. Polikarpov, A. D. Andricopulo and R. C. Garratt, J. Chem. Inf. Model., 2009, 49, 2606–2616 CrossRef CAS PubMed.
- U. Egner, J. Kratzschmar, B. Kreft, H. D. Pohlenz and M. Schneider, ChemBioChem, 2005, 6, 468–479 CrossRef CAS.
- A. Lange, M. Gunther, F. M. Buttner, M. O. Zimmermann, J. Heidrich, S. Hennig, S. Zahn, C. Schall, A. Sievers-Engler, F. Ansideri, P. Koch, M. Laemmerhofer, T. Stehle, S. A. Laufer and F. M. Boeckler, J. Am. Chem. Soc., 2015, 137, 14640–14652 CrossRef CAS PubMed.
- M. H. Kolar and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef CAS PubMed.
- B. Furie and B. C. Furie, Cell, 1988, 53, 505–518 CrossRef CAS.
- X. Puente, G. Ordóñez and C. López-Otín, in The Cancer Degradome, ed. D. Edwards, G. Høyer-Hansen, F. Blasi and B. Sloane, Springer New York, 2008, DOI:10.1007/978-0-387-69057-5_1, ch. 1, pp. 3–15.
- M. Drag and G. S. Salvesen, Nat. Rev. Drug Discovery, 2010, 9, 690–701 CrossRef CAS PubMed.
- L. Hedstrom, Chem. Rev., 2002, 102, 4501–4523 CrossRef CAS PubMed.
- M. J. Page and E. Di Cera, J. Biol. Chem., 2008, 283, 30010–30014 CrossRef CAS.
- J. J. Perona and C. S. Craik, Protein Sci., 1995, 4, 337–360 CrossRef CAS.
- J. C. Ngo, L. Jiang, Z. Lin, C. Yuan, Z. Chen, X. Zhang, H. Yu, J. Wang, L. Lin and M. Huang, Curr. Drug Targets, 2011, 12, 1729–1743 CrossRef CAS.
- G. Spraggon, C. Phillips, U. K. Nowak, C. P. Ponting, D. Saunders, C. M. Dobson, D. I. Stuart and E. Y. Jones, Structure, 1995, 3, 681–691 CrossRef CAS.
- K. Dano, P. A. Andreasen, J. Grondahl-Hansen, P. Kristensen, L. S. Nielsen and L. Skriver, Adv. Cancer Res., 1985, 44, 139–266 CrossRef CAS.
- C. Yuan, L. Chen, E. J. Meehan, N. Daly, D. J. Craik, M. Huang and J. C. Ngo, BMC Struct. Biol., 2011, 11, 30–42 CrossRef CAS.
- J. Joossens, O. M. Ali, I. El-Sayed, G. Surpateanu, P. Van der Veken, A.-M. Lambeir, B. Setyono-Han, J. A. Foekens, A. Schneider, W. Schmalix, A. Haemers and K. Augustyns, J. Med. Chem., 2007, 50, 6638–6646 CrossRef CAS.
- M. Zhu, V. M. Gokhale, L. Szabo, R. M. Munoz, H. Baek, S. Bashyam, L. H. Hurley, D. D. Von Hoff and H. Han, Mol. Cancer Ther., 2007, 6, 1348–1356 CrossRef CAS.
- M. Venkatraj, J. Messagie, J. Joossens, A.-M. Lambeir, A. Haemers, P. Van der Veken and K. Augustyns, Bioorg. Med. Chem., 2012, 20, 1557–1568 CrossRef CAS PubMed.
- M. E. Frederickson and D. M. Gordon, Proc. R. Soc. B, 2007, 274, 1117–1123 CrossRef.
- C. W. West, M. Adler, D. Arnaiz, D. Chen, K. Chu, G. Gualtieri, E. Ho, C. Huwe, D. Light, G. Phillips, R. Pulk, D. Sukovich, M. Whitlow, S. Yuan and J. Bryant, Bioorg. Med. Chem. Lett., 2009, 19, 5712–5715 CrossRef CAS.
- B. C. Doak, R. S. Norton and M. J. Scanlon, Pharmacol. Ther., 2016, 167, 28–37 CrossRef CAS PubMed.
- G. Zhao, C. Yuan, T. Wind, Z. Huang, P. A. Andreasen and M. Huang, J. Struct. Biol., 2007, 160, 1–10 CrossRef CAS PubMed.
- W. Otwinoski and W. Minor, Methods Enzymol., 1997, 276, 307–326 Search PubMed.
- CCP4, Acta Crystallogr., 1994, D50, 760–763 Search PubMed.
- L. Jiang, K. A. Botkjaer, L. M. Andersen, C. Yuan, P. A. Andreasen and M. Huang, Biochem. J., 2013, 449, 161–166 CrossRef CAS.
- P. Emsley and K. Cowtan, Acta Crystallogr D Biol Crystallogr, 2004, 60, 2126–2132 CrossRef.
- W. L. Delano, 2002.
- R. Kumari, R. Kumar, C. Open Source Drug Discovery and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- J. Li, L. Jiang and X. Zhu, J. Mol. Model., 2015, 21, 177 CrossRef PubMed.
- S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- G. X. Zhao, L. G. Jiang, C. B. Bian, C. Yuan, Z. X. Huang and M. D. Huang, Chin. J. Struct. Chem., 2009, 28, 253–259 Search PubMed.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS.
- M. R. Scholfield, C. M. Zanden, M. Carter and P. S. Ho, Protein Sci., 2013, 22, 139–152 CrossRef CAS.
- A. R. Voth and P. S. Ho, Curr. Top. Med. Chem., 2007, 7, 1336–1348 CrossRef CAS PubMed.
- J. J. Liao, J. Med. Chem., 2007, 50, 409–424 CrossRef CAS.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed.
- S. Roehrig, A. Straub, J. Pohlmann, T. Lampe, J. Pernerstorfer, K. H. Schlemmer, P. Reinemer and E. Perzborn, J. Med. Chem., 2005, 48, 5900–5908 CrossRef CAS.
- K. J. Stauffer, P. D. Williams, H. G. Selnick, P. G. Nantermet, C. L. Newton, C. F. Homnick, M. M. Zrada, S. D. Lewis, B. J. Lucas, J. A. Krueger, B. L. Pietrak, E. A. Lyle, R. Singh, C. Miller-Stein, R. B. White, B. Wong, A. A. Wallace, G. R. Sitko, J. J. Cook, M. A. Holahan, M. Stranieri-Michener, Y. M. Leonard, J. J. Lynch, Jr., D. R. McMasters and Y. Yan, J. Med. Chem., 2005, 48, 2282–2293 CrossRef CAS.
- M. J. Hartshorn, C. W. Murray, A. Cleasby, M. Frederickson, I. J. Tickle and H. Jhoti, J. Med. Chem., 2005, 48, 403–413 CrossRef CAS.
- P. E. Sanderson, K. J. Cutrona, D. L. Dyer, J. A. Krueger, L. C. Kuo, S. D. Lewis, B. J. Lucas and Y. Yan, Bioorg. Med. Chem. Lett., 2003, 13, 161–164 CrossRef CAS.
- T. J. Tucker, S. F. Brady, W. C. Lumma, S. D. Lewis, S. J. Gardell, A. M. Naylor-Olsen, Y. W. Yan, J. T. Sisko, K. J. Stauffer, B. J. Lucas, J. J. Lynch, J. J. Cook, M. T. Stranieri, M. A. Holahan, E. A. Lyle, E. P. Baskin, I. W. Chen, K. B. Dancheck, J. A. Krueger, C. M. Cooper and J. P. Vacca, Abstr. Pap. Am. Chem. Soc., 1998, 216, U207 Search PubMed.
- M. Nazare, D. W. Will, H. Matter, H. Schreuder, K. Ritter, M. Urmann, M. Essrich, A. Bauer, M. Wagner, J. Czech, M. Lorenz, V. Laux and V. Wehner, J. Med. Chem., 2005, 48, 4511–4525 CrossRef CAS PubMed.
- H. Matter, M. Nazare, S. Gussregen, D. W. Will, H. Schreuder, A. Bauer, M. Urmann, K. Ritter, M. Wagner and V. Wehner, Angew. Chem., Int. Ed., 2009, 48, 2911–2916 CrossRef CAS.
- B. O. Villoutreix and O. Sperandio, Curr. Opin. Struct. Biol., 2010, 20, 168–179 CrossRef CAS PubMed.
- I. Melnikova, Nat. Rev. Drug Discovery, 2009, 8, 353–354 CrossRef CAS PubMed.
- A. R. Fersht, J. P. Shi, J. Knilljones, D. M. Lowe, A. J. Wilkinson, D. M. Blow, P. Brick, P. Carter, M. M. Y. Waye and G. Winter, Nature, 1985, 314, 235–238 CrossRef CAS.
- E. Aubert, E. Espinosa, I. Nicolas, O. Jeannin and M. Fourmigue, Faraday Discuss., 2017, 203, 389–406 RSC.
- J. Fanfrlik, M. Kolar, M. Kamlar, D. Hurny, F. X. Ruiz, A. Cousido-Siah, A. Mitschler, J. Rezac, E. Munusamy, M. Lepsik, P. Matejicek, J. Vesely, A. Podjarny and P. Hobza, ACS Chem. Biol., 2013, 8, 2484–2492 CrossRef CAS PubMed.
- R. Wilcken, X. Liu, M. O. Zimmermann, T. J. Rutherford, A. R. Fersht, A. C. Joerger and F. M. Boeckler, J. Am. Chem. Soc., 2012, 134, 6810–6818 CrossRef CAS PubMed.
- M. Frederickson, O. Callaghan, G. Chessari, M. Congreve, S. R. Cowan, J. E. Matthews, R. McMenamin, D. M. Smith, M. Vinkovic and N. G. Wallis, J. Med. Chem., 2008, 51, 183–186 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, Fig. S1 and S2 and Table S1–S4. See DOI: 10.1039/c8ra03145b |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2018 |