
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new promising nucleating agent for polymer foaming: effects of hollow molecular-sieve particles on polypropylene supercritical CO2 microcellular foaming
Chenguang Yang abc,
Mouhua Wanga,
Zhe Xinga,
Quan Zhaoa,
Minglei Wanga and
Guozhong Wu*ac
abc,
Mouhua Wanga,
Zhe Xinga,
Quan Zhaoa,
Minglei Wanga and
Guozhong Wu*ac
aShanghai Institute of Applied Physics, Chinese Academy of Sciences, Jialuo Road 2019, Jiading, Shanghai 201800, China. E-mail: wuguozhong@sinap.ac.cn
bUniversity of China Academy of Sciences, Beijing, 100049, China
cSchool of Physical Science and Technology, ShanghaiTech University, Shanghai, 200031, China
First published on 4th June 2018
Abstract
Because polypropylene (PP) foam normally exhibits nonuniform cell size and cracked cellular structure, a narrow cell-size distribution and a well-defined morphology are always the focus of PP foaming technology. In this work, hollow molecular-sieve (MS) particles were applied as a potential nucleating agent in supercritical carbon dioxide (scCO2) foaming of PP. It was observed that the addition of MS particles largely narrowed the cell-size distribution. The resultant PP/MS foams exhibited significant concurrent enhancement in their cell density and mechanical properties: the cell density increased remarkably, by approximately 10 times, and the tensile strength increased from 6.1 MPa to 12.6 MPa. The hollow-structure MS particles resulted in a higher heterogeneous nucleation efficiency in the PP foaming process. We believe that the trapping of CO2 in the hollow holes of MS particles largely increased the solubility CO2 in PP and a number of gas cavities were formed. The existence of gas cavities reduced the energy barrier of heterogeneous nucleation, favoring the formation of a well-defined cellular structure. Additionally, the regular-hexagon shape of the cells might endow the PP foam with better mechanical properties compared with a circular cell shape.
1. Introduction
Polymer foam has been widely used in various industrial applications because of its good performance. Besides their excellent properties, the reduction of raw material cost is a significant advantage in using these materials.1–3 Polypropylene (PP) foam is one of the most promising materials because of its outstanding overall performance, such as high toughness, low electrical conductivity, excellent chemical resistance, easy recycling, and high thermal stability for a variety of applications.4–10 However, owing to its weak melt strength and melt elasticity, the properties of the resulting PP foams are poor. The reason for this is that the foam structure (cell walls) is not sufficiently strong to bear the extensional force applied during cell growth and the cells are prone to coalescing and collapsing during CO2 release from the melt PP matrix. Consequently, the foam exhibits nonuniform cell distribution and a high open-cell content,11–14 which are unfavorable for practical applications.Foams with a uniform cell size and high closed-cell content may have excellent mechanical properties. The foaming mechanism is a so-called phase separation process by supercritical CO2 (scCO2), which is caused by a quick change in the temperature and pressure of the homogeneous polymer/CO2 system.1,15 During the foaming process, the nucleation stage has a significant influence on the morphology of the final cells. To obtain a better micropore structure, many nano-inorganic materials, including clay,14,16 carbon nanotubes,17 carbon nanofibers,18 and nanosilica,19 which are efficient in heterogeneous nucleation, have been widely used in the foaming of different polymers. Heterogeneous nucleation has a lower energy barrier and can promote the simultaneous formation of embryos, thereby reducing the average cell size and narrowing the cell-size distribution.20 A small cell size, narrow cell-size distribution, and high cell density indicate good tensile properties of foamed polymers.
The low energy barrier has been widely known to be a critical concern in the mechanism of heterogeneous nucleation, and all parameters associated with this issue can affect nucleation.1 Hollow molecular sieves (MSs) are inorganic materials with many unique properties, such as hollow structure, high surface area, large pore volume, and tunable nanometer pore size.21–23 Owing to these properties, hollow MSs have been widely applied in many fields, such as catalysis, separation and purification, adsorption, polymer modification, and antibacterial materials.21,23 Hollow MSs may be excellent nucleating agents for the scCO2-assisted foaming process, according to the principles of the heterogeneous nucleation mechanism and the major factors affecting the nucleation efficiency. The existence of a high specific area and a large pore volume increases the solubility of the blowing agent and the nanometer pores may trap CO2 to form gas cavities during the foaming process, which can considerably lower the critical free energy of nucleation. In this study, microcellular foaming technology using scCO2 was applied to PP foaming, and a hollow MS was selected as a nucleation agent. Our target is to fabricate PP foams with small cell size, high cell density, and uniform cell morphology, as well as good mechanical properties. Additionally, the influence of cell density, cell size, local stress caused by hollow MS particles, and cell shape on the tensile property was investigated in detail.
2. Materials and methods
2.1 Materials
PP T03 (pellets, isotactic) with a density of 0.91 g cm−3 and a melt flow index of 3.1 g/10 min (230 °C/2.16 kg) was purchased from Sinopec Shanghai Chemical Co. MS particles (ZSM-5, particle size: 100–400 nm) were purchased from Shanghai Shen Tan Chemical New Material Co. Ltd. Carbon dioxide with a purity of 99.95% was supplied by Xiangkun Special Gases of Shanghai.2.2 Composite sample preparation
The PP pellets and MS particles were vacuum-dried at 60 °C for 4 h before they were mixed. A series of blends of PP with MS particles with contents of 0.5, 2.0, and 5.0 wt% were made using two-screw extruder (Thermo Haake PolyDrive 7, Germany). PP/MS sheets with 1 mm thickness were prepared by hot pressing. These samples were coded as PP/MS(0.5%), PP/MS(2.0%), and PP/MS(5.0%), respectively.2.3 Foaming process
A PP/MS sheet sample (4–5 g) was placed in an autoclave and then the autoclave was pressurized with CO2 using a high-pressure liquid pump, the parameters of the foaming device being previously described in the literature.7–10,24,25 The system was kept at 154 °C and 20 MPa for 2 h. Then the vessel was depressurized to vent CO2 in less than 10 s. After that, the sample was removed from the vessel and allowed to cool to room temperature.2.4 Sample characterization
A NETZSCH STA 449 F3 Jupiter differential scanning calorimeter (DSC) equipped with a data station was used for scanning the melting transitions of the samples in aluminum pans. The samples were heated from 25 to 200 °C at a heating rate of 10 °C min−1 under an argon flow (20 ml min−1). The degree of crystallization was calculated using eqn (1), as follows:
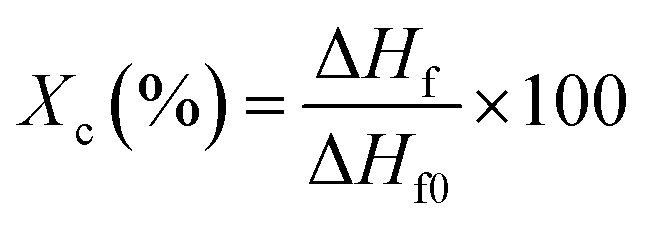 | (1) |
X-ray diffraction patterns were collected by a Bruker D8 Discover apparatus (Bruker, Germany) with Cu Kα as the radiation source (λ = 1.5418 Å). It was operated at 40 kV and 40 mA and the scanning range was 10–50° at a scanning rate of 2° min−1.
The hollow structure and particle size of MSs were visualized using a transmission electron microscope (TEM) with 200 kV field emission (Tecnai G2 F20 S-TWIN, USA). For TEM imaging, particle dispersions diluted in ethanol were deposited on the carbon side of a carbon/copper grid.
The morphology of the PP/MS foams was examined using a scanning electron microscope (SEM; Zeiss MERLIN Compact 14184, Germany). Samples were immersed in liquid nitrogen for 2 min, fractured, and mounted on stubs. They were then sputter-coated with gold to prevent charging during the test.
2.5 Morphology observation of the foams
Microstructural morphology of the PP/MS foams was characterized by measuring the cell density and average cell size. Image Pro-plus software was used to analyze the SEM images. The average diameter of the cells in the micrographs, D, was calculated using eqn (2):
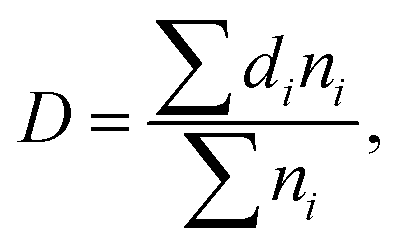 | (2) |
The volume expansion ratio for each sample was calculated as the ratio of the density of the original sample ρs to the measured density of the foam sample ρf. The densities (ρf) of foamed samples were determined using the Archimedes law involving weighing polymer foam in water with a sinker using an electronic analytical balance (HANG-PING FA2104), calculated using eqn (3):
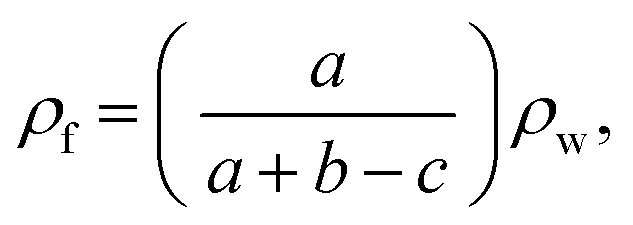 | (3) |
The cell density (N) was determined by the number of cells per unit volume of the foam, calculated using eqn (4):
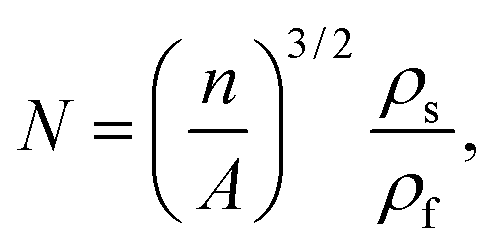 | (4) |
2.6 Tensile testing
Tensile tests of the foams were carried out using a universal testing machine (Instron 5943, USA). The foam samples were cut into specimens of dimensions 2 mm × 4 mm × 22 mm, and the specimens were measured at room temperature in accordance with ASTM D-638 at a speed of 50 mm min−1.3. Results and discussion
The MS particles were compounded with PP via melt-processing at 220 °C using a twin-screw extruder. First, the MS particle size and MS dispersion in the PP matrix before foaming were characterized. Then, the effect of the MS particles on the cell morphology of the PP/MS foams was investigated based on the change of the crystallinity of the PP/MS composites.3.1 MS particle dispersion in PP matrix
Fig. 1 shows images of the MS particles, and the fracture surfaces of pure PP and the resultant PP/MS (2.0%) composites. First, a nanometer-size (∼200 nm) hollow hole can be observed in the MS particle. It is also evident that PP/MS (2.0%) has many well-scattered multi-MS particles and MS aggregates in the PP matrix after melt compounding. The dispersion of Si from the MS is observed in the energy-dispersive spectrometry image. There is a uniform dispersion of Si in the PP matrix, which also indicates a good dispersion of MS particles in the PP matrix.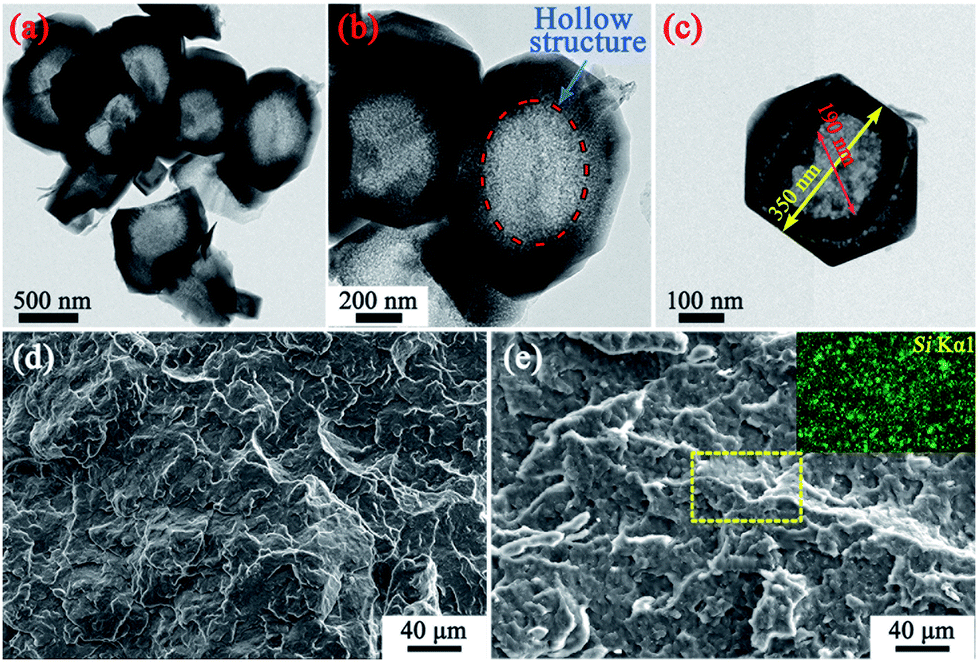 | ||
| Fig. 1 TEM micrographs of the MS particles (a–c); SEM images showing fractured surfaces of the PP (d) and PP/MS (2.0%) (e) samples, and the dispersion of Si in the PP matrix. | ||
3.2 Influence of MS on the scCO2 foaming behavior of PP
Fig. 2 shows the influence of MS addition on the cell morphologies and cell-size distributions of PP and PP/MS foams. The samples were saturated at 20 MPa and foamed at 154 °C for 10 s. As expected, the pure PP foam presented poor cellular structure and obvious cracked cells.27 In contrast, PP/MS (0.5%), PP/MS (2.0%), and PP/MS (5.0%) foams exhibited more uniform cell distribution, smaller cell size, a regular-polygon cell shape, and well-defined cell walls. The cells were strongly contacted by the junction regions of three contacting cells of the different-content PP/MS foams. The outstanding structural parameters of PP/MS foams indicate good mechanical properties for many applications.8,10,27 It can be clearly seen in Fig. 2 that the cell-size distribution of the PP/MS foam becomes narrower than that of the pure PP foam, which indicates that the presence of MS particles improves the cellular structure of PP/MS foams greatly, resulting in a uniform cell size in the obtained foams. The nanometer-size hollow holes in an MS may trap CO2 to form gas cavities during the foaming process, which can lower the critical free energy of nucleation, thus helping to stabilize the cell size. Moreover, the cell-size distribution becomes narrower as the MS loading increases. We believe that the presence of MS particles also suppressed the cell growth during the foaming process.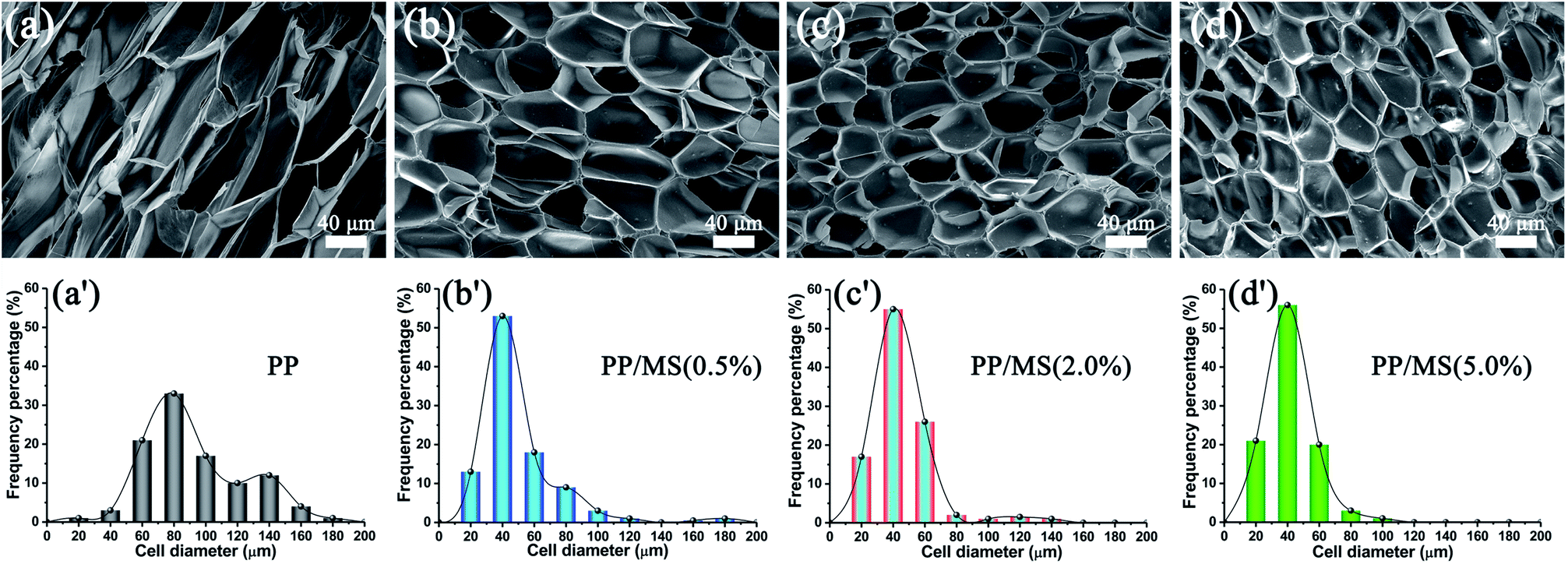 | ||
| Fig. 2 SEM micrographs and cell-size distributions of foamed PP (a), PP/MS (0.5%) (b), PP/MS (2.0%) (c), and PP/MS (5.0%) (d) saturated at 20 MPa and foamed at 154 °C. | ||
Fig. 3 summarizes the properties (average cell size and cell density) of the cellular structure of the PP/MS composite foams as a function of MS content. The figure shows that the average cell size decreased greatly from 86.3 μm, the size of the PP foam, to 30.7 μm, the size of the PP/MS (0.5%) foam. There was a slight decrease in the average cell size of the PP/MS foams as the MS particle content increased from 0.5% to 5.0%. On the other hand, the cell density increased remarkably from 6.9 × 106 cells per cm3 in the pure PP foam to 5.2 × 107 cells per cm3 in the PP/MS (5.0%) foam, indicating that the addition of MSs enhanced the nucleation of PP during the foaming process. The addition of MSs into PP significantly increases the storage modulus of PP/MS, which improves the stiffness of the compounding materials,28 whereas the existence of gas cavities caused by the holes in the MS particles may considerably lower the free energy of heterogeneous nucleation. The obtained high stiffness restricted cell growth, led to an obvious decrease in cell size, and combined with the enhanced heterogeneous nucleation during the saturation and foaming process, which greatly increased the cell density of PP/MS foams with different MS contents.
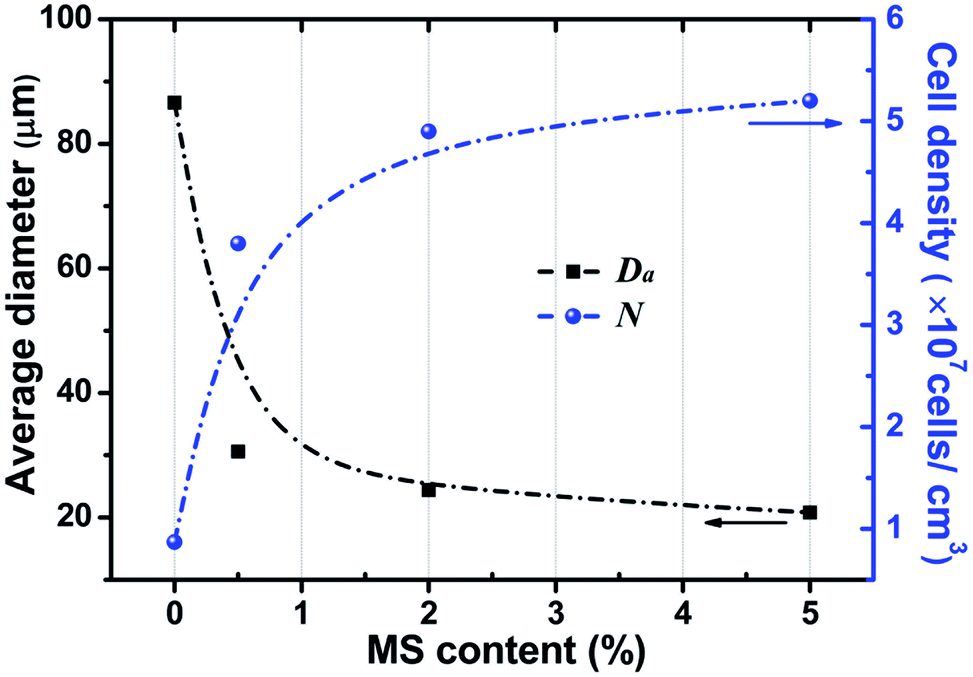 | ||
| Fig. 3 Average cell size and cell density of PP and PP/MS nanocomposite foams saturated at 20 MPa and foamed at 154 °C for 10 s. | ||
3.3 Influence of crystallite size on the foaming behavior of PP/MS
Fig. 4(a) shows the DSC curves of unfoamed PP and PP/MS. It can be seen that the melting point of the samples decreases as the MS loading increases from 0% to 5.0%. Moreover, the melting point and crystallinity are shown in Table 1. All these data were collected in the second scan. The melting points decreased with an increase of the MS content from 0% to 5.0% and the melting point of PP/MS (5.0%) was 164.5 °C. The obtained crystallinity of PP/MS decreased from 46.1% to 40.7% as the MS loading increased. The reason for this is that the introduction of MSs reduced the crystal size of PP, and an increase in the MS content tended to reduce the crystal size of PP gradually. Additionally, the MSs might also destroy the crystal integrity of PP in the crystalline regions during the heat-treatment process. The X-ray diffraction patterns of PP, PP/MS (0.5%), PP/MS (2.0%), and PP/MS (5.0%) are shown in Fig. 4(b). All four curves show the typical diffraction pattern for pure α-PP.29 The peaks at 2θ = 14.1°, 17.1°, 18.6°, 21.3°, and 22.3° correspond to the primary diffraction of the (1 1 0), (0 4 0), (1 3 0), (1 1 1), and (1 3 1) crystal planes of the α-crystal, respectively.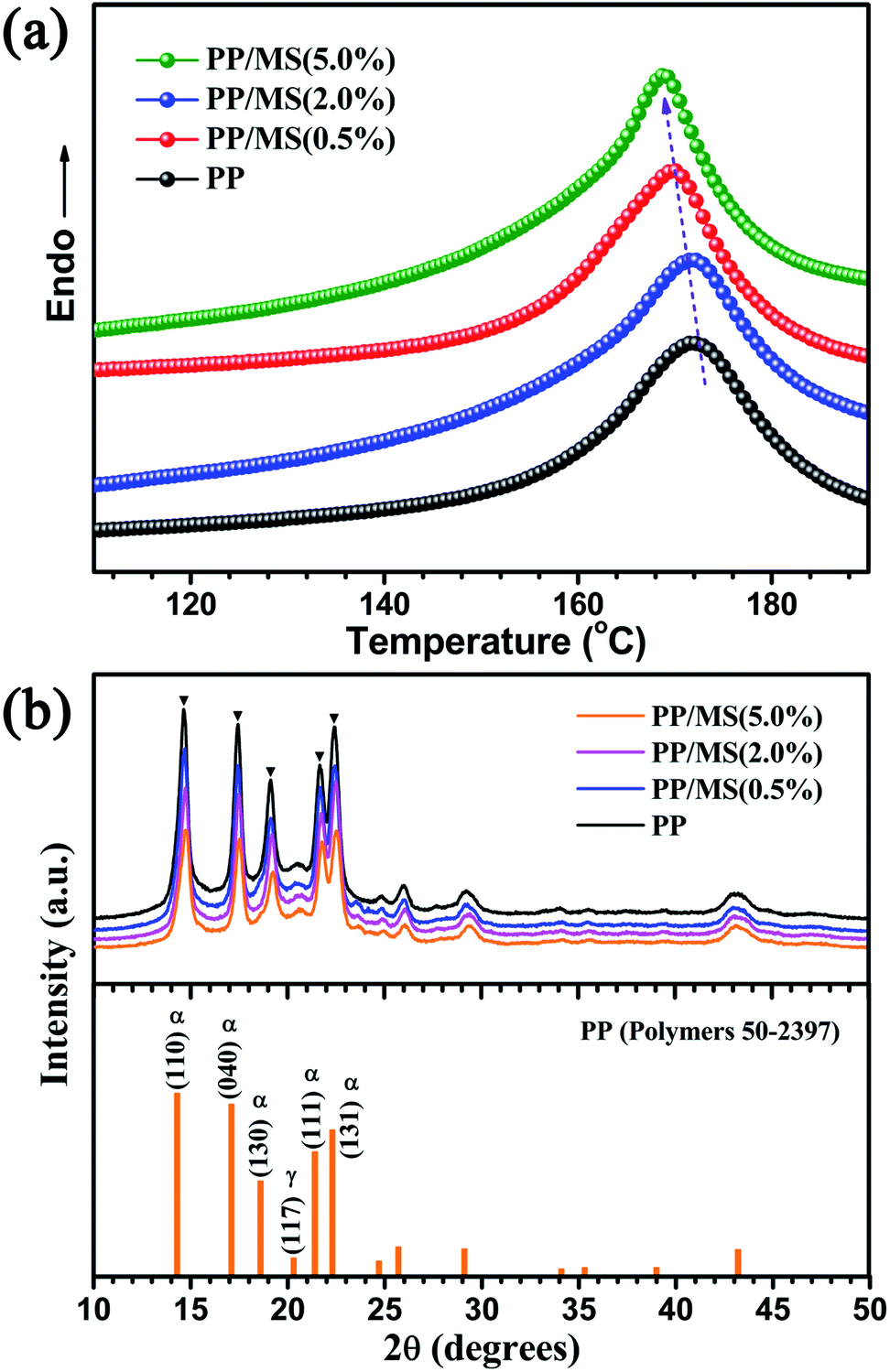 | ||
| Fig. 4 DSC curves and X-ray diffraction patterns of PP and PP/MS samples with different MS content. | ||
| Sample | PP | PP/MS (0.5%) | PP/MS (2.0%) | PP/MS (5.0%) |
|---|---|---|---|---|
| Melting point (°C) | 168.4 | 167.7 | 165.9 | 164.5 |
| Crystallinity (%) | 46.1 | 43.3 | 42.5 | 40.7 |
In the patterns of all the samples, a small peak at 2θ = 20.2°, corresponding to the (1 1 7) plane of the γ-crystal of PP, can also be seen, indicating that pure PP contains a small amount of the γ-phase. It can be clearly seen that the intensity of the diffraction peaks corresponding to the (1 1 0), (0 4 0), (1 3 0), (1 1 1), and (1 3 1) planes decreases as the MS loading in the PP matrix increases, which indicates a decrease in the crystal size of PP. In previous studies, a small crystal size and low crystallinity enhanced the formation of a uniform cellular structure.8,10,28
Furthermore, previous studies have demonstrated that the energy barrier mainly determines the nucleation rate in polymer foaming by scCO2. It is known that nanometer-particle-induced heterogeneous cell nucleation has a lower energy barrier than homogeneous nucleation during the foaming process. Therefore, we speculate that the addition of hollow MS particles caused a different cell nucleation mechanism in pure PP foam and PP/MS composite foams, based on the cell nucleation mechanism recently proposed by Leung et al.30 and Wong et al.31 Owing to the lower energy barrier needed for heterogeneous nucleation than homogeneous nucleation in the PP foaming process, the addition of a nucleating agent, such as hollow MS particles, lowers the free energy barrier, and the nanoparticles act as heterogeneous nucleation sites during the foaming process. Furthermore, the introduction of MSs significantly decreases the crystal growth rate of PP under saturated CO2. Therefore, the tiny crystal domains induced cell nucleation of PP/MS during the foaming process. We believe that both the decreased energy barrier and the enhanced cell nucleation endow PP/MS foams with well-defined cellular structure. Thus, the average cell diameter decreases and the cell density increases in the presence of these particles. It is interesting to note that the existence of nanometer-sized hollow holes in particles may result in a lower free energy barrier in the gas cavities, favoring heterogeneous nucleation. This is demonstrated in the schematic of Fig. 5.
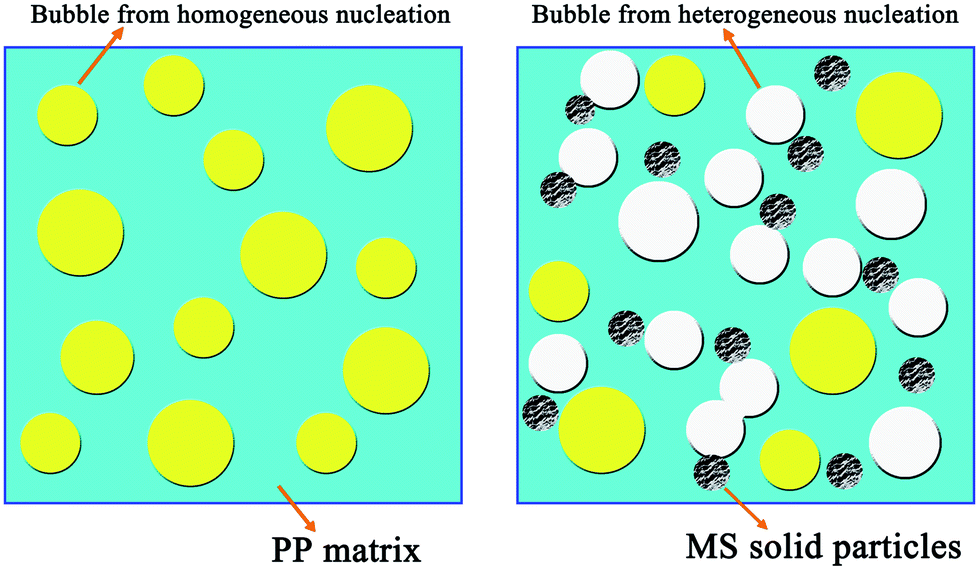 | ||
| Fig. 5 Schematic showing the nucleation process during foaming for pure PP and PP/MS composites. | ||
3.4 Influence of cellular structure of foams on their mechanical properties
PP foam is considered as one of the most attractive polymer foams, and the mechanical properties of a material determine the applications of the material. The stress–strain curves for the pure PP foam and PP/MS foams prepared at 154 °C and 20 MPa are shown in Fig. 6. The stress strength of the PP/MS foam improved significantly from 6.1 MPa to 12.6 MPa, and the tensile strains of the PP/MS foams also increased substantially, reaching 260%. With an increase in cell density, neighboring cell walls begin to touch each other, which decreases the stress concentrations on cell walls,27 and a higher tensile stress is obtained.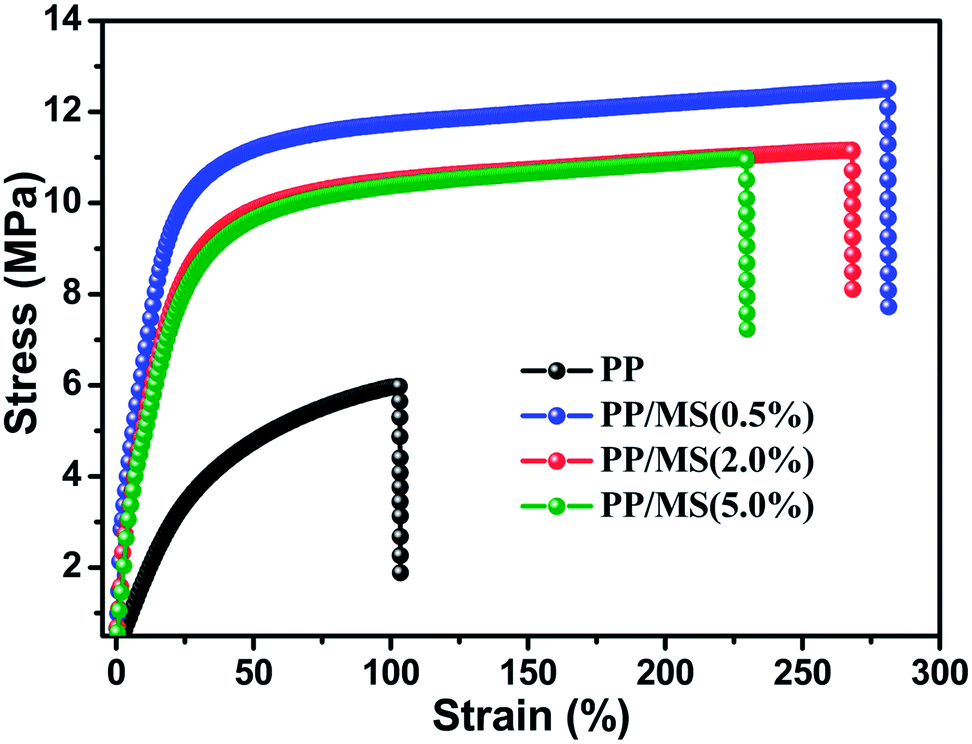 | ||
| Fig. 6 Strain–stress curves of foamed pure PP and foamed PP/MS with different MS contents at 154 °C and 20 MPa. | ||
It is well known that well-defined cellular structures are favorable in terms of the mechanical properties of polymer foams.10,32 According to the results mentioned above, the addition of MS improved the morphology of the PP/MS foams greatly. Therefore, the resultant good mechanical properties further proved that better microstructure significantly increases the tensile stress and tensile strain at break, which is preferred for potential applications.33,34
However, the tensile results of the PP foam and PP/MS foams showed that the PP/MS (2.0%) and PP/MS (5.0%) foams had lower tensile stress and tensile strain compared to the PP/MS (0.5%) foam, which were prepared under the same foaming conditions. The PP/MS (2.0%) and PP/MS (5.0%) foams had a higher cell density and smaller cell size than PP/MS (0.5%), whereas the measured mechanical properties exhibited the opposite behavior. Further measurements of the foam parameters were conducted to explain this interesting phenomenon. The cell wall thickness and foam bulk density (Table 2) increased as the loading of MS particles increased. In previous studies,8,10,27,35,36 thick cell walls and large bulk density of the foam indicated better mechanical properties. Therefore, these results cannot explain this behavior.
| Foam sample | PP | PP/MS (0.5%) | PP/MS (2.0%) | PP/MS (5.0%) |
|---|---|---|---|---|
| Cell wall thickness (μm) | 1.61 | 2.40 | 2.73 | 2.81 |
| Bulk density (g cm−3) | 0.10 | 0.16 | 0.18 | 0.22 |
We turn our attention to the effect of MS particles on the mechanical properties of the PP foams. Fig. 7 shows SEM micrographs of PP/MS foams with different MS contents obtained at 154 °C and 20 MPa. It can be clearly seen in the magnified micrographs that many MS aggregates appear in the cell walls and junction regions of three contacting cells, as noted by the arrows in Fig. 7. Moreover, the number of MS aggregates increased as the loading of MS particles increased. Given the interface bonding between the PP matrix and the MS aggregates, it may be easy to form local stress around the aggregate region during the stretching process, which is easier to break, leading to a poor tensile strength. We believe that the amount of added inorganic particles had a significant influence on the prepared polymer foams using scCO2, i.e., on their cell size, cell density, and mechanical properties. Therefore, determining the best amount of additives is necessary to improve polymer foams, and 0.5 wt% was found to be the optimum MS particle content to improve the cellular structure of PP foams with good mechanical properties in this study.
 | ||
| Fig. 7 SEM micrographs of the dispersion of MS particles in the foamed samples, (a) PP/MS (0.5%), (b) PP/MS (2.0%), and (c) PP/MS (5.0%). | ||
It must be noted that the cell shape of the polymer foam may also have an influence on its tensile strength. Fig. 8 shows SEM micrographs of PP, PP/MS (0.5%), PP/MS (2.0%), and PP/MS (5.0%) foams. Interestingly, we found that the PP/MS (0.5%) foam had a regular-hexagon cell shape, while the cell shapes of the PP/MS (2.0%) and PP/MS (5.0%) foams were approximate elliptical shapes. The difference in cell shape may have a considerable influence on the tensile strength during the stretching process. Fig. 9 depicts a schematic of the cellular deformation during the tensile testing process. The blue parts are the contact parts between cells in the non-stretched stage. It can be clearly seen that hexagonal cells have larger contact areas than circular cells. In the stretching stage, the hexagonal cells had reduced stress concentrations between cells because of the large contact areas, resulting in a uniform deformation in the stretching direction. Neighboring cell walls begin to touch each other and the foams are able to undergo plastic deformation so that uniform variations in stress are achieved during the stretching process. However, the stress concentrations on the junction regions of three contacting cells may be enhanced for the small contact parts between elliptical cells and they become “egg-like” at an intermediate deformation before fracture, combining with the local stress variation around the MS aggregates during the stretching process, leading to cell wall fracture with a decrease in tensile strength. Therefore, the PP/MS (0.5%) foam with regular cell shapes had better mechanical properties than the pure PP, PP/MS (2.0%), and PP/MS (5.0%) foams.
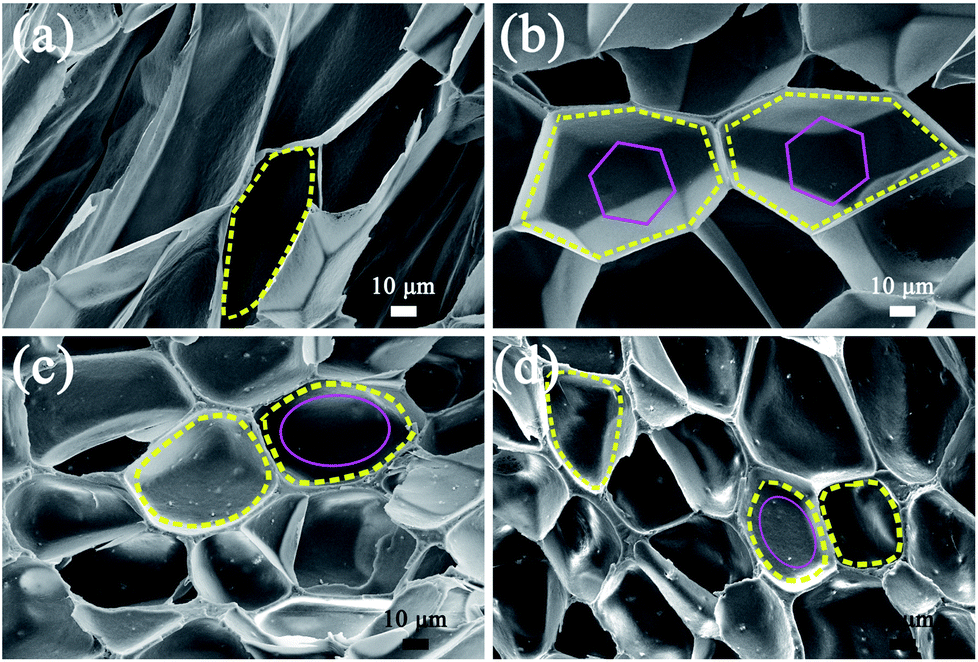 | ||
| Fig. 8 SEM micrographs of PP (a), PP/MS (0.5%) (b), PP/MS (2.0%) (c), and PP/MS (5.0%) (d) with different cell shapes. | ||
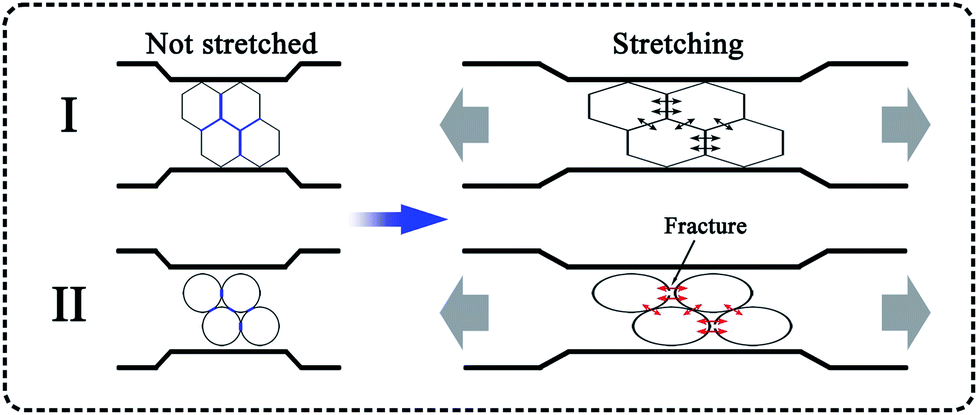 | ||
| Fig. 9 Schematic diagram of cell stretching during tensile testing: I, hexagonal cell; II, circular cell. | ||
4. Conclusions
In summary, the microcellular foaming behavior of PP and PP/MS samples was investigated using scCO2 as the physical blowing agent. The prepared pure PP foam had poor cellular structure and tensile strength. However, the PP/MS foams had small cell sizes and high cell densities with narrow cell-size distributions, which greatly improved their mechanical properties. The presence of MS particles decreased the crystal size of PP, and gas cavities caused by the hollow MS structure reduced the free energy barrier of heterogeneous nucleation, which endowed PP/MS foams with well-defined cellular structure. The results showed that the cell size decreased and the cell density increased greatly as the MS loading increased, and the optimum MS addition amount was 0.5%. The local stress around the aggregate region in the foams reduced the tensile strength during the stretching process. Furthermore, the regular-hexagon cell shape of the PP foam might provide better mechanical properties than a circular cell shape.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 11079048).Notes and references
- J. Yang, L. Huang, Y. Zhang, F. Chen, P. Fan, M. Zhong and S. Yeh, Ind. Eng. Chem. Res., 2013, 52, 14169–14178 CrossRef.
- Z. Li, Y. Jia and S. Bai, RSC Adv., 2018, 8, 2880–2886 RSC.
- J. E. Lee, Y. Guo, R. E. Lee and S. N. Leung, RSC Adv., 2017, 7, 48712–48722 RSC.
- L. J. Lee, C. C. Zeng, X. Cao, X. M. Han, J. Shen and G. J. Xu, Compos. Sci. Technol., 2005, 65, 2344–2363 CrossRef.
- L. Chen, D. Rende, L. S. Schadler and R. Ozisik, J. Mater. Chem. A, 2013, 1, 3837–3850 Search PubMed.
- Y. Kim, C. B. Park, P. Chen and R. B. Thompson, Polymer, 2013, 54, 841–845 CrossRef.
- C.-G. Yang, M.-H. Wang, M.-X. Zhang, X.-H. Li, H.-L. Wang, Z. Xing, L.-F. Ye and G.-Z. Wu, Molecules, 2016, 21, 1660 CrossRef PubMed.
- C. Yang, Z. Xing, M. Zhang, Q. Zhao, M. Wang and G. Wu, Radiat. Phys. Chem., 2017, 141, 276–283 CrossRef.
- C. Yang, X. Zhe, M. Zhang, M. Wang and G. Wu, Radiat. Phys. Chem., 2017, 131, 35–40 CrossRef.
- C. G. Yang, Z. Xing, Q. Zhao, M. H. Wang and G. Z. Wu, J. Appl. Polym. Sci., 2018, 135, 45809 CrossRef.
- G. B. James, J. Cell. Plast., 1978, 14, 341–345 CrossRef.
- C. B. Park and L. K. Cheung, Polym. Eng. Sci., 1997, 37, 1–10 Search PubMed.
- W. Zhai, H. Wang, J. Yu, J. Dong and J. He, Polym. Eng. Sci., 2008, 48, 1312–1321 Search PubMed.
- W. Zhai, T. Kuboki, L. Wang, C. B. Park, E. K. Lee and H. E. Naguib, Ind. Eng. Chem. Res., 2010, 49, 9834–9845 CrossRef.
- X. Liao, H. Xu, S. Li, C. Zhou, G. Li and C. B. Park, RSC Adv., 2015, 5, 106981–106988 RSC.
- S. K. Yeh, Y. C. Liu, W. Z. Wu, K. C. Chang, W. J. Guo and S. F. Wang, J. Cell. Plast., 2013, 49, 119–130 CrossRef.
- Y. L. Yang and M. C. Gupta, Nano Lett., 2005, 5, 2131–2134 CrossRef PubMed.
- J. Shen, C. C. Zeng and L. J. Lee, Polymer, 2005, 46, 5218–5224 CrossRef.
- S. Q. Liu, R. Eijkelenkamp, J. Duvigneau and G. J. Vancso, ACS Appl. Mater. Interfaces, 2017, 9, 37929–37940 Search PubMed.
- W. T. Zhai, J. Yu, L. C. Wu, W. M. Ma and J. S. He, Polymer, 2006, 47, 7580–7589 CrossRef.
- A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef PubMed.
- G. Horvath and K. Kawazoe, J. Chem. Eng. Jpn., 1983, 16, 470–475 CrossRef.
- D.-Y. Koh, B. A. McCool, H. W. Deckman and R. P. Lively, Science, 2016, 353, 804–807 CrossRef PubMed.
- J. I. Velasco, C. Morhain, A. B. Martínez, M. A. Rodríguez-Pérez and J. A. de Saja, Polymer, 2002, 43, 6813–6819 CrossRef.
- Z. Xing, G. Wu, S. Huang, S. Chen and H. Zeng, J. Supercrit. Fluids, 2008, 47, 281–289 CrossRef.
- Z. Xing, M. Wang, G. Du, T. Xiao, W. Liu, Q. Dou and G. Wu, J. Supercrit. Fluids, 2013, 82, 50–55 CrossRef.
- J.-B. Bao, A. N. Junior, G.-S. Weng, J. Wang, Y.-W. Fang and G.-H. Hu, J. Supercrit. Fluids, 2016, 111, 63–73 CrossRef.
- G. Ji, W. Zhai, D. Lin, Q. Ren, W. Zheng and D. W. Jung, Ind. Eng. Chem. Res., 2013, 52, 6390–6398 CrossRef.
- L. Cui, P. Wang, Y. Zhang, L. Zhang, Y. Chen, L. Wang, L. Liu and X. Guo, RSC Adv., 2017, 7, 42783–42791 RSC.
- S. N. Leung, A. Wong, L. C. Wang and C. B. Park, J. Supercrit. Fluids, 2012, 63, 187–198 CrossRef.
- A. Wong and C. B. Park, Chem. Eng. Sci., 2012, 75, 49–62 CrossRef.
- R. Verdejo, R. Stämpfli, M. Alvarez-Lainez, S. Mourad, M. Rodriguez-Perez, P. Brühwiler and M. Shaffer, Compos. Sci. Technol., 2009, 69, 1564–1569 CrossRef.
- C. B. Park, A. H. Behravesh and R. D. Venter, Polym. Eng. Sci., 1998, 38, 1812–1823 Search PubMed.
- K. A. Arora, A. J. Lesser and T. J. McCarthy, Polym. Eng. Sci., 1998, 38, 2055–2062 Search PubMed.
- D. Miller and V. Kumar, Polymer, 2011, 52, 2910–2919 CrossRef.
- J. Zhao, Z. Wang, H. Wang and G. Zhou, RSC Adv., 2017, 7, 36662–36669 RSC.
| This journal is © The Royal Society of Chemistry 2018 |