
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient access to amides of the carborane carboxylic acid [1-(COOH)–CB11H11]−†
Yunjun Shen‡
,
Kai Zheng‡,
Rakesh Dontha,
Yani Pan,
Jiyong Liu and
Simon Duttwyler *
*
Department of Chemistry, Zhejiang University, Zheda Road 38, 310027 Hangzhou, P.R. China. E-mail: duttwyler@zju.edu.cn
First published on 19th June 2018
Abstract
The preparation of the carborane acid chloride [1-(COCl)–CB11H11]− from the carboxylic acid [1-(COOH)–CB11H11]− is reported. This acid chloride exhibits remarkable inertness towards moisture and can be stored under ambient conditions for several months. Reaction with amines affords secondary and tertiary carborane amides [1-(CONR1R2)–CB11H11]− in moderate to high yields under mild conditions. Two of the amide products were characterized by X-ray crystallography in addition to spectroscopic analysis. Preliminary studies show that the amides can be reduced to the corresponding amines and that the acid chloride has the potential to serve as a starting material for carborane ester formation.
Introduction
Owing to their unique steric and electronic properties, boron-based clusters have been the focus of a growing body of studies that are concerned with their fundamental properties, new methods of their preparation and their applications. Carboranes based on the scaffolds [CB11H12]− and C2B10H12 are icosahedral clusters in which one or two B–H vertices are replaced by C–H (Fig. 1a). Their three-dimensional delocalization of electron density is frequently called σ aromaticity, in reference to the π aromaticity of organic arenes; it leads to exceptional chemical and thermal stability.1,2 A large number of applications of carboranes have emerged over the past decade; their utility has become evident in ligand design,3 supramolecular4 and medicinal5 chemistry, as well as fluorescence/phosphorescence6 and materials science.7 Weakly coordinating halogenated derivatives of 1 are the anions of choice as counterions for cationic reactive intermediates and of highly active catalysts.8 Provided the practical versatility of such boron cage compounds, expedient methods that provide access to tailor-made derivatives are highly desirable. In many cases, the focus lies on the construction of inorganic–organic hybrid molecules, i.e., coupling of a cluster with an organic molecule.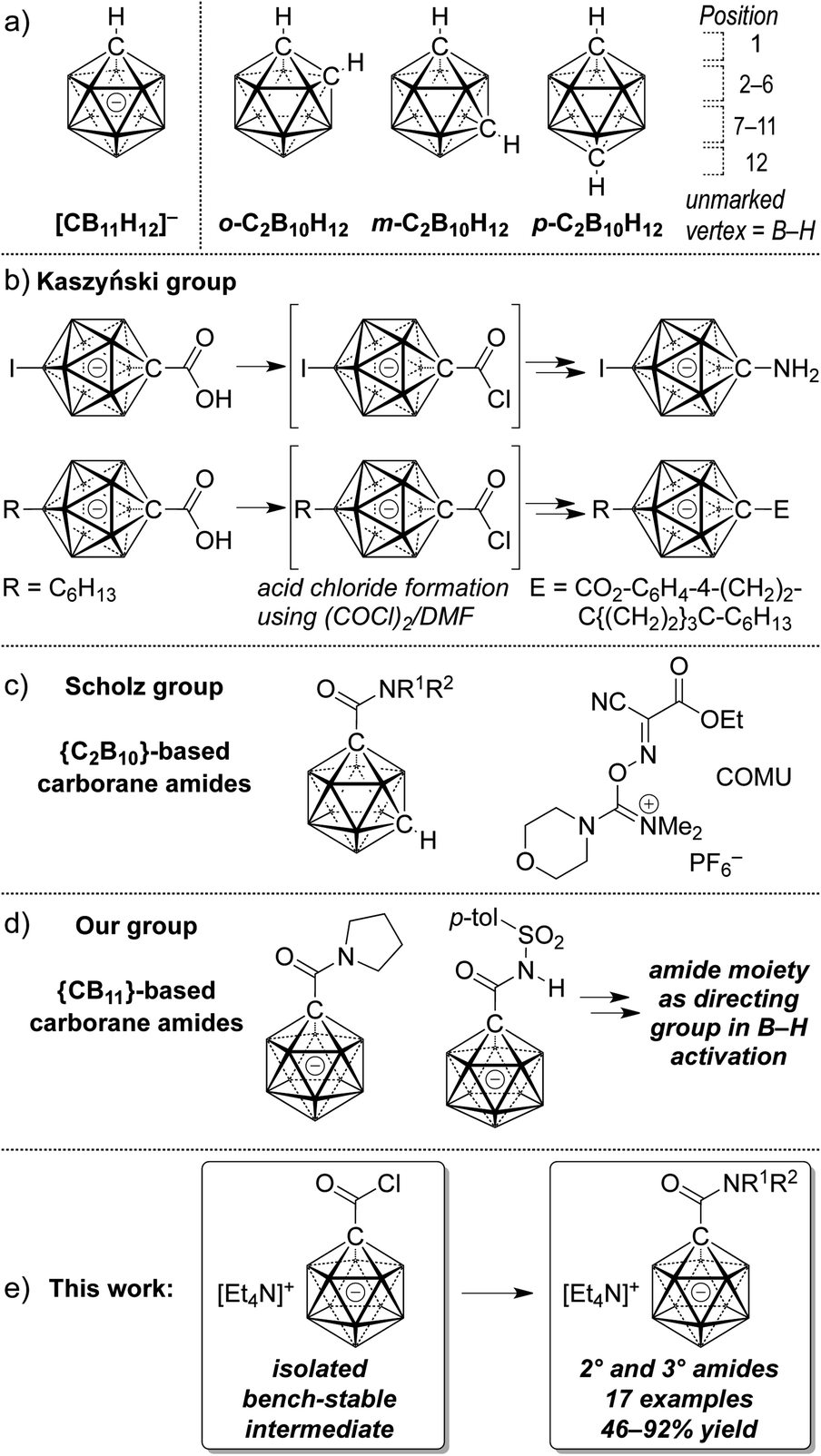 | ||
| Fig. 1 General structure of icosahedral carboranes (a), reported carborane esters and carboxamides (b–d) and outline of the present study (e). | ||
Early reports on the formation of neutral dicarba-closo-dodecaborane carboxylic acid derivatives stem from Kahl and coworkers.9 Starting from carboxylic acids, acid chlorides were prepared using phosphorous pentachloride; the isolation of 1,2-dicarbo-closo-dodecaborane-1-carbonyl chloride by distillation was reported in 1999.9b Subsequent transformations involved the synthesis of carborane carboxylic acid esters. Kaszyński and coworkers have synthesized a large number of monocarba-closo-dodecaborate-based materials based on C1 vertex ester functionalization.10 Their protocols used oxalyl chloride/dimethylformamide to convert the C1-carboxylic acids to acid chloride intermediates that were not isolated but directly treated with nucleophiles (Fig. 1b).
With respect to medicinal chemistry in particular, carboranes have been used in boron neutron capture therapy, and they have also been demonstrated to be potent pharmacophores, acting as agonists or inhibitors of enzymes.11 The amide group is one of the most important functional groups in organic chemistry and biochemistry, and amide moieties are present in peptides, synthetic polymers, many natural products and marketed drugs.12 It is in this context that the Scholz group has recently reported on the synthesis of dicarbaborane amides (Fig. 1c).13 Their protocol relied on the use of the reagent (1-cyano-2-ethoxy-2-oxoethylideneaminooxy)-(dimethylamino)morpholinocarbenium hexafluorophosphate (COMU), which allowed the coupling of carborane carboxylic acids with amines. Our group's interest in the functionalization of anionic carboranes led us to probe amide moieties as directing groups for B–H activation mediated by transition metals. We found that the carborane carboxylic acid [1-(COOH)–CB11H11]− can be transformed to pyrrolidine and tosyl amides after activation by a combination of oxalyl chloride and dimethyl formamide (Fig. 1d).14 In the current study we show that the corresponding acid chloride is an intermediate that can be isolated in high yield and stored for a prolonged time under ambient conditions. It undergoes substitution with a variety of amines to give secondary and tertiary amides in moderate to high yields (Fig. 1e). X-ray crystal structures and additional transformations are presented as well.
Results and discussion
At the outset of our study, we decided to investigate the activation of [1-(COOH)–CB11H11]− more thoroughly. Treatment of the acid with 1 equivalent of oxalyl chloride and catalytic amounts of dimethyl formamide in dichloromethane caused clean formation of the acid chloride 1 within 30 min at 25 °C, as indicated by 11B NMR spectroscopy and ESI-mass spectrometry (Scheme 1). This species was subsequently successfully isolated by either a classical work-up including removal of volatiles or by simple precipitation upon addition of hexane to the reaction mixture (see the ESI† for details). Both procedures furnished 1 in 87% yield. Acid chloride 1 exhibits high inertness towards moisture; [Et4N][1], stored as a powder in a closed vial under air, remained unchanged over several months. The hydrolysis of [Et4N][1] in a mixture of acetonitrile/water (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) was studied by NMR spectroscopy, and the details of the monitoring are provided in the ESI file (Fig. S1 and S2†). A comparison of the 11B{1H} NMR spectra of [1-(COOH)–CB11H11]− and 1 showed that these two species can be distinguished clearly based on the chemical shifts of their B12 and B2–6/7–11 positions (Fig. 2a and b).
:1 v/v) was studied by NMR spectroscopy, and the details of the monitoring are provided in the ESI file (Fig. S1 and S2†). A comparison of the 11B{1H} NMR spectra of [1-(COOH)–CB11H11]− and 1 showed that these two species can be distinguished clearly based on the chemical shifts of their B12 and B2–6/7–11 positions (Fig. 2a and b).
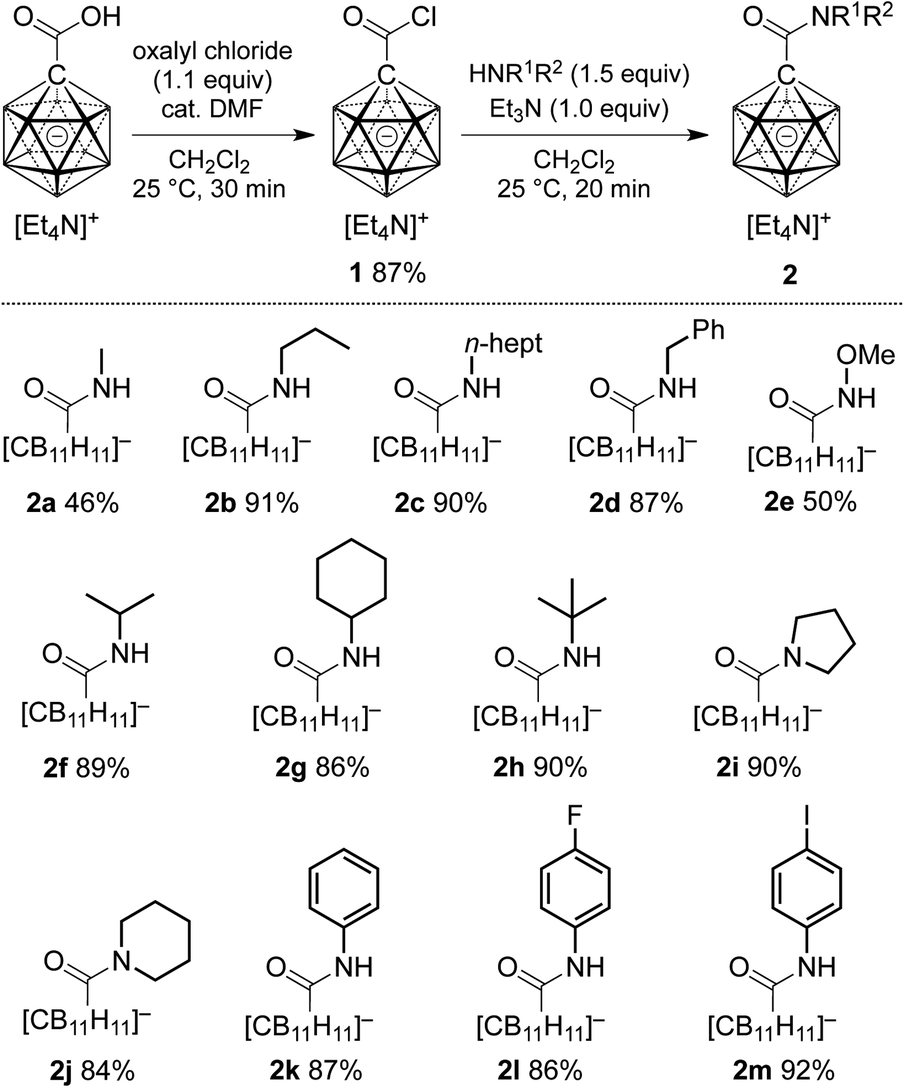 | ||
| Scheme 1 Preparation of carboxylic acid chloride 1 and subsequent synthesis of carborane amides 2. Yields are isolated yields with respect to 1. | ||
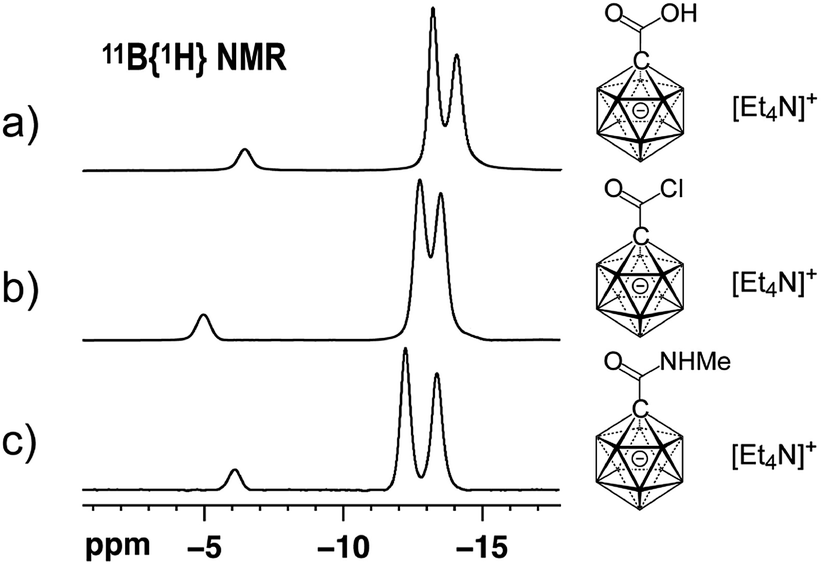 | ||
| Fig. 2 11B{1H} NMR spectra of [Et4N][1-(COOH)–CB11H11] (a), [Et4N][1] (b) and [Et4N][2a] (c) (acetone-d6, 22 °C, 160 MHz for 11B). | ||
We next probed the reactivity of 1 towards amines with the aim to prepare a series of carborane amides. Reaction with amines took place at a much higher rate than with water. Addition of 1.5 equivalents of amine cleanly afforded the corresponding amide in dichloromethane within 20 min at 25 °C; one equivalent of triethylamine was used to neutralize the HCl byproduct (Scheme 1). Primary unbranched amines afforded products 2a–e in yields of 46–91%. Sterically more congested amines afforded the isopropyl, cyclohexyl and tert-butyl derivatives 2f–h, while pyrrolidine and piperidine gave tertiary amides 2i and 2j. Anilines proved to be viable sub-strates as well, as demonstrated by the formation of 2k–m. Compounds 2f–m were obtained in high yields of 84–92%. Isolation of amides 2 involved either simple precipitation or purification by silica gel column chromatography. In their 11B NMR spectra, all products exhibited a characteristic B12 resonance at −6 to −7 ppm; as a representative example, the 11B{1H} NMR spectrum of 2a is depicted in Fig. 2c.
Cage halogenation affects the properties of boron clusters (e.g., polarity, coordinating ability, solubility) and offers the possibility of further functionalization by transition metal-catalyzed cross coupling.2a,2b,15 Amide formation was probed starting from brominated and iodinated carborane carboxylic acids 3 (Scheme 2). Applying reaction conditions identical to the ones for the synthesis of 2, amides 4a–d were obtained in yields of 81–91%. In these cases, the acid chloride was precipitated from the initial reaction mixture, collected by filtration and used directly in the amide formation step.
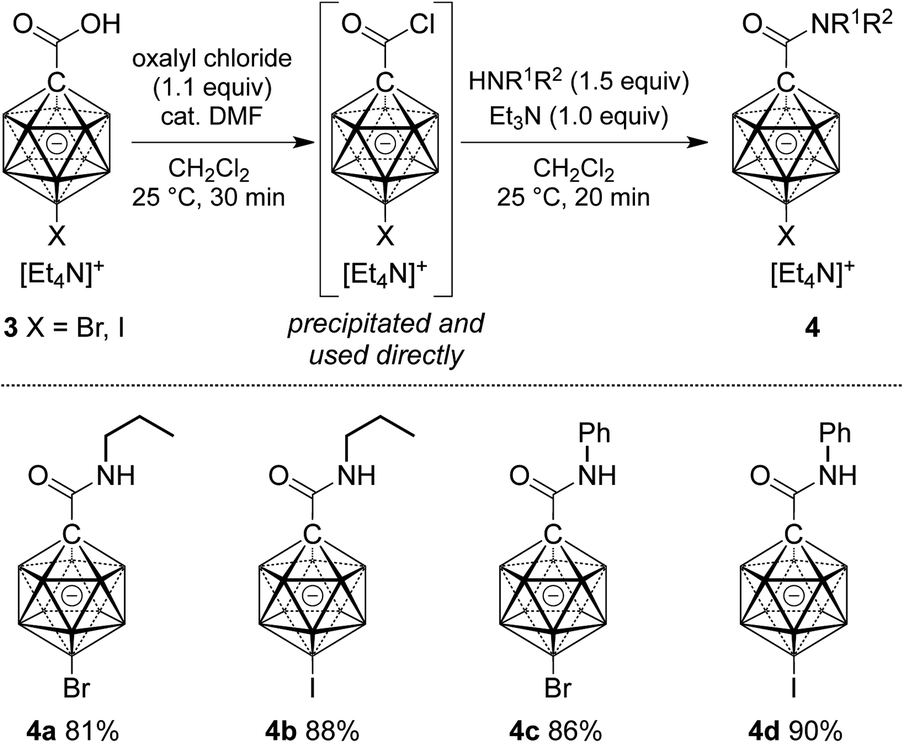 | ||
| Scheme 2 Synthesis of carborane amides 4 starting from halogenated carborane precursors. Yields are isolated yields with respect to 3. | ||
X-ray crystallography revealed the solid-state structures of 2e and 2m. Single crystals of the composition [Na][Et4N][2e]2 were obtained from an acetonitrile solution in the presence of one equivalent of Na+ by slow evaporation (see the ESI† for details). The unit cell contains two {[Na]2[Et4N]2[2e]4}2 dimers, both of which feature a center of inversion and coordination of the Na+ centers by oxygen and nitrogen astoms (Fig. 3 and S1†). The [Et4N]+ cations are well-separated from the anions. The different carboxamides exhibit similar geometries, and the structural parameters of one of them will be summarized (Fig. 4a). Generally, there is a pronounced resemblance to typical organic amides. Observed distances (Å) for 2e are C1–C2 1.525(8), C2–O1 1.214(8) and C2–N1 1.339(9). The sum of angles around C2 is 360.0(6)°, and the torsion angle O2–N1–C2–C1 of 177.1(5)° indicates coplanarity of the entire amide moiety. Solvent layering of an acetone solution of 2m with hexane gave single crystals of the composition [Et4N][2m] (Fig. 4b and S2†). The distances (Å) and angles are similar to those of 2e, namely, C1–C2 1.506(8), C2–O1 1.223(8), C2–N1 1.359(8), sum of angles around C2 360.0(6)° and torsion angle C3–N1–C2–C1 178.7(6)°.
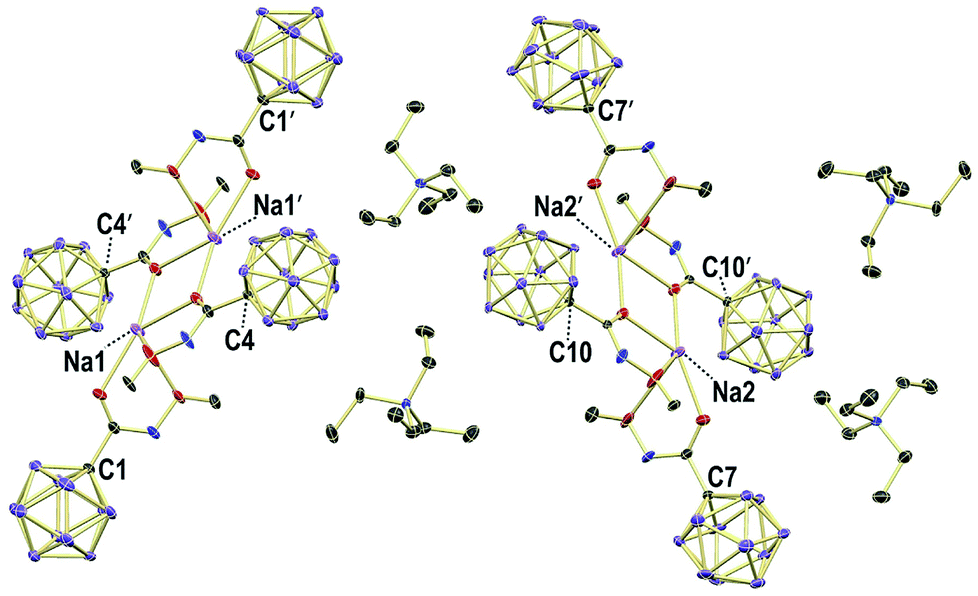 | ||
| Fig. 3 Packing of 2e in crystals of the composition Na[Et4N][2e]2; H atoms omitted for clarity; 30% displacement ellipsoids. | ||
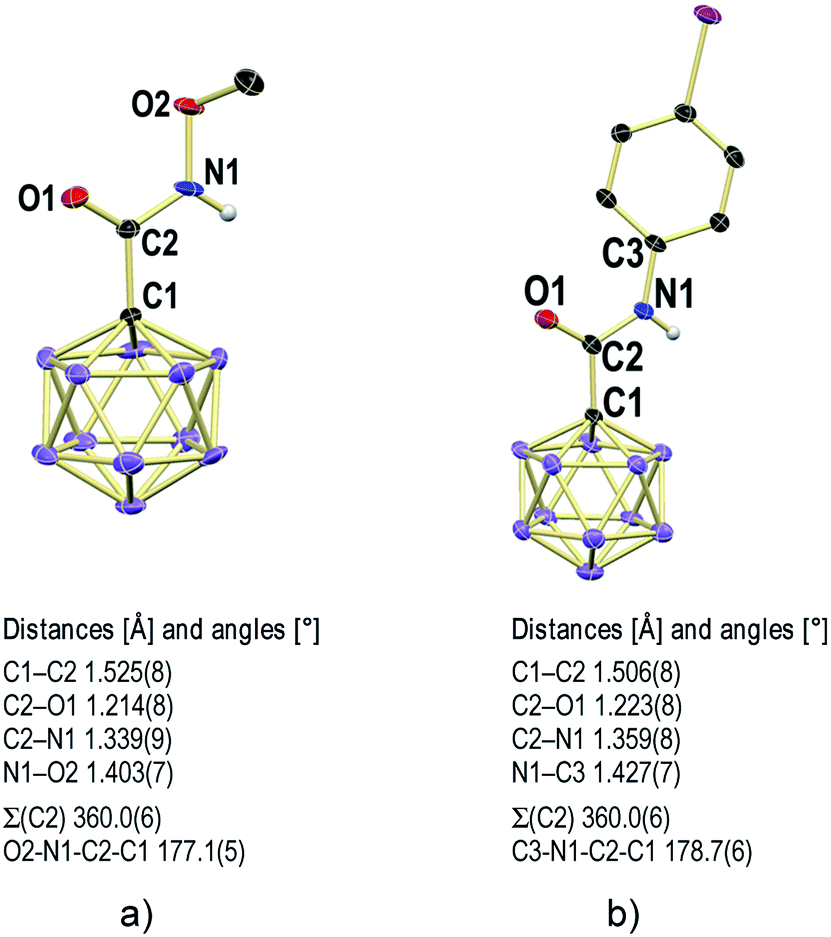 | ||
| Fig. 4 X-ray crystal structures of 2e (a) and 2m (b). Cations and H atoms except for N–H omitted for clarity; 30% displacement ellipsoids. | ||
Finally, the potential of 1 as a precursor of carborane esters and the reduction of two selected amides 2 were investigated. As anticipated from the slow reaction of 1 with water, ester formation required more forcing conditions than amide formation. Heating a solution of 1 in methanol at 50 °C for 5 h gave methyl ester 5 in 91% yield (Scheme 3a). Treatment of 2i and 2j with an excess of lithium aluminum hydride caused clean reduction to the corresponding amines (Scheme 3b). Isolation under acidic conditions afforded products 6a and 6b in as N-protonated zwitterions in yields of 88% and 91%, respectively.
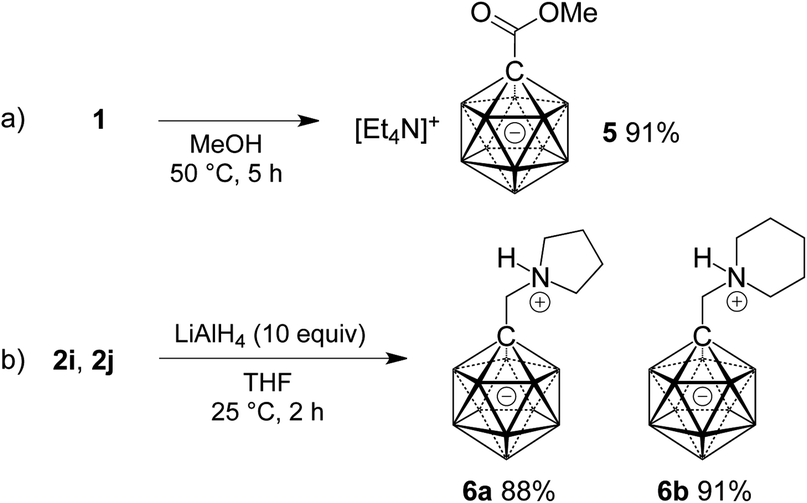 | ||
| Scheme 3 Ester formation starting from 1 (a) and reduction of amides 2i and 2j (b). | ||
Conclusions
In conclusion, the results of this study demonstrate that the carborane acid chloride 1 can be conveniently prepared and stored in the form of [Et4N][1] under ambient conditions without noticeable decomposition. In combination with amines it affords secondary and tertiary carborane amides 2 under mild conditions and generally in high yields. Additional transformations such as ester formation and reduction of products 2 to amines underscore the potential of 1 as a versatile starting material towards C1-substituted derivatives of the monocarba-closo-dodedaborate anion. We are currently exploring transformations of 1 with other nucleophiles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (grant 21472166), the Chinese “1000 Young Talents Plan”, and the National Basic Research Program of China (973 Project, grant 2015CB856500).Notes and references
- (a) R. N. Grimes, Carboranes, Elsevier, Amsterdam, 3rd edn, 2016 Search PubMed; (b) N. S. Hosmane, Boron Science: New Technologies and Applications, Taylor & Francis/CRC, Boca Raton, 2011 CrossRef.
- (a) C. Douvris and J. Michl, Chem. Rev., 2013, 113, PR179–PR233 CrossRef PubMed; (b) C. Knapp, Weakly Coordinating Anions: Halogenated Borates and Dodecaborates in Comprehensive Inorganic Chemistry II, Elsevier, Amsterdam, 2013, vol. 1, pp. 651–679 Search PubMed; (c) R. N. Grimes, Dalton Trans., 2015, 44, 5939–5956 RSC; (d) J. Poater, M. Solà and F. Teixidor, Chem.–Eur. J., 2016, 22, 7437–7443 CrossRef PubMed; (e) P. Melichar, D. Hnyk and J. Fanfrlík, Phys. Chem. Chem. Phys., 2018, 20, 4666–4675 RSC; (f) J. C. Axtell, L. M. A. Saleh, E. A. Qian, A. I. Wixtrom and A. M. Spokoyny, Inorg. Chem., 2018, 57, 2333–2350 CrossRef PubMed.
- (a) M. Finze, J. A. P. Sprenger and B. B. Schaack, Dalton Trans., 2010, 39, 2708–2716 RSC; (b) A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef PubMed; (c) Z.-J. Yao and G.-X. Jin, Coord. Chem. Rev., 2013, 257, 2522–2535 CrossRef; (d) A. El-Hellani and V. Lavallo, Angew. Chem., Int. Ed., 2014, 53, 4489–4493 CrossRef PubMed; (e) M. J. Asay, S. P. Fisher, S. E. Lee, F. S. Tham, D. Borchardt and V. Lavallo, Chem. Commun., 2015, 51, 5359–5362 RSC; (f) L. E. Riley, A. P. Y. Chan, J. Taylor, W. Y. Man, D. Ellis, G. M. Rosair, A. J. Welch and I. B. Sivaev, Dalton Trans., 2016, 45, 1127–1137 RSC; (g) J. Estrada, C. A. Lugo, S. G. McArthur and V. Lavallo, Chem. Commun., 2016, 52, 1824–1826 RSC; (h) A. L. Chan, J. Estrada, C. E. Kefalidis and V. Lavallo, Organometallics, 2016, 35, 3257–3260 CrossRef; (i) S. P. Fisher, A. El-Hellani, F. S. Tham and V. Lavallo, Dalton Trans., 2016, 45, 9762–9765 RSC; (j) J. Holmes, C. M. Pask, M. A. Fox and C. E. Willans, Chem. Commun., 2016, 52, 6443–6446 RSC; (k) Y.-P. Zhou, S. Raoufmoghaddam, T. Szilvási and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 12868–12872 CrossRef PubMed; (l) F. Šembera, J. Plutnar, A. Higelin, Z. Janoušek, I. Císařová and J. Michl, Inorg. Chem., 2016, 55, 3797–3806 CrossRef PubMed; (m) P. Coburger, J. Schulz, J. Klose, B. Schwarze, M. B. Sárosi and E. Hey-Hawkins, Inorg. Chem., 2017, 56, 292–304 CrossRef PubMed; (n) C. Selg, W. Neumann, P. Lönnecke, E. Hey-Hawkins and K. Zeitler, Chem.–Eur. J., 2017, 23, 7932–7937 CrossRef PubMed; (o) J. Estrada and V. Lavallo, Angew. Chem., Int. Ed., 2017, 56, 9906–9909 CrossRef PubMed.
- (a) H. Jude, H. Disteldorf, S. Fischer, T. Wedge, A. M. Hawkridge, A. M. Arif, M. F. Hawthorne, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2005, 127, 1231–12139 CrossRef PubMed; (b) S.-L. Huang, L.-H. Weng and G.-X. Jin, Dalton Trans., 2012, 41, 11657–11662 RSC; (c) L. Kobr, K. Zhao, Y. Shen, R. K. Shoemaker, C. T. Rogers and J. Michl, Adv. Mater., 2013, 25, 443–448 CrossRef PubMed; (d) R. D. Kennedy, V. Krungleviciute, D. J. Clingerman, J. E. Mondloch, Y. Peng, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, J. T. Hupp, T. Yildirim, O. K. Farha and C. A. Mirkin, Chem. Mater., 2013, 25, 3539–3543 CrossRef; (e) Y.-F. Han and G.-X. Jin, Acc. Chem. Res., 2014, 47, 3571–3579 CrossRef PubMed; (f) C. E. Housecroft, J. Organomet. Chem., 2015, 798, 218–228 CrossRef; (g) D. J. Clingerman, W. Morris, J. E. Mondloch, R. D. Kennedy, A. A. Sarjieant, C. Stern, J. T. Hupp, O. K. Farha and C. A. Mirkin, Chem. Commun., 2015, 51, 6521–6523 RSC; (h) S. Rodríguez-Hermida, M. Y. Tsang, C. Vignatti, K. C. Stylianou, V. Guillerm, J. Pérez-Carvajal, F. Teixidor, C. Viñas, D. Choquesillo-Lazarte, C. Verdugo-Escamilla, I. Peral, J. Juanhuix, A. Verdaguer, I. Imaz, D. Maspoch and J. Giner Planas, Angew. Chem., Int. Ed., 2016, 55, 16049–16053 CrossRef PubMed; (i) M. Y. Tsang, S. Rodríguez-Hermida, K. C. Stylianou, F. Tan, D. Negi, F. Teixidor, C. Viñas, D. Choquesillo-Lazarte, C. Verdugo-Escamilla, M. Guerrero, J. Sort, J. Juanhuix, D. Maspoch and J. Giner Planas, Cryst. Growth Des., 2017, 17, 846–857 CrossRef.
- (a) I. B. Sivaev and V. V. Bregadze, Eur. J. Inorg. Chem., 2009, 1433–1450 CrossRef; (b) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef PubMed; (c) M. Scholz and E. H-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef PubMed; (d) D. Gabel, Pure Appl. Chem., 2015, 87, 173–179 Search PubMed; (e) Z. J. Leśnikowski, J. Med. Chem., 2016, 59, 7738–7758 CrossRef PubMed.
- (a) Y.-J. Cho, S.-Y. Kim, M. Cho, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 18, 9702–9708 RSC; (b) B. H. Choi, J. H. Lee, H. Hwang, K. M. Lee and M. H. Park, Organometallics, 2016, 35, 1771–1777 CrossRef; (c) B. P. Dash, R. Satapathy, E. R. Gaillard, J. A. Maguire and N. S. Hosmane, J. Am. Chem. Soc., 2016, 132, 6578–6587 CrossRef PubMed; (d) S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC; (e) J. C. Axtell, K. O. Kirlikovali, P. I. Djurovich, D. Jung, V. T. Nguyen, B. Munekiyo, A. T. Royappa, A. L. Rheingold and A. M. Spokoyny, J. Am. Chem. Soc., 2016, 138, 15758–15765 CrossRef PubMed; (f) R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed; (g) H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef PubMed; (h) A. Ferrer-Ugalde, J. Cabrera-González, E. J. Juárez-Pérez, F. Teixidor, E. Pérez-Inestrosa, J. M. Montenegro, R. Sillanpää, M. Haukka and R. Núñez, Dalton Trans., 2017, 46, 2091–2104 RSC; (i) N. V. Nghia, J. Oh, J. Jung and M. H. Lee, Organometallics, 2017, 36, 2573–2580 CrossRef; (j) N. Shin, S. Yu, J. H. Lee, H. Hwang and K. M. Lee, Organometallics, 2017, 36, 1522–1529 CrossRef; (k) M. Hailmann, N. Wolf, R. Renner, B. Hupp, A. Steffen and M. Finze, Chem.–Eur. J., 2017, 23, 11684–11693 CrossRef PubMed.
- (a) K. Tanaka and Y. Chujo, Macromol. Rapid Commun., 2012, 33, 1235–1255 CrossRef PubMed; (b) E. G. Cansu-Ergun and A. Cihaner, Mater. Chem. Phys., 2013, 143, 387–392 CrossRef; (c) J. Pecyna, D. Pociecha and P. Kaszyński, J. Mater. Chem. C, 2014, 2, 1585–1591 RSC; (d) T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizunl, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem., Int. Ed., 2014, 53, 3173–3177 CrossRef PubMed; (e) S. G. McArthur, L. Geng, J. Guo and V. Lavallo, Inorg. Chem. Front., 2015, 2, 1101–1104 RSC; (f) J. Pecyna, P. Kaszyński, B. Ringstrand, D. Pociecha, S. Pakhomov, A. G. Douglass and V. G. Young Jr, Inorg. Chem., 2016, 55, 4016–4025 CrossRef PubMed; (g) R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC; (h) R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef PubMed; (i) S. G. McArthur, R. Jay, L. Geng, J. Guo and V. Lavallo, Chem. Commun., 2017, 53, 4453–4456 RSC; (j) Y. Zhu and N. S. Hosmane, J. Organomet. Chem., 2017, 849–850, 286–292 CrossRef.
- (a) K.-C. Kim, C. A. Reed, D. W. Elliott, L. J. Mueller, F. Tham, L. Lin and J. B. Lambert, Science, 2002, 297, 825–827 CrossRef PubMed; (b) T. Kato and C. A. Reed, Angew. Chem., Int. Ed., 2004, 43, 2908–2911 CrossRef PubMed; (c) C. Douvris and O. V. Ozerov, Science, 2008, 31, 1188–1190 CrossRef PubMed; (d) O. Allemann, S. Duttwyler, P. Romanato, K. K. Baldridge and J. S. Siegel, Science, 2011, 332, 574–577 CrossRef PubMed; (e) C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef PubMed; (f) Y. Shoji, N. Tanaka, K. Mikami, M. Uchiyama and T. Fukushima, Nat. Chem., 2014, 6, 498–503 CrossRef PubMed; (g) B. Shao, A. L. Bagdasarian, S. Popov and H. M. Nelson, Science, 2017, 355, 1403–1407 CrossRef PubMed.
- (a) S. B. Kahl, Tetrahedron Lett., 1990, 31, 1517–1520 CrossRef; (b) R. A. Kasar, G. M. Knudsen and S. T. Kahl, Inorg. Chem., 1999, 38, 2936–2940 CrossRef PubMed.
- (a) A. Jankowiak, J. Kanazawa, P. Kaszyński, R. Takita and M. Uchiyama, J. Organomet. Chem., 2013, 747, 195–200 CrossRef; (b) J. Pecyna, D. Pociecha and P. Kaszyński, J. Mater. Chem. C, 2014, 2, 1585–1591 RSC; (c) J. Pecyna, R. P. Denicola, H. M. Gray, B. Ringstrand and P. Kaszyński, Liq. Cryst., 2014, 41, 1188–1198 CrossRef; (d) A. Jankowiak, A. Sivaramamoorthy, D. Pociecha and P. Kaszyński, RSC Adv., 2014, 4, 53907–53914 RSC.
- For selected recent publications on carboranes in medicinal chemistry, see the following references and also ref. 5e: (a) M. B. Sarosi, W. Neumann, T. P. Lybrand and E. Hey-Hawkins, J. Chem. Inf. Model., 2017, 57, 2056–2067 CrossRef PubMed; (b) R. Kawasaki, Y. Sasaki and K. Akiyoshi, Biochem. Biophys. Res. Commun., 2017, 483, 147–152 CrossRef PubMed; (c) K. Ohta, T. Ogawa and Y. Endo, Bioorg. Med. Chem. Lett., 2017, 27, 4030–4033 CrossRef PubMed; (d) I. Takeuchi, K. Nomura and K. Makino, Colloids Surf., B, 2017, 159, 360–365 CrossRef PubMed; (e) A. Kaise, K. Ohta, C. Shirata and Y. Endo, Bioorg. Med. Chem., 2017, 25, 6417–6426 CrossRef PubMed; (f) A. Kaise, K. Ohta and Y. Endo, Bioorg. Med. Chem., 2017, 25, 6371–6378 CrossRef PubMed; (g) D. C. Tribovane and M. S. Scholz, Chem. Phys. Lipids, 2018, 210, 149–154 CrossRef PubMed; (h) S. C. Kohler, S. Vandati, M. S. Scholz and M. Wiese, Eur. J. Med. Chem., 2018, 146, 483–500 CrossRef PubMed; (i) G. Calabrese, A. Daou, E. Barbu and J. Tsibouklis, Drug Discovery Today, 2018, 23, 63–75 CrossRef PubMed; (j) T. Lutzenburg, I. Neundorf and M. Scholz, Chem. Phys. Lipids, 2018, 213, 62–67 CrossRef PubMed.
- J. Zabicky, Chemistry of the Amides (Patai's Chemistry of Functional Groups), John Wiley & Sons, New York, 1970 Search PubMed.
- M. Scholz and L. M. Wingen, Inorg. Chem., 2017, 56, 5510–5513 CrossRef PubMed.
- (a) Y. Shen, Y. Pan, J. Liu, T. Sattasathuchana, K. K. Baldridge and S. Duttwyler, Chem. Commun., 2016, 53, 176–179 RSC; (b) Y. Shen, Y. Pan, K. Zhang, X. Liang, B. Spingler and S. Duttwyler, Dalton Trans., 2017, 46, 3135–3140 RSC; (c) Y. Shen, J. Liu, T. Sattasathuchana, K. K. Baldridge and S. Duttwyler, Eur. J. Inorg. Chem., 2017, 4420–4424 CrossRef.
- For cross coupling reactions of carboranes, see the following reference and references cited therein: R. M. Dziedzic, L. M. A. Saleh, J. C. Axtell, J. L. Martin, S. L. Stevens, A. T. Royappa, A. L. Rheingold and A. M. Spokoyny, J. Am. Chem. Soc., 2016, 138, 9081–9084 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures and spectroscopic data. CCDC 1829932 and 1829933 for 2e and 2m. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra03067g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |