
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePractical, mild and efficient electrophilic bromination of phenols by a new I(III)-based reagent: the PIDA–AlBr3 system†
Yuvraj Satkar a,
Velayudham Ramadoss‡
a,
Pradip D. Nahide‡a,
Ernesto García-Medinaa,
Kevin A. Juárez-Ornelasa,
Angel J. Alonso-Castrob,
Ruben Chávez-Riverac,
J. Oscar C. Jiménez-Halla*a and
César R. Solorio-Alvarado*a
a,
Velayudham Ramadoss‡
a,
Pradip D. Nahide‡a,
Ernesto García-Medinaa,
Kevin A. Juárez-Ornelasa,
Angel J. Alonso-Castrob,
Ruben Chávez-Riverac,
J. Oscar C. Jiménez-Halla*a and
César R. Solorio-Alvarado*a
aUniversidad de Guanajuato, Departamento de Química, División de Ciencias Naturales y Exactas, Campus Guanajuato, Cerro de la Venada S/N, 36040, Guanajuato, Gto., México. E-mail: csolorio@ugto.mx; jjimenez@ugto.mx
bUniversidad de Guanajuato, Departamento de Farmacia, División de Ciencias Naturales y Exactas, Campus Guanajuato, Noria alta S/N, 36050, Guanajuato, Gto., México
cUniversidad Michoacana de San Nicolás de Hidalgo, Facultad de Químico Farmacobiología, Tzintzuntzan 173, col. Matamoros, Morelia, Mich., México
First published on 15th May 2018
Abstract
A practical electrophilic bromination procedure for phenols and phenol–ethers was developed under efficient and very mild reaction conditions. A broad scope of arenes was investigated, including the benzimidazole and carbazole core as well as analgesics such as naproxen and paracetamol. The new I(III)-based brominating reagent PhIOAcBr is operationally easy to prepare by mixing PIDA and AlBr3. Our DFT calculations suggest that this is likely the brominating active species, which is prepared in situ or isolated after centrifugation. Its stability at 4 °C after preparation was confirmed over a period of one month and no significant loss of its reactivity was observed. Additionally, the gram-scale bromination of 2-naphthol proceeds with excellent yields. Even for sterically hindered substrates, a moderately good reactivity is observed.
Introduction
Aryl bromides are compounds of great importance in organic chemistry.1 They appear or are frequently used in materials science,2 agrochemicals,3 natural compounds4 and pharmaceuticals5 (Fig. 1). Moreover, they constitute important building blocks for C–C bond formation, in the transition-metal-free procedures6 or metal-catalyzed Suzuki,7 Stille8,37 and Negishi9 cross-coupling reactions as well as the Mizoroki–Heck10 olefination and the Sonogashira11 alkynylation.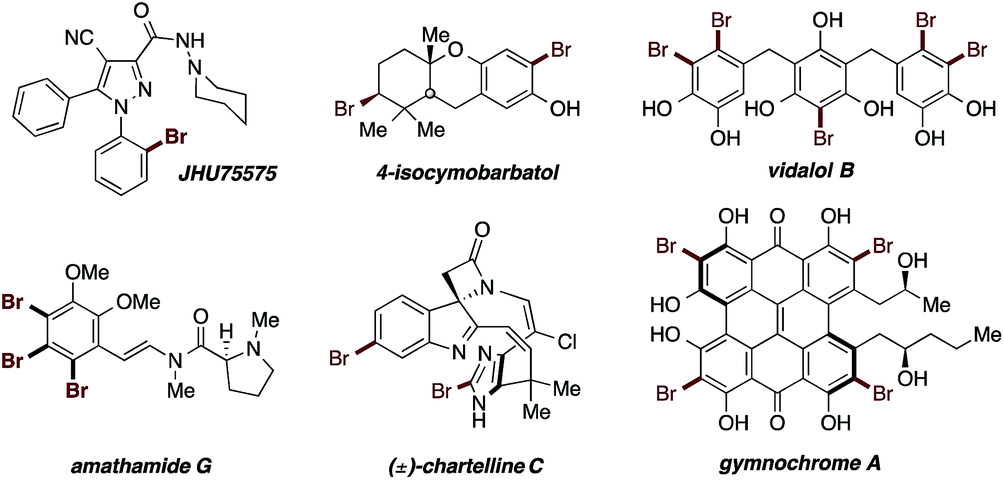 | ||
| Fig. 1 Examples highlighting the aryl bromide core relevance. | ||
To date, several procedures for the bromination of arenes have been reported. Among the most representative of these, metal-catalyzed bromination reactions using Ru,12 Rh,13 V,14 Cu,15 Pd16 or Au17 are excellent examples. On the other hand, Br2- and NBS-based brominating procedures are broadly used in the presence of organocatalysts,18 ionic liquids,19 TMSCl,20 supercritical CO2 (ref. 21) or Fe2O3–zeolite22 as additives or reaction media. Regarding the reagent-economy procedures, the oxidation of different bromide salts is a widely exploited tool. Thus, a range of different oxidant systems, such as HBr–Selectfluor®,23 HBr–H2O2,24 HBr–DMSO25 or KBr–I2O5 (ref. 26) have been successfully utilized. Finally, in the context of this work, bromide salt oxidation by I(III) reagents is a particularly elegant and useful bromination strategy. In this regard, the work of Braddock,27 Zhou28 and Evans29 should be emphasized (Scheme 1). Herein we describe an economical procedure developed for the efficient electrophilic bromination of the phenolic core, whereby an I(III) reagent oxidizes the bromine atoms of AlBr3. A number of representative advantages of this strategy over those previously published are highlighted, such as its easy handling, fast in situ formation of the brominating reagent and mild, non-toxic as well as operationally simple procedure.
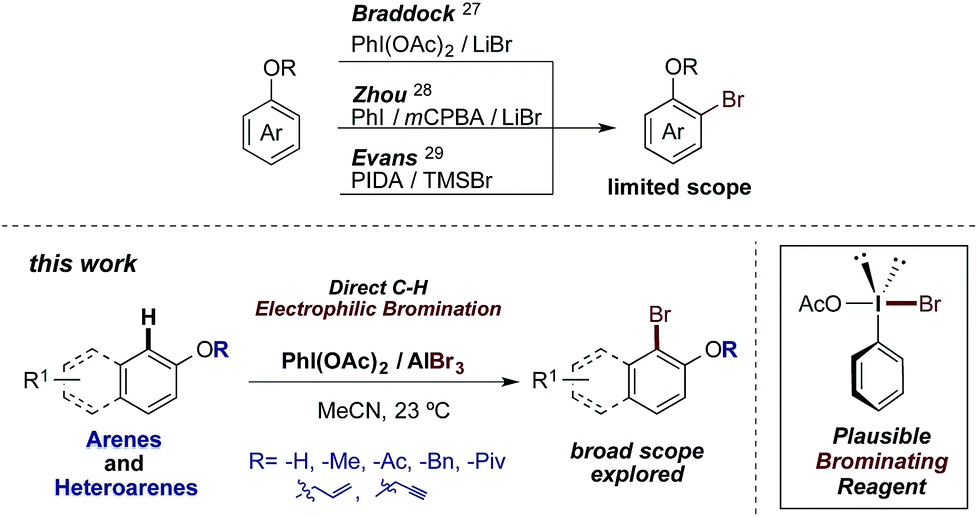 | ||
| Scheme 1 I(III)-based bromination methods. | ||
Results
Recently, we described an efficient procedure for the chlorination of electron-rich arenes using the PIFA-AlCl3 system (PIFA = PhI(OTFA)2).30 These results inspired us to extend our oxidative halogenation strategy to the bromination of arenes. In this way we decided to test the hypothesis by initially mixing PIFA and AlBr3, in order to effect the bromination of 2-naphthol. Our optimization results are summarized in Table 1.
|
|||||
|---|---|---|---|---|---|
| Entry | I(III) (equiv.) | AlBr3 equiv. | Solvent | Temp. (°C) | Yielda (%) |
| a All reactions were carried out without the use of inert atmosphere. Isolated yields are described. n.r. = no reaction observed. PIFA = PhI(OTFA)2 or [bis(trifluoroacetoxy)iodo]benzene. PIDA = PhI(OAc)2 or (diacetoxyiodo)ben-zene. c.m.r. = complex reaction mixture. | |||||
| 1 | PIFA (0.5) | 2.4 | MeCN | 23 | 28 |
| 2 | PIFA (1.0) | 2.4 | MeCN | 23 | 81 |
| 3 | PIFA (1.0) | 2.4 | MeCN | 40 | 65 |
| 4 | PIFA (1.5) | 2.4 | MeCN | 23 | 84 |
| 5 | PIFA (2.0) | 2.4 | MeCN | 23 | 78 |
| 6 | PIFA (1.0) | 2.4 | DCM | 23 | c.r.m. |
| 7 | PIDA (1.2) | 2.4 | MeCN | 23 | 93 |
| 8 | PIDA (1.0) | 1.0 | MeCN | 23 | 32 |
| 9 | PIDA (1.0) | 1.5 | MeCN | 23 | 44 |
| 10 | PIDA (1.0) | 2.0 | MeCN | 23 | 73 |
| 11 | PIDA (1.5) | 2.4 | MeCN | 23 | 70 |
| 12 | PIDA (1.0) | 2.4 | MeCN | 40 | 72 |
| 13 | PIDA (1.0) | 2.4 | DCM | 23 | c.r.m. |
| 14 | PIDA (1.0) | 2.4 | DCE | 23 | c.r.m. |
| 15 | PIDA (1.0) | 2.4 | THF | 23 | 57 |
| 16 | — | 1.0 | MeCN | 23 | n.r. |
| 17 | — | 2.0 | MeCN | 23 | n.r. |
We started the optimization by using 0.5 and 1.0 equiv. of PIFA and 2.4 equivalents of aluminium tribromide in acetonitrile at room temperature. As expected, we found that the reaction gave 1-bromo-2-naphthol in yields of 28% and 81%, respectively (entries 1 and 2). These experiments validated our hypothesis, and in principle, we could extend our previously developed oxidative halogenation procedure to the bromination of arenes. It was also evident that at least a stoichiometric amount of the I(III) reagent is necessary as oxidant for completion of the reaction. Then, maintaining the aluminium tribromide amount at 2.4 equivalents and keeping acetonitrile as solvent, one equivalent of PIFA at 40 °C was tested (entry 3), leading to a decreased yield (65%). By using 1.5 equivalents of PIFA we obtained a yield of 84% (entry 4), which is slightly higher than entry 2. The use of two equivalents of PIFA gave a lower yield, corresponding to 78% (entry 5). When the solvent was changed to dichloromethane, a very complex reaction mixture was observed (entry 6). At this point, entry 2 represented a promising result. However, we realized that while PIFA and PIDA ((diacetoxyiodo)benzene) are structurally similar reagents, commercial PIDA is ca. one-third the price of PIFA. Thus, we tested the conditions used in entry 2 with PIDA as oxidant. Gratifyingly, this reaction provided an excellent 93% yield (entry 7). The use of 1, 1.5 and 2 equivalents of aluminium tribromide (entries 8–10) did not lead to completion of the reaction presumably due to lower bromine concentration compared to entry 7 and gave moderate to poor yields (ca. 44% to 73%). We determined that 2.4 equivalents was thus the optimal amount of aluminium tribromide. Further attempts to optimize PIDA, solvent or temperature (entries 11–15), gave lower yields or complex reaction mixtures. Finally, control experiments, performed by testing only aluminium tribromide, did not produce any observable reaction. This set of experiments suggested that entry 7 is the best choice of conditions for the oxidative halogenation of arenes. To the best of our knowledge, this is the first report describing an oxidation of the bromine atoms of AlBr3 to a “Br+” equivalent using an I(III) reagent. Additionally, the change of PIFA (6 USD per g) to PIDA as the oxidant makes the reaction a synthetically and economically attractive procedure, since the starting materials are cheap: AlBr3 (2 USD per g) and PIDA (1.9 USD per g).
With the optimal conditions in hand, we proceeded to test the scope and limitations of our oxidative bromination protocol (Scheme 2).
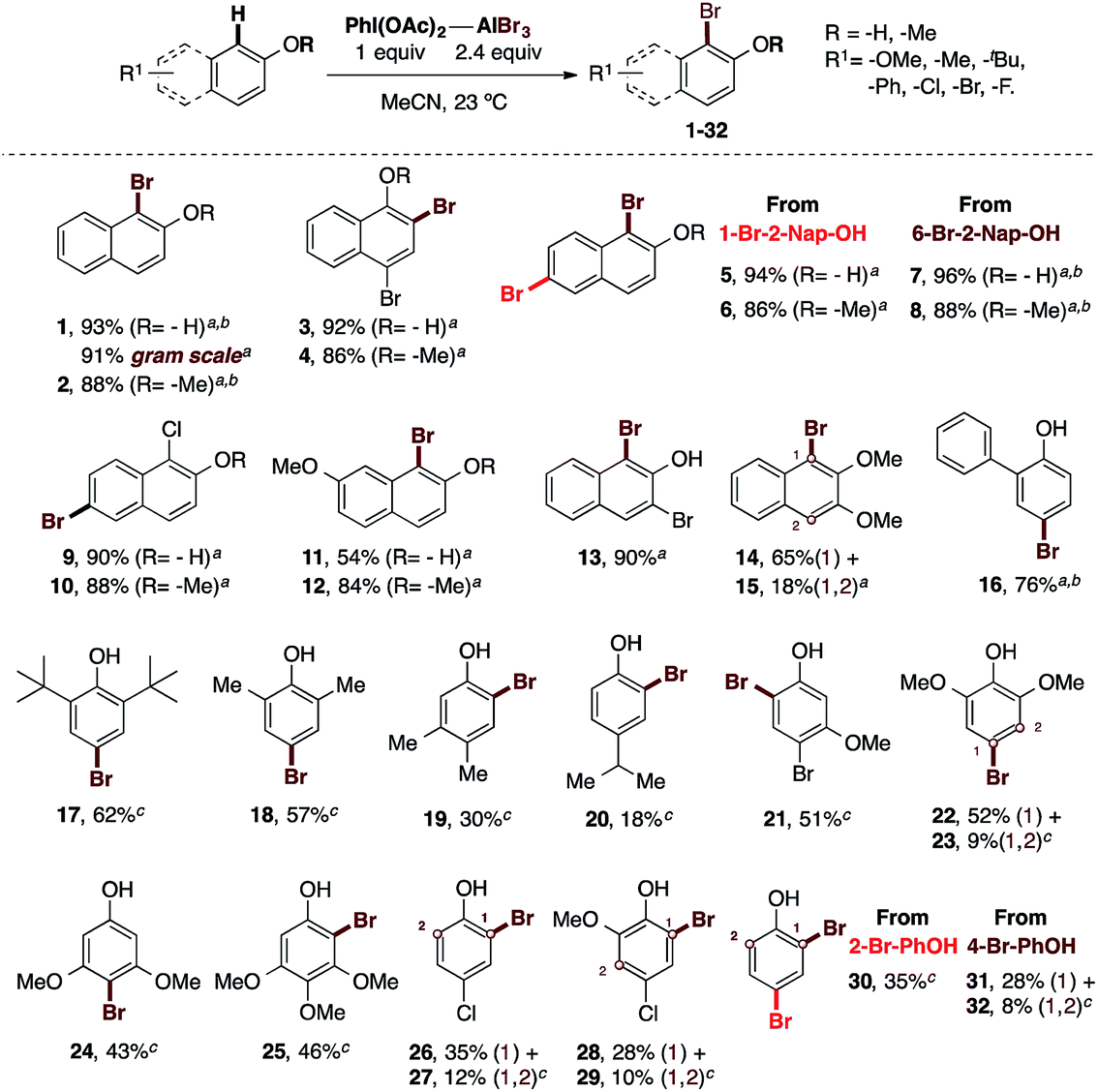 | ||
| Scheme 2 Scope of the oxidative bromination at the phenol core using the PIDA–AlBr3 system. Isolated yields are described. The reactions were carried out in the presence of 1.2 equiv. of an I(III) source as oxidant, 2.4 equiv. of AlBr3 in MeCN at 23 °C, open flask and without inert atmosphere. aReactions were carried out using PIDA as oxidant (general procedure A, see ESI†). bReactions were carried out using PIFA as oxidant (general procedure B, see ESI†). cReactions were carried out using the general procedure C (see ESI†). The newly-formed carbon–bromine bond is highlighted in red. | ||
A range of electron-rich and electron-poor phenolic substrates was tested, as well as some of their methyl-ether derivatives. Thus, 2-naphthol was brominated in 93% yield to obtain 1, and this reaction also gave an excellent 91% yield when performed on a gram-scale. This experiment demonstrated the scalability and efficiency of our procedure. 2-Methoxynaphthalene was also brominated in 88% yield to afford 2. In addition, electron-poor naphthol derivatives containing bromine and chlorine were successfully brominated (3 to 10). The bromination of 4-bromo-1-naphthol and its methyl-ether led to 3 and 4 in 86% and 92% yields, respectively. Starting from 1-bromo-2-naphthol or 6-bromo-2-naphthol, 94% and 96% yields were obtained for 5 and 7, respectively. Their corresponding methyl-ethers furnished the desired brominated derivatives in 86% to 88% yields for compounds 6 and 8, respectively. Also the bromination of 1-chloro-2-naphthol synthesized by our procedure,30 and its methyl-ether, proceeded in 90% and 88% yields, respectively. The monobromination of the electron-rich 7-methoxy-2-naphthol yielded 11 in 54% and the corresponding methyl-ether was monobrominated in 84% yield to afford 12. The 3-bromo-2-naphthol was brominated in excellent 90% yield to get 13. In the same way the 2,3-dimethoxynaphthalene was brominated to afford a separable mixture of 14 and 15 in 65% and 18% respectively. This set of experiments highlight the efficacy of our protocol applied to the bromination of naphthols. The further scope of the reaction was tested with different mono-annular phenols. Thereby, electron-rich phenols containing various aryl or alkyl groups such as phenyl, methyl, iso-propyl or tert-butyl, were successfully brominated. Compound 16 containing a phenyl ring was obtained in 76% yield. The bulky 2,6-di-tert-butylphenol provided 17 in 62% yield. We observed that the more hindered the substituents of the phenolic core are, the lower the yield is. Additionally, the typical para regioselectivity was obtained. In comparison, the less bulky but also less electron-rich 2,6-dimethylphenol furnished 18 in 57% yield. The weakly electron-donating 3,4-dimethylphenol produced the ortho-brominated derivative 19 in 30% yield. This example shows a dramatic decrease in yield, which clearly indicates that the reaction at the para position is chemically preferred and the steric hindrance in the proximity of the hydroxyl group also affects our procedure. In this regard, the bromination of 4-isopropoxyphenol was achieved in 18% yield to obtain 20, confirming our hypothesis. Thus, we identified that bulky substituents at the para position with respect to the hydroxyl group may also affect the yield. On the other hand, the presence of one, two or three strongly electron-donating methoxy groups afford moderate yields of 43% to 52% for 21 to 25. Finally, to complete this scope, a number of electron-deficient phenols were examined. Accordingly, 2-bromophenol was brominated in 35% yield to provide 30. The bromination of 4-bromophenol provided 31 in 28% yield and the bis-brominated derivative 32 in 8% yield as a separable mixture of products from a complex reaction. Similarly low yields were observed for chloro derivative. This way, 4-chlorophenol gave 26 and 27 in 35% and 12% of yield. Also the bromination of 4-chloro-2-methoxynaphthol provide 28 and 29 in 28% and 10% yields, respectively.
These results clearly delineate the scope and limitations of our protocol. The procedure is excellent for naphthols, moderately good for electron-rich mono-annular phenols, but struggles with bulky para-substituted phenols as well as those electron-poor phenols.
However, it is important to highlight the following advantages of our protocol: (1) the reagent-economy, (2) the operational ease of handling, (3) the absence of the need for an inert atmosphere, (4) the absence of the need for reagent activation, as required in the known NBS-based procedures, and (5) excellent yields are shown in general for naphthols.
Subsequently, we tested the scope of this procedure with further compounds to identify the tolerance of the reaction to different functional groups and heterocycles, which could negatively impact our brominating procedure at the naphthol moiety (Scheme 3).
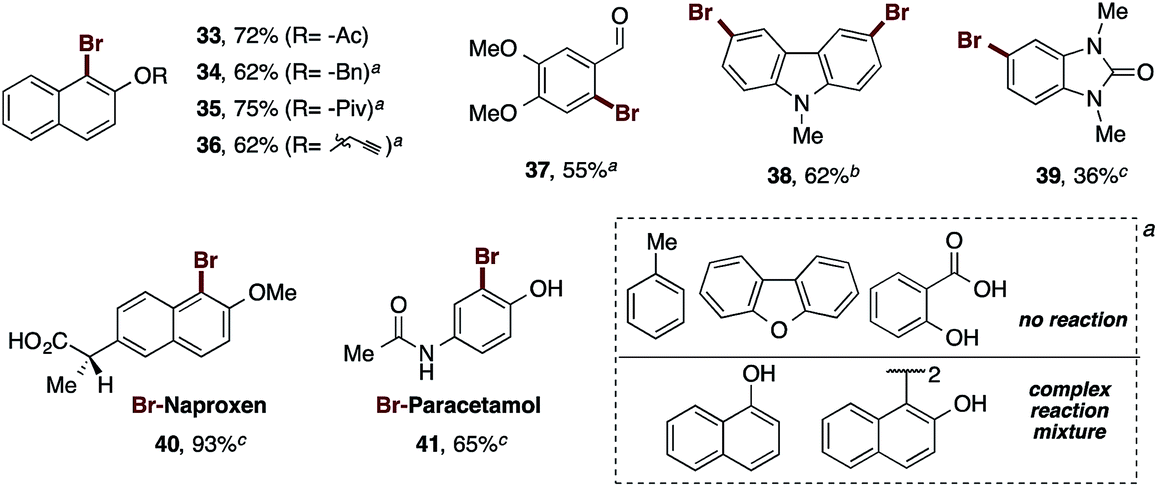 | ||
| Scheme 3 Scope of the functional groups and heterocycles used in the bromination reaction by the PIDA–AlBr3 system. Isolated yields are shown. aReactions were carried out in the presence of 1.2 equiv. of PIDA and 2.4 equiv. of AlBr3 at 80 °C. bReactions were carried out in the presence of 2.4 equiv. of PIDA and 4.8 equiv. of AlBr3 at 80 °C. cReactions were carried out in the presence of 1.2 equiv. of PIDA and 2.4 equiv. of AlBr3 at 23 °C. | ||
In this regard, the functionalization of 2-naphthol at the oxygen with the benzyl and propargyl groups was explored, in addition to acyls of assorted sizes. Thereby, the acetyl derivative yielded the corresponding brominated naphthol 33 in 72% yield. The presence of a benzyl group allowed the bromination reaction to obtain 34 to proceed in 62% yield. In this case, the amounts of the oxidant and bromine source had to be doubled, and heating to 80 °C was necessary for complete consumption of the starting material. The pivaloyl and propargyl naphthol derivatives afforded the desired compounds 35 and 36 in 75% and 62% yields, respectively, under the previous conditions.31
These tests demonstrate the excellent reactivity of our reagent, which provided good yields for sterically hindered naphthols even in the presence of bulky groups such as pivaloyl. The increased amounts of PIDA and AlBr3, and the increase of the temperature to 80 °C, still represent mild reaction conditions, and maintain the high reactivity of our brominating reagent. At this point, we tested the reaction with mono-annular phenols bearing other functional groups. Thus, the formyl group was tolerated in the phenolic core, furnishing product 37 in 55% yield. The carboxylic acid and the methyl derivatives failed under our bromination conditions. Likewise, heterocycles such as N-methylcarbazole, and N,N′-dimethylbenzimidazolone were brominated in 62% and 36% to yield 38 and 39 respectively. The dibenzofuran did not react. Finally, we sought to directly brominate some common pharmacologically active analgesics as their sodium salts. The sodium salt of naproxen gave rise to its corresponding bromo-derivative 40 in excellent yield (93%), while the sodium salt paracetamol was brominated to provide 41 in 65% yield. Examples such as 1-naphthol and the 2-naphthol dimer provided complex reaction mixtures.
During the exploration of the functional group scope of this reaction, we observed an interesting reactivity of the allyl and ester functionalities (eqn (1) and (2)).
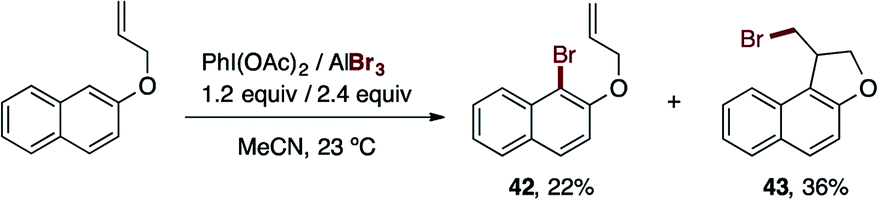 | (1) |
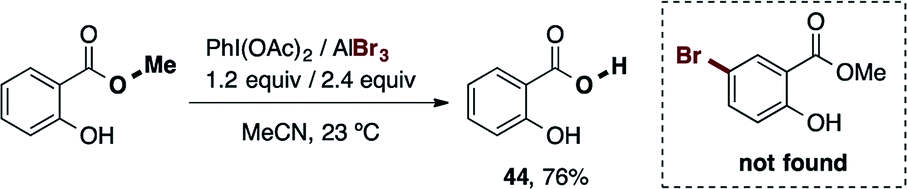 | (2) |
Specifically, the bromination of 2-(allyloxy)naphthalene (eqn (1)) afforded two compounds, the expected brominated compound 42 (22% yield) and the naphthofuran 43 (36% yield). The former product is the result of the cationic-π bromocyclization induced by our reagent. This is a very important feature which provides the possibility of applying our procedure in polyene bromocyclizations.32 While the obtained yield for 43 was not particularly high, the optimization of this cyclization reaction is a possibility for future work. On the other hand (eqn (2)), when we attempted the bromination reaction of the salicylic acid methyl-ester, the corresponding carboxylic acid 44 was obtained in 76% yield. This reaction represents a novel and mild procedure to transform the ester into carboxylic acids via halolysis.
We then carried out attempts to synthesize and characterize a plausible active brominating species from this reaction.33 Thereby, PIDA and AlBr3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) were mixed and stirred for 30 minutes at room temperature. After centrifugation and solvent concentration, we obtained a yellow-orange solid with a strong smell of bromine. Several attempts to obtain tractable 1H and 13C NMR spectra showed only decomposition, as judged from the complexity of the observed NMR signals. This observation is presumably a result of decomposition due to thermal sensitivity. Nevertheless, the evidence obtained while isolating this new solid and its organoleptic characteristics suggested to us that this is presumably the active species in the reaction. With this hypothesis in mind, we decided to test this solid as the plausible brominating species (PhIOAcBr) in the bromination of 2-naphthol. Also, to evaluate the possible thermal sensitivity of the reagent, we carried out the bromination reaction over the period of one month by comparing two storage temperatures: 23 °C and 4 °C (Scheme 4).
:2) were mixed and stirred for 30 minutes at room temperature. After centrifugation and solvent concentration, we obtained a yellow-orange solid with a strong smell of bromine. Several attempts to obtain tractable 1H and 13C NMR spectra showed only decomposition, as judged from the complexity of the observed NMR signals. This observation is presumably a result of decomposition due to thermal sensitivity. Nevertheless, the evidence obtained while isolating this new solid and its organoleptic characteristics suggested to us that this is presumably the active species in the reaction. With this hypothesis in mind, we decided to test this solid as the plausible brominating species (PhIOAcBr) in the bromination of 2-naphthol. Also, to evaluate the possible thermal sensitivity of the reagent, we carried out the bromination reaction over the period of one month by comparing two storage temperatures: 23 °C and 4 °C (Scheme 4).
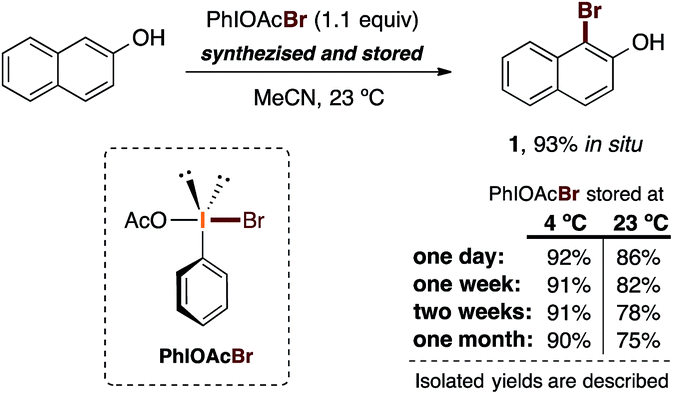 | ||
| Scheme 4 Experimental study on the thermal stability and reactivity of the proposed active species PhIOAcBr for our developed bromination procedure. | ||
To our delight, using 1.1 equiv. of the presumed active brominating species (PhIOAcBr) in acetonitrile at 23 °C provided an excellent yield (92%) of 1. This reaction was carried out the day after the synthesis of the reagent, which was stored at 4 °C before use. When the reaction was attempted with a reagent stored at 23 °C for one day, we obtained a lower yield (86%). In general, bromination reactions with reagents stored at 4 °C gave essentially the same high yields (91% to 90%) even after one month of storage. In the reactions carried out with the reagent stored at 23 °C, notably diminished but still encouraging yields were observed (86% to 75%) over a period of one month.
These reactions potentially implicate PhIOAcBr as the plausible brominating active species, which can be synthesized in situ or by mixing PIDA with AlBr3, followed by centrifugation and isolation. Moreover, theoretical calculations were carried out to provide preliminary support for the identity of the active species. The enthalpy and Gibbs free energy of the reaction between PIDA and AlBr3 in acetonitrile at 23 °C were calculated at the (smd:acetonitrile)M08-hx/(LANL08d, G-311G*) level34 and suggest the favorable formation of PhIOAcBr (eqn (3)).
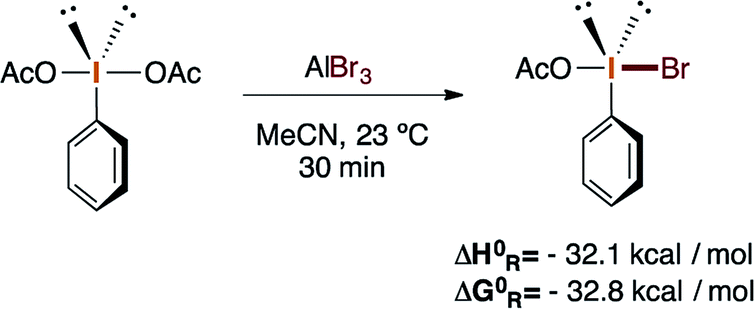 | (3) |
In light of these results, additional calculations are currently underway in our group to establish a plausible reaction mechanism for this novel procedure. Meanwhile, we have provided several arguments pointing to PhIOAcBr35 being the active brominating species in our protocol.
Based on the experimental work presented here, and the known chemistry of I(III) reagents,36 we have compiled the following mechanic proposal (Scheme 5).
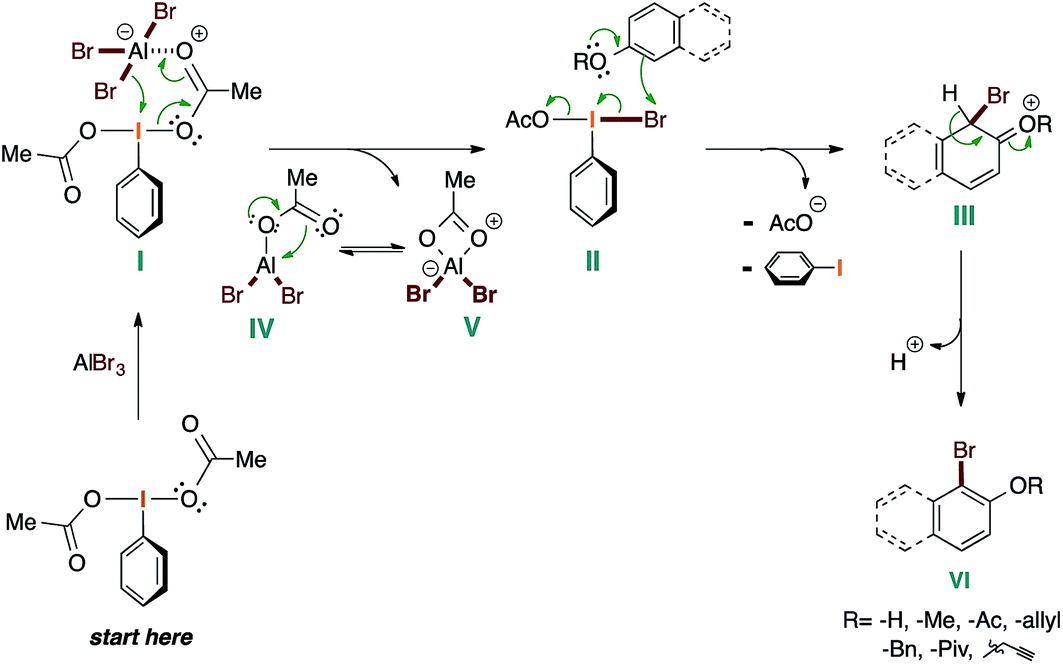 | ||
| Scheme 5 Mechanic proposal for the electrophilic bromination mediated by the PIDA–AlBr3 system. | ||
The mechanism starts with the coordination of the AlBr3 to PIDA in order to provide I. This generates the plausible active brominating species II and releases the aluminate V, which is in equilibrium with IV. At this point, the corresponding phenolic molecule attacks to II, giving rise to III with the concomitant loss of the acetate anion and iodobenzene by reductive elimination. Finally, the spontaneous aromatization of III affords the desired brominated phenol derivative VI.
Conclusions
In summary, we have developed a new and efficient I(III)-based electrophilic bromination procedure for phenols, phenol–ethers and some heterocycles. Our protocol uses aluminium tribromide as ligand activator for the reaction and as a source of bromine atoms. The reaction is a reagent-economic procedure since its reagents PIDA and AlBr3 are inexpensive. The reagent is additionally easy to handle and the reactions proceed under very mild reaction conditions (r.t., open flask). Reagent activation is not necessary and excellent yields, mainly for naphthols, are observed. Our experimental and theoretical work suggests that PhIAcOBr(II) is most likely the active brominating species, which can be prepared in situ or synthesized and isolated. This reagent is stable at least for one month, without losing its reactivity, if stored at 4 °C. Our reagent shows excellent reactivity across a broad scope of functional groups in bis- and mono-annular phenols as well as heterocycles. Additionally, analgesics like naproxen or paracetamol were brominated with our procedure.To the best of our knowledge this is the first report of the oxidation of the bromine atoms in AlBr3 to a “Br+” equivalent and their application to the bromination of phenolic cores. This strategy presents a clear advantage over those using peroxides, since it is not easy to control oxidation processes involving the latter reagents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to CONACyT (CB-2013/220836) for financial support. We acknowledge the facilities of the DCNyE, the Chemistry Department, and the National Laboratory UG-CONACyT (LACAPFEM) of the University of Guanajuato for full characterization. We thank CONACyT for fellowships to Y.·S., P. D. N., V. R. and K. A. J. O. We also thank Dr Ramón Zapata for providing samples of Naproxen and Paracetamol and L. Yera for kind contribution.Notes and references
- Organic Bromine and Iodine Compounds, The Handbook of Environmental Chemistry, ed. Nelson A. H., Springer, Berlin, 2003, vol. 3R Search PubMed.
- M. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef.
- (a) C. Wagner, M. El Omari and G. M. König, J. Nat. Prod., 2009, 72, 540–543 CrossRef PubMed; (b) C. S. Neumann, D. G. Fujimori and C. T. Walsh, Chem. Biol., 2008, 15, 99–109 CrossRef PubMed; (c) K. Benkendroff, Mar. Drugs, 2013, 11, 1370–1398 CrossRef PubMed.
- (a) G. W. Gribble, Chem. Soc. Rev., 1999, 28, 335–346 RSC; (b) For synthesis of 4-isocymobarbatol see: S. A. Snyder, D. S. Treitler and A. P. Brucks, J. Am. Chem. Soc., 2010, 132, 14303–14314 CrossRef PubMed; (c) For isolation of vidalol B see: N. Sitachitta and J. Gerwick, J. Nat. Prod., 1998, 61, 681–684 CrossRef PubMed; (d) For isolation of amathamide G see: H. Kang and W. Fenical, Tetrahedron Lett., 1997, 38, 941–944 CrossRef; (e) For total synthesis of (±)-chartelline C see: P. S. Baran, R. A. Shenvi and C. A. Mistos, Angew. Chem., Int. Ed., 2005, 44, 3714–3717 CrossRef PubMed; (f) P. S. Baran and R. A. Shenvi, J. Am. Chem. Soc., 2006, 128, 14028–14029 CrossRef PubMed; (g) for gymnochrome A see: B. D. Morris and M. R. Prinsep, J. Org. Chem., 1998, 63, 9545–9547 CrossRef.
- (a) M. Gobinath, N. Subramanian and V. Alagarsamy, J. Saudi Chem. Soc., 2015, 19, 282–286 CrossRef; (b) A. Jain, L. S. Duvvuri, S. Farah, N. Beyth, A. J. Domb and W. Khan, Adv. Healthcare Mater., 2014, 3, 1969–1985 CrossRef PubMed; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef PubMed; (d) A. S. Christophersen, Toxicol. Lett., 2000, 127, 112–113 Search PubMed; (e) For synthesis of JHU75575 see: S. R. Donohue, R. F. Dannals, C. Halldin and V. W. Pike, J. Med. Chem., 2011, 54, 2961–2970 CrossRef PubMed.
- F. Hu, M. Patel, F. Luo, C. Flach, R. Mendelson, E. Garfunkel, H. He and M. Szostak, J. Am. Chem. Soc., 2015, 137, 14473–14480 CrossRef PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef.
- J. H. Li, Y. Liang and Y. X. Xie, Tetrahedron, 2005, 61, 7289–7293 CrossRef.
- E. Negishi, Acc. Chem. Res., 1982, 15, 340–348 CrossRef.
- C. B. Ziegler and R. F. Heck, J. Org. Chem., 1978, 34, 2941–2946 CrossRef.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef PubMed.
- Q. Yu, L. Hu, Y. Wang, S. Zheng and J. Huang, Angew. Chem., Int. Ed., 2015, 54, 15284–15288 CrossRef PubMed.
- (a) T. Zhang, X. Qi, S. Liu, R. Bai, C. Liu and Y. Lan, Chem.–Eur. J., 2017, 23, 2690–2699 CrossRef PubMed; (b) P. Zhang, L. Hong, G. Li and R. Wang, Adv. Synth. Catal., 2015, 357, 345–349 CrossRef.
- (a) F. Mendoza, R. Ruíz-guerrero, C. Hernández-Fuentes, P. Molina, M. Norzagaray-Campos and E. Reguere, Tetrahedron Lett., 2016, 57, 5644–5648 CrossRef; (b) S. M. Islam, R. A. Molla, A. S. Roy, K. Ghosh, N. Salam, M. A. Iqubal and K. Tuhina, J. Organomet. Chem., 2014, 761, 169–178 CrossRef.
- (a) W. Liu, J. Chen, R. Jin, D. Xu, Y. Li, F. Ba, G. Gu, Y. Kuang and H. Guo, Org. Chem. Front., 2016, 3, 852–855 RSC; (b) X.-E. Li, W. Wu, X.-H. Fan and L.-M. Yang, RSC Adv., 2013, 3, 12091–12095 RSC.
- X. Sun, G. Shan, Y. Sun and Y. Rao, Angew. Chem., Int. Ed., 2013, 52, 4440–4444 CrossRef PubMed.
- F. Mo, J. M. Yan, D. Qiu, F. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 2028–2032 CrossRef PubMed.
- For NBS-organocatalyst see: Y. Xiong, F. Tan and Y.-Y. Yeung, Org. Lett., 2017, 19, 4243–4246 CrossRef PubMed.
- (a) For NBS in ionic liquid, see: S. R. Pingali, M. Madhav and B. S. Jursic, Tetrahedron Lett., 2010, 51, 1383–1385 CrossRef; (b) For Br2 in ionic liquid, see: Y.-L. Ren, B. Wang, X.-E. Tian, S. Zhao and J. Wang, Tetrahedron Lett., 2010, 56, 6452–6455 CrossRef.
- For NBS-TMSCl see: T. Mainbukaew, B. Thongsornkleeb, J. Tummatorn, A. Bunrit and S. Ruchirawat, Synlett, 2014, 25, 1769–1775 CrossRef.
- For Br2-supercritical CO2 see: T. Delgado-Abad, M.-J. Ferrer, J. Reig-López, R. Mello, R. Acerete, G. Asensio and M. E. González-Núñez, RSC Adv., 2014, 4, 51016–51021 RSC.
- For Br2 on Fe2O3–zeolites see: Y. Nishina and K. Takami, Green Chem., 2012, 14, 2380–2383 RSC.
- D. Liang, X. Li, C. Wang, Q. Dong, B. Wang and H. Wang, Tetrahedron Lett., 2016, 57, 5390–5394 CrossRef.
- A. Podgoršek, S. Stavber, M. Zupan and J. Iskra, Tetrahedron, 2009, 65, 4429–4439 CrossRef.
- S. Song, X. Sun, X. Li, Y. Yuan and N. Jiao, Org. Lett., 2015, 17, 2886–2889 CrossRef PubMed.
- J. Hou, Z. Li, X.-D. Xia and Z.-Q. Liu, Synth. Commun., 2014, 44, 181–187 CrossRef.
- D. C. Braddock, G. Cansell and S. A. Hermitage, Synlett, 2004, 3, 461–464 CrossRef.
- Z. Zhou and H. He, Synthesis, 2011, 2, 207–209 CrossRef.
- P. A. Evans and T. A. Brandt, J. Org. Chem., 1997, 62, 5321–5326 CrossRef.
- P. D. Nahide, V. Ramadoss, K. Juárez-Ornelas, Y. Satkar, R. Ortiz-Alvarado, J. M. J. Cervera-Villanueva, A. J. Alonso-Castro, J. R. Zapata-Morales, M. A. Ramírez-Morales, A. Ruiz-Padilla, M. A. Deveze-Álvarez and C. R. Solorio-Alvarado, Eur. J. Org. Chem., 2018, 485–493 CrossRef.
- (a) An extensive search in the literature reveals that this described route (O-functionalization and bromination) is opposite that most commonly used for the synthesis of 29–32. This supports the excellent reactivity and the good tolerance to bulky functional groups of our developed bromination procedure; ; (b) The latent α-bromination in acetyl derivative, as well as the benzylic bromination in benzyl derivative or bromination in triple bond for propargyl example was not observed; this also showed the chemoselectivity of our procedure.
- Excellent examples of reagents that induce halocyclization: (a) For Et2SBr·SbCl5Br: S. A. Snyder and D. S. Treitler, Angew. Chem., Int. Ed., 2009, 121, 8039–8043 CrossRef; (b) For CDSC, IDSI and BDSB: S. A. Snyder, D. S. Treitler and A. P. Brucks, J. Am. Chem. Soc., 2010, 132, 14303–14314 CrossRef PubMed.
- See ESI† for a detailed preparation procedure.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016, see ESI† for further details.
- PhIOAcBr has been previously postulated as a I(III)-based brominating active species, see ref. 26. However, in this reference, there is a very narrow scope and there is no evidence to support its formation, stability, reactivity or physicochemical characteristics.
- (a) V. Zhdankin, ARKIVOC, 2009, i, 1–62 Search PubMed; (b) V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523–2584 CrossRef PubMed.
- P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed.Angew. Chem., Int. Ed., 2004, 43, 4707–4734 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02982b |
| ‡ These two authors contributed equally to the paper. |
| This journal is © The Royal Society of Chemistry 2018 |