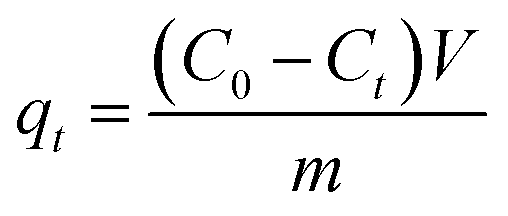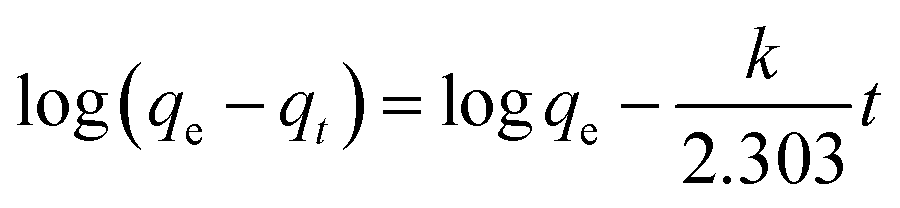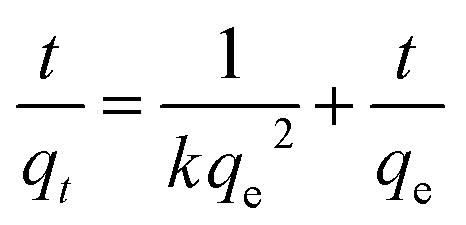DOI:
10.1039/C8RA02964D
(Paper)
RSC Adv., 2018,
8, 21644-21650
Preparation of MoS2-based polydopamine-modified core–shell nanocomposites with elevated adsorption performances
Received
6th April 2018
, Accepted 4th June 2018
First published on 13th June 2018
Abstract
New molybdenum disulfide (MoS2)-based core–shell nanocomposite materials were successfully prepared through the self-assembly of mussel-inspired chemistry. Characterization by Fourier transform infrared, thermogravimetric analysis, scanning electron microscope and transmission electron microscopy revealed that the surface of the flaked MoS2 was homogeneously coated with a thin layer of polydopamine (PDA). Dye adsorption performances of the synthesized MoS2–PDA nanocomposites were investigated at different pH values and reaction times. Compared with pure MoS2 nanosheets, the obtained core–shell nanocomposites showed elevated adsorption performances and high stability, indicating their potential applications in wastewater treatment and composite materials.
1. Introduction
Over the past several decades, graphene has been attracting a great deal of attention due to the unusual properties associated with its ultrathin structure.1–7 Among other two-dimensional layer materials,8–11 molybdenum disulfide (MoS2) is one of the most attractive and has a similar structure to graphite.12–16 MoS2 has a sandwich structure consisting of two layers of sulfur atoms and a middle layer of molybdenum atoms, which are bonded by van der Waals forces.17–20 MoS2 nanosheets have many excellent physical and chemical properties, and demonstrate important applications in sensors,21–24 optoelectronic devices,25,26 catalysis27,28 and other fields.29 In addition, MoS2 nanocomposites have attracted wide study in the field of polymer nanocomposites.30–33 For example, Li et al. investigated polyethylene glycol (PEG)-modified MoS2 surfaces,34 and the obtained products showed good removal efficiency toward some dyes.
In recent years, mussel-inspired chemistry has become a hot research topic in materials science, chemistry and other fields.35–37 In the marine environment, mussels can secrete proteins through their feet, which have excellent adhesion and good biocompatibility. Dopamine, as an imitation mussel-protein material, has strong adhesion properties through a complex self-assembly process to form a polydopamine (PDA) coating with various functions.38 The formed PDA layer can be used to modify the surface of inorganic and organic materials, so it demonstrates wide prospects for application in the fields of separation membranes, adsorbent materials, biomedical materials, biological binders and so on.39–42 Now core–shell materials have shown great potential applications in biology, electricity, catalysis and so on.43–46 For example, Zhao et al. synthesized core–shell diamond-based nanocomposites, exhibiting high activity and high catalytic performance.47 Although the catalytic performance of prepared composite materials may seem ideal, there are still some deficiencies, such as high cost and harsh operating conditions. Thus, mussel-inspired chemistry has the advantages of mild reaction conditions, a wide range of applications and diverse functions, etc. On the other hand, core–shell nanocomposites have unique structural characteristics, integrating the properties of both internal and external materials.
In this work, we used PDA to modify the surface of MoS2 nanosheets to synthesize core–shell nanocomposites. The as-prepared composites were characterized by a series of morphological and spectral characterization techniques. The results showed that we had successfully prepared MoS2–PDA polymeric materials. The obtained MoS2–PDA composites were used as adsorbents for the removal of methylene blue and Safranine T, and they showed enhanced adsorption ability.
2. Materials and methods
2.1 Materials
Molybdenum disulfide (MoS2, 99.9%) nanosheets were obtained from Huajing Powdery Material Science and Technological Co., Ltd. (Hunan, China). Hydroxyphenethylamine hydrochloride (dopamine, 98%) and tris(hydroxymethyl)aminomethane hydrochloride (Tris–HCl, 99%) were purchased from Aladdin Chemicals and Alfa Aesar Chemicals (Shanghai, China). Methylene blue (MB) and Safranine T (ST) were obtained from Tianjin KaiTong Chemical Reagent and Sinopharm Chemical Reagent Co., Ltd. without further purification. Deionized (DI) water was used to prepare aqueous solutions in all experiments.
2.2 Fabrication of the MoS2–PDA nanocomposites
The synthesis of core–shell MoS2–PDA nanocomposite was undertaken according to the method given in previous literature.48 Briefly, 300 mg of MoS2 nanosheets were first dispersed in 100 mL of Tris buffer (10 mM, pH = 8.5) with sonication for 10 minutes, and then 200 mg of dopamine was added to the above solution with stirring. After stirring at room temperature in the dark for 12 h, 24 h, and 48 h, the obtained composite products were washed several times with water and ethanol, respectively, then centrifuged at 6000 rpm for 10 min. Finally, the products were freeze-dried for 2–3 days for the next experiments.
2.3 Adsorption performance test
In order to investigate the dye adsorption performances of MoS2 nanosheets and MoS2–PDA nanocomposites, we selected MB and ST as model dyes to complete the experiment. 10 mg of MoS2 nanosheets and 10 mg of the prepared MoS2–PDA nanocomposites were added to 100 mL of MB solution (8 mg L−1) and ST solution (30 mg L−1), respectively. After different adsorption time intervals, the samples were centrifuged and the supernatant analyzed by a UV-Vis spectrophotometer (664 nm for MB; 530 nm for ST). In addition, we also studied the effect of MoS2 and MoS2–PDA on MB adsorption with different initial solution pH values. The pH of the initial solution was adjusted to values of 2–11 by adding diluted aqueous HCl solution or NaOH solution.20 All experiments were carried out at room temperature under dark conditions. The amount of adsorbed dye per unit mass of adsorbent qt (mg g−1) at time t (min) is calculated from the following formula:| |
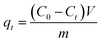 | (1) |
where C0 is the initial concentration of the adsorption solution (mg L−1), Ct is the concentration of the adsorption solution at time t (mg L−1), m is the total MoS2 sample added (g), and V is the volume of the adsorption solution (L).
2.4 Characterization
The microstructures of the samples were obtained using a field-emission scanning electron microscope (SEM) (S-4800II, Hitachi, Japan) equipped with an energy dispersive X-ray spectroscope (EDS) and a transmission electron microscope (TEM) (HT7700, Hitachi High-Technologies Corporation, Japan) with an accelerating voltage of 20 kV. Thermogravimetric analysis (TGA) was performed by a NETZSCH STA 409 PC Luxx simultaneous thermal analyzer (Netzsch Instruments Manufacturing Co, Ltd, Germany). BET measurements (NOVA 4200-P, US) were taken to characterize the specific surface areas and pore diameter distribution. Fourier transform infrared (FTIR) spectra were accomplished on a Thermo Nexus 470 FT-IR spectrometer (KBr disk). X-ray photoelectron spectroscopy (XPS) was performed using a Bragg diffraction setup (SMART LAB, Rigaku, Japan) with an Al Kα X-ray source. The adsorption experiments were monitored using a Shimadzu UV2550 spectrophotometer. All experimental processes were undertaken in a beaker under dark conditions. The synthesized composite materials were completely dewatered using an FD-1C-50 freeze dryer (Beijing Boli Experimental Instruments Co., Ltd., China). All aqueous solutions were prepared with water purified in a double-stage Millipore Milli-Q Plus purification system.
3. Results and discussion
3.1 Characterization of nanocomposites
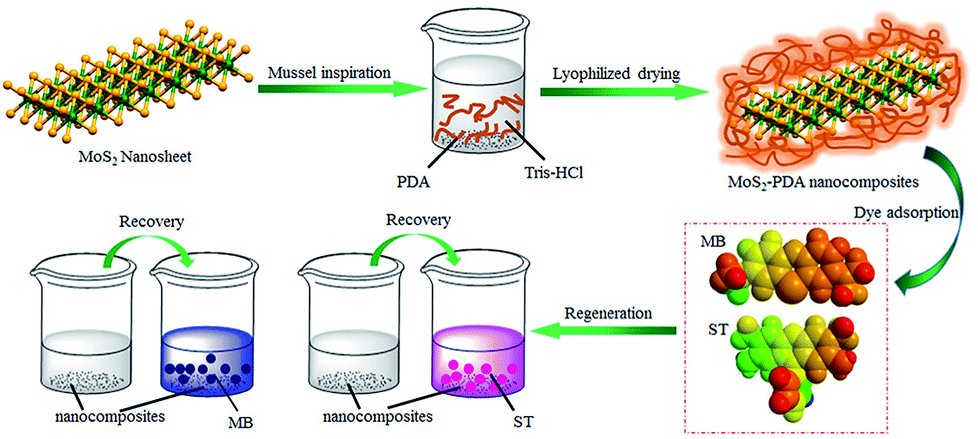
Firstly, Fig. 1 shows the entire experimental process, including the preparation of MoS2–PDA and the adsorption experiments of the obtained nanocomposites. First, MoS2–PDA nanocomposites were obtained by modification of MoS2 nanosheets with a PDA layer in Tris buffer solution and dried by centrifugation. Then two model dyes (MB and ST) were used to evaluate the adsorption capacity of the prepared composite materials.
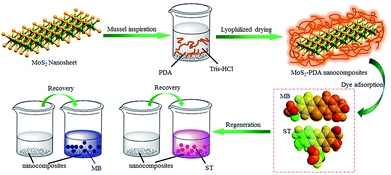 |
| | Fig. 1 Schematic illustration of the fabrication of MoS2–PDA composites by mussel-inspired chemistry. | |
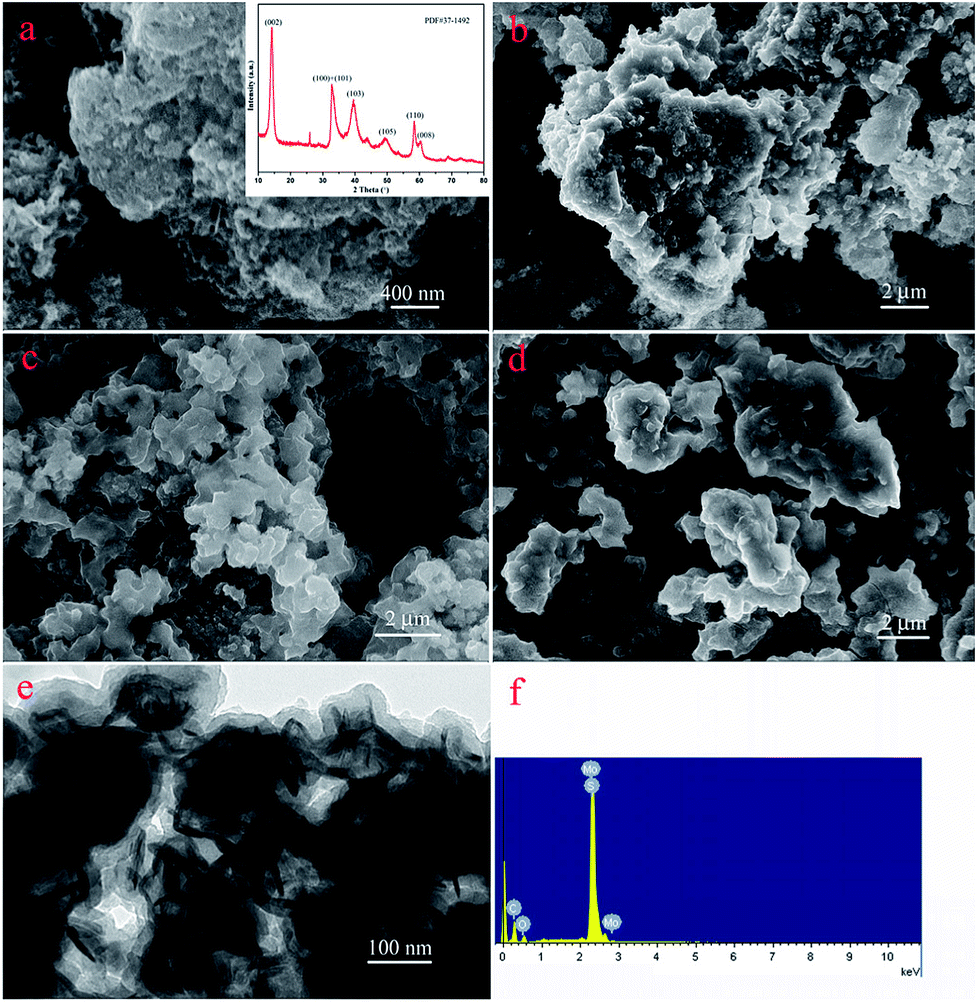
The nanostructures of the obtained materials were investigated by SEM and TEM, as shown in Fig. 2. Obviously, compared with the original MoS2 (Fig. 2a), with an increase in modification time, the layer thickness of PDA coated on the MoS2 surface increased and showed a tendency to aggregate together, as shown in Fig. 2b–d. In addition, Fig. 2e represents the TEM image of the MoS2–PDA composite with a modification time of 48 h, which was named MoS2–PDA-48. In the TEM image, it was obvious that the outer edge of the MoS2 nanosheets demonstrated some ultra-thin layer structures, which indicated that the PDA layer had been successfully anchored on the surface of the MoS2 nanosheets. Moreover, the typical XRD pattern of MoS2 (the inset image of Fig. 2a) and the EDX pattern of MoS2–PDA-48 (Fig. 2f) also provided obvious characteristics and suggested the above speculations.
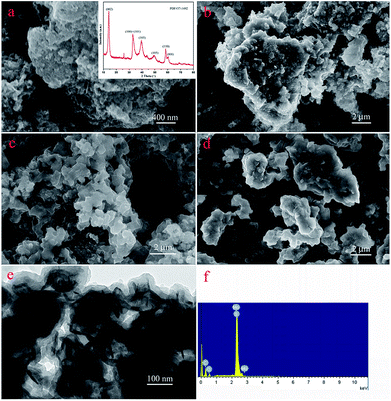 |
| | Fig. 2 SEM and TEM images of as-prepared materials: (a) SEM image and inserted XRD data of MoS2; (b–d) SEM images of core–shell MoS2–PDA composites after different modification times (12, 24, and 48 h); (e) and (f) TEM image and EDX spectra of MoS2–PDA-48. | |
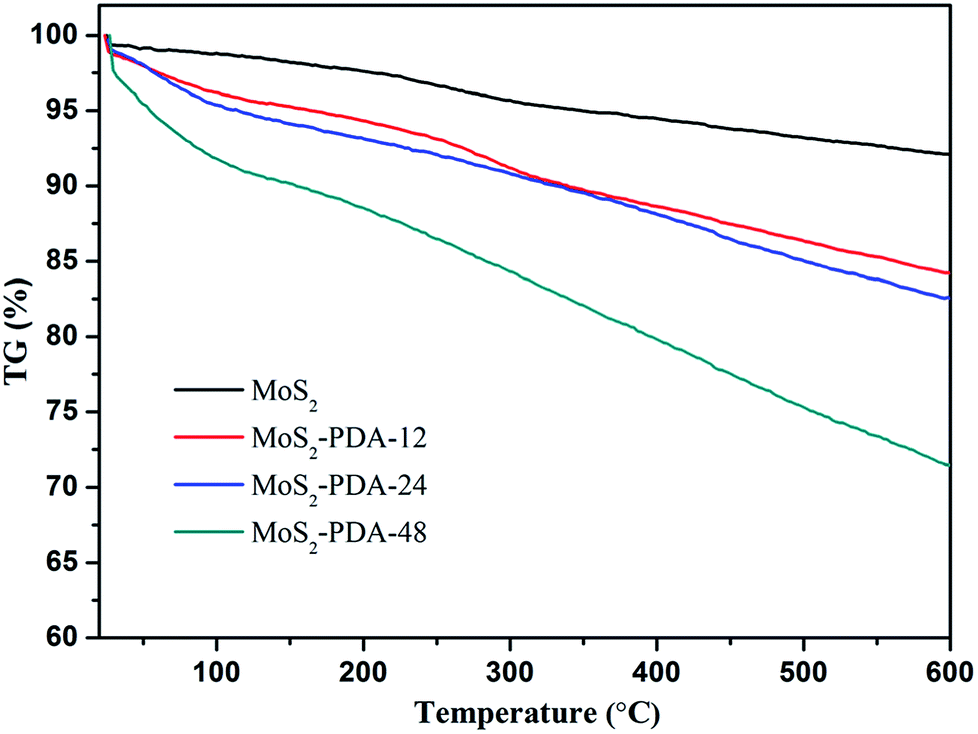
In addition, the thermal stability of MoS2 and the obtained MoS2–PDA composites were characterized and are shown in Fig. 3. According to the TG curves, MoS2 showed a low mass loss (about 7.9%) from room temperature to 600 °C, which indicated good thermal stability. The mass losses of the MoS2–PDA composites took the values of 15.8%, 17.4%, and 28.6% with different modification times (12 h, 24 h, and 48 h, respectively). The obtained results indicated that the PDA layer had been successfully coated onto the MoS2 surface and the amount of PDA component in the composites increased with an increase in modification time.
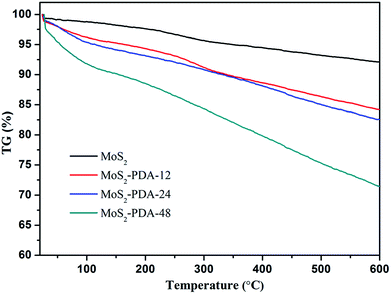 |
| | Fig. 3 TG curves of MoS2 nanosheets and the obtained MoS2–PDA nanocomposites. | |
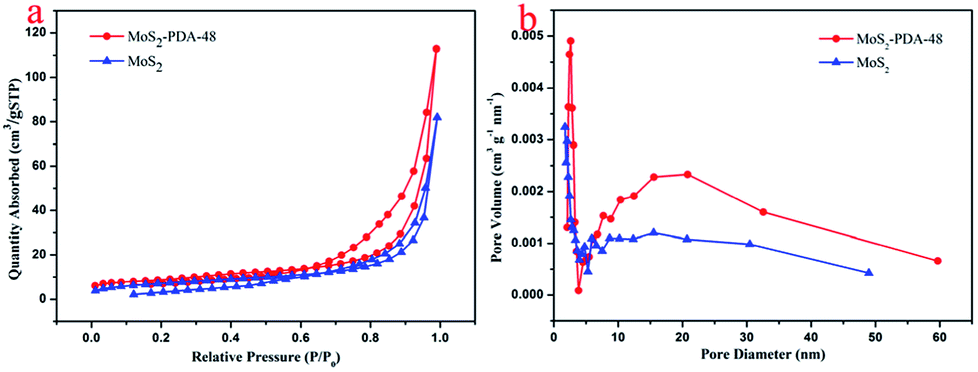
In order to study the microstructure characteristics of MoS2 and MoS2–PDA-48, Fig. 4a shows the BET measurements using N2 adsorption–desorption isotherms. The adsorption–desorption isotherms of both MoS2 and MoS2–PDA-48 belonged to type IV isotherms. The hysteresis loop of MoS2 has an abrupt increase in the amount of adsorption at P/P0 = 0.6–0.95, which is characteristic of a mesoporous material. For the MoS2–PDA-48 nanocomposites, a hysteresis loop could be observed at pressure P/P0 = 0.8–0.95 that indicated a mesoporous structure. The pore size distribution curves were obtained according to the BET method and are shown in Fig. 4b. The pore size distribution of MoS2–PDA-48 showed a larger mesoporous size of 22.84 nm, compared to 19.62 nm for MoS2. The data for BET surface area and total pore volume are listed in Table 1. The specific surface area of MoS2–PDA-48 was calculated to be 30.57 m2 g−1, which was much larger than that of MoS2 (25.83 m2 g−1). The higher specific surface area can increase the number of active sites on the surface of the MoS2–PDA composite and enhance the chances of dye molecules anchoring at active sites, thus making adsorbents with better adsorption properties.
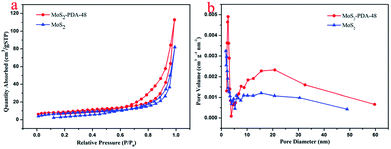 |
| | Fig. 4 (a) Adsorption–desorption isotherms and (b) pore size distribution of MoS2 and the MoS2–PDA-48 nanocomposite. | |
Table 1 Physical data of MoS2 and the MoS2–PDA-48 nanocomposite
| Sample |
Specific surface area (m2 g−1) |
Average pore diameter (nm) |
Pore volume (cm3 g−1) |
| MoS2 |
25.83 |
19.62 |
0.127 |
| MoS2–PDA-48 |
30.57 |
22.84 |
0.175 |
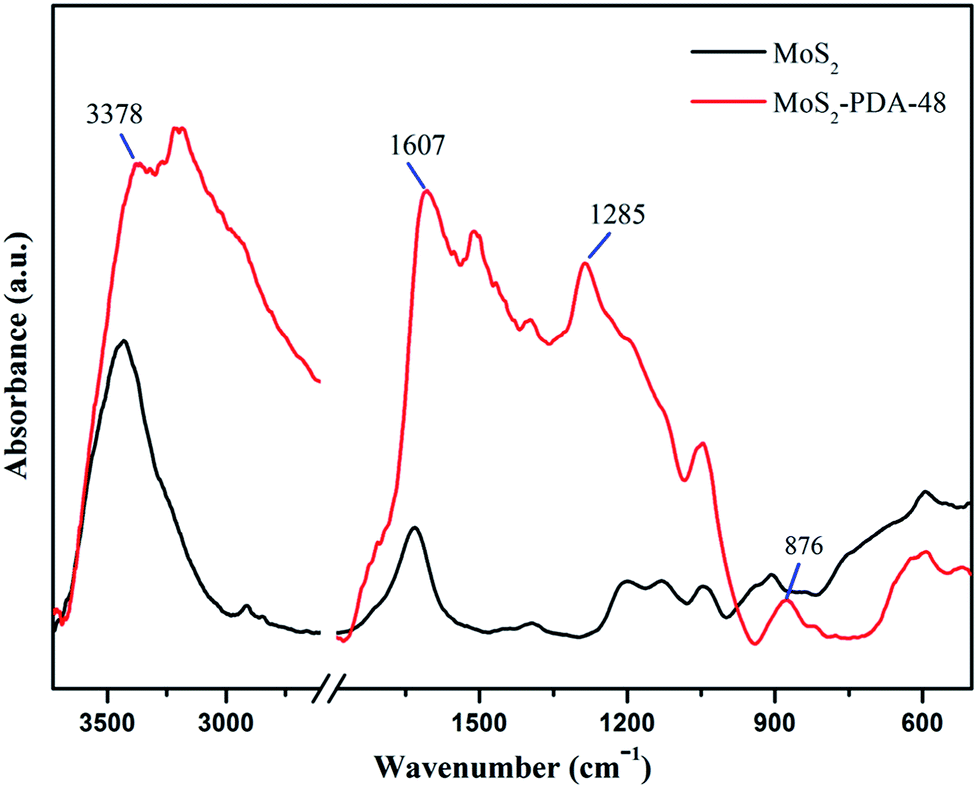
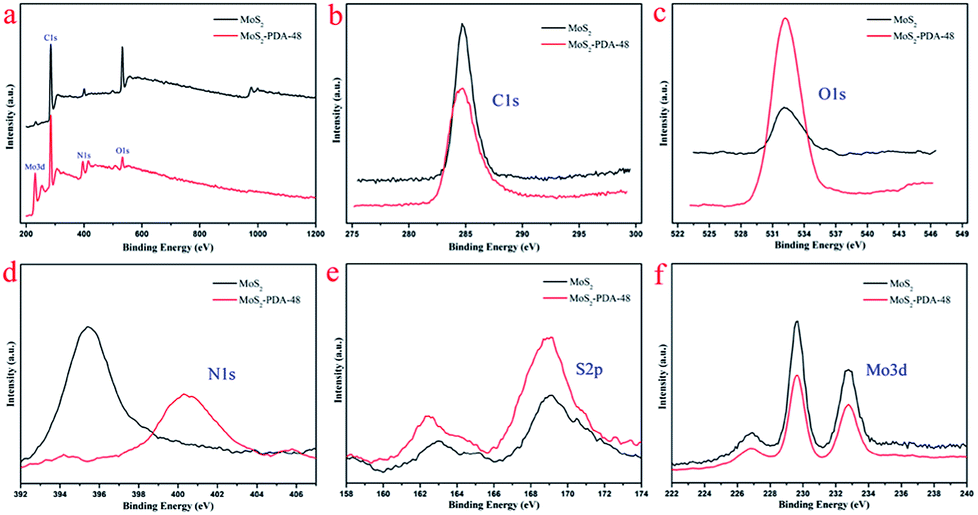
FT-IR spectra of MoS2 and the MoS2–PDA-48 composite are demonstrated in Fig. 5. The curve of MoS2–PDA-48 shows a characteristic band at 3378 cm−1, which could be ascribed to the stretch vibrations of –NH2, –NH–, and –OH.49–55 The characteristic peak at 1607 cm−1 could be deduced as a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration in the benzene ring. At the same time, the weak peaks around 1285 and 876 cm−1 could be assigned to C–OH stretching vibration from the catechol groups and C–O stretching, respectively, which indicate that the surface of MoS2 was successfully modified by a PDA layer.56–59 In addition, the XPS analysis (Fig. 6) demonstrates the composition of MoS2 and MoS2–PDA-48. Fig. 6a demonstrates the characteristic peaks in the curve of the MoS2–PDA-48 composite, such as Mo (3d), O (1s), C (1s), and N (1s). Additionally, the high resolution XPS spectra are shown in Fig. 6b–f. Compared with pure MoS2, the relative intensity of C 1s of MoS2–PDA-48 decreased and the O 1s peak increased significantly (Fig. 6b and c). In the meantime, it was clear that the relative intensity of the N 1s of MoS2–PDA-48 at 400 eV increased obviously in Fig. 6d. In addition, as shown in Fig. 6e and f, the relative intensity of Mo 3d and S 2p in MoS2–PDA-48 was significantly reduced compared with the MoS2 nanosheets, which also indicates that the PDA layer was successfully coated onto the surface of the MoS2 nanosheets. On the other hand, the relative elemental analysis data based on XPS analysis is shown in Table 2. The atom percentages of Mo and S in the table fell from 13.99% and 27.71% to 3.51% and 9.11%, respectively. At the same time, the atomic percentage of N in MoS2–PDA-48 accounted for 7.58%, indicating successful PDA modification on the surface of the MoS2 nanosheets.
C stretching vibration in the benzene ring. At the same time, the weak peaks around 1285 and 876 cm−1 could be assigned to C–OH stretching vibration from the catechol groups and C–O stretching, respectively, which indicate that the surface of MoS2 was successfully modified by a PDA layer.56–59 In addition, the XPS analysis (Fig. 6) demonstrates the composition of MoS2 and MoS2–PDA-48. Fig. 6a demonstrates the characteristic peaks in the curve of the MoS2–PDA-48 composite, such as Mo (3d), O (1s), C (1s), and N (1s). Additionally, the high resolution XPS spectra are shown in Fig. 6b–f. Compared with pure MoS2, the relative intensity of C 1s of MoS2–PDA-48 decreased and the O 1s peak increased significantly (Fig. 6b and c). In the meantime, it was clear that the relative intensity of the N 1s of MoS2–PDA-48 at 400 eV increased obviously in Fig. 6d. In addition, as shown in Fig. 6e and f, the relative intensity of Mo 3d and S 2p in MoS2–PDA-48 was significantly reduced compared with the MoS2 nanosheets, which also indicates that the PDA layer was successfully coated onto the surface of the MoS2 nanosheets. On the other hand, the relative elemental analysis data based on XPS analysis is shown in Table 2. The atom percentages of Mo and S in the table fell from 13.99% and 27.71% to 3.51% and 9.11%, respectively. At the same time, the atomic percentage of N in MoS2–PDA-48 accounted for 7.58%, indicating successful PDA modification on the surface of the MoS2 nanosheets.
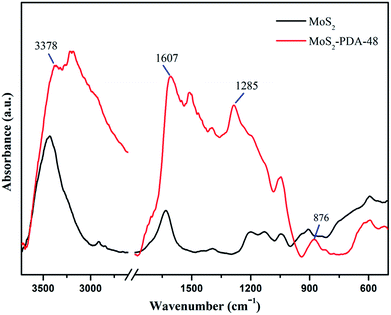 |
| | Fig. 5 IR spectra of MoS2 and the MoS2–PDA-48 composite. | |
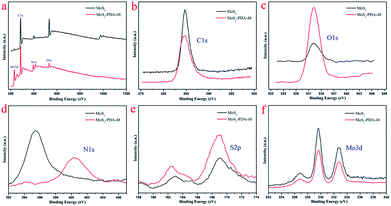 |
| | Fig. 6 (a) XPS survey spectra of MoS2 and MoS2–PDA-48; high resolution scans of different elements: (b) C 1s; (c) O 1s; (d) N 1s; (e) S 2p; (f) Mo 3d. | |
Table 2 Elemental contents (%) of MoS2 and MoS2–PDA-48 based on XPS analysis
| Samples |
Mo [at%] |
S [at%] |
C [at%] |
O [at%] |
N [at%] |
| MoS2 |
13.99 |
27.71 |
28.94 |
26.10 |
3.26 |
| MoS2–PDA-48 |
3.51 |
9.11 |
52.56 |
27.24 |
7.58 |
3.2 Adsorption performances
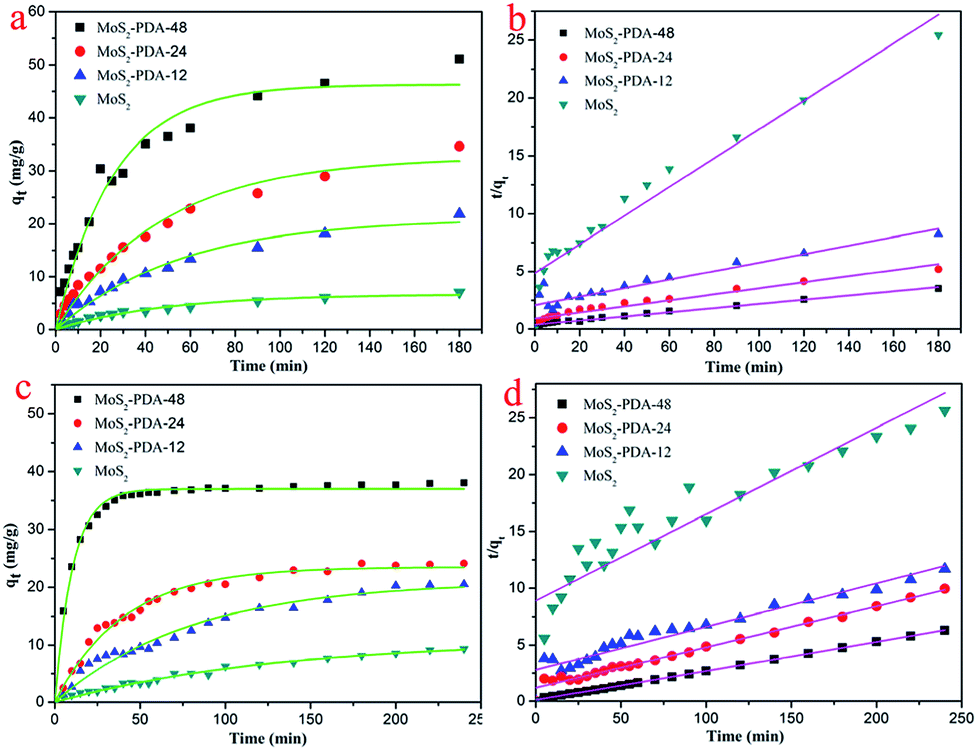
In order to study the adsorption properties of the prepared MoS2–PDA nanocomposites, two model dyes (MB and ST) were used in this research work. The concentrations of residual dyes at different reaction time intervals were monitored at different absorbance wavelengths (664 nm for MB; 530 nm for ST). Fig. 7 shows the results of the adsorption kinetics of MoS2 nanosheets and MoS2–PDA nanocomposites for the removal of MB and ST. It can obviously be observed from Fig. 7a and c that the dye adsorption capacities of the MoS2–PDA nanocomposites increased markedly compared to pure MoS2 in the equilibrium state, and the adsorption effect of MoS2–PDA-48 was the best. The detailed kinetic parameters are shown in Table 3. It should be noted that PDA is a kind of hydrophilic and adhesive material. After PDA anchored onto the surface of the MoS2 nanosheets, the core–shell nanostructures formed demonstrated a porous surface with a number of enhanced active sites to improve the adsorption performance of the composites.
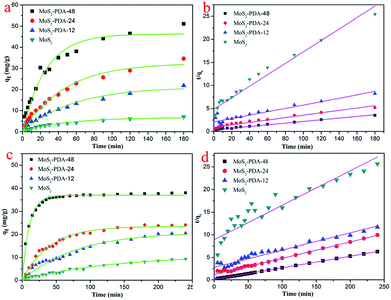 |
| | Fig. 7 Adsorption kinetics curves of MB (a and b) and ST (c and d) onto MoS2 nanosheets and prepared MoS2–PDA nanocomposites with different reaction times. | |
Table 3 Kinetic parameters of the MoS2 and MoS2–PDA composites for MB and ST adsorption
| MB |
Pseudo-first-order model |
Pseudo-second-order model |
| qe (mg g−1) |
R2 |
k1 (min−1) |
qe (mg g−1) |
R2 |
k2 (g mg−1 min−1) |
| MoS2 |
6.7309 |
0.9772 |
0.0208 |
8.0567 |
0.9340 |
0.0032 |
| MoS2–PDA-12 |
21.1223 |
0.9763 |
0.0181 |
27.2183 |
0.8321 |
6.46 × 10−4 |
| MoS2–PDA-24 |
32.5736 |
0.9753 |
0.0212 |
38.2409 |
0.9372 |
7.33 × 10−4 |
| MoS2–PDA-48 |
46.2684 |
0.9665 |
0.0389 |
54.9451 |
0.9801 |
8.84 × 10−4 |
| Safranine T |
Pseudo-first-order model |
Pseudo-second-order model |
| qe (mg g−1) |
R2 |
k1 (min−1) |
qe (mg g−1) |
R2 |
k2 (g mg−1 min−1) |
| MoS2 |
10.9608 |
0.9865 |
0.0077 |
13.1268 |
0.8101 |
6.52 × 10−4 |
| MoS2–PDA-12 |
23.4885 |
0.9878 |
0.0251 |
26.2192 |
0.9231 |
5.18 × 10−4 |
| MoS2–PDA-24 |
21.0367 |
0.9714 |
0.0127 |
27.7239 |
0.9845 |
0.0011 |
| MoS2–PDA-48 |
36.9899 |
0.9929 |
0.0959 |
38.610 |
0.9996 |
0.0063 |
In addition, the adsorption kinetic process can be described by classical kinetic models as follows:
The pseudo-first-order model can be represented by eqn (2):
| |
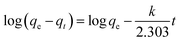 | (2) |
The pseudo-second-order model can be represented by eqn (3):
| |
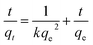 | (3) |
where
qe is the equilibrium adsorption capacity, mg g
−1;
qt is the adsorption capacity at time
t, mg g
−1; the
k1 and
k2 values are the kinetic rate constants. Kinetic results (
Table 3) could be better characterized by a pseudo-first-order model with a correlation coefficient (
R2 > 0.9665) or a pseudo-second-order model with a correlation coefficient (
R2 > 0.8101). Comparing the above adsorption data, it could easily be observed that MoS
2–PDA-48 showed the best adsorption capacity, which could be related to the rich functional groups on the surface of PDA. And the ability to remove MB seemed higher than that for ST. In addition, the gradual increase in the maximum adsorption indicated that the experimental method used in this study is feasible.
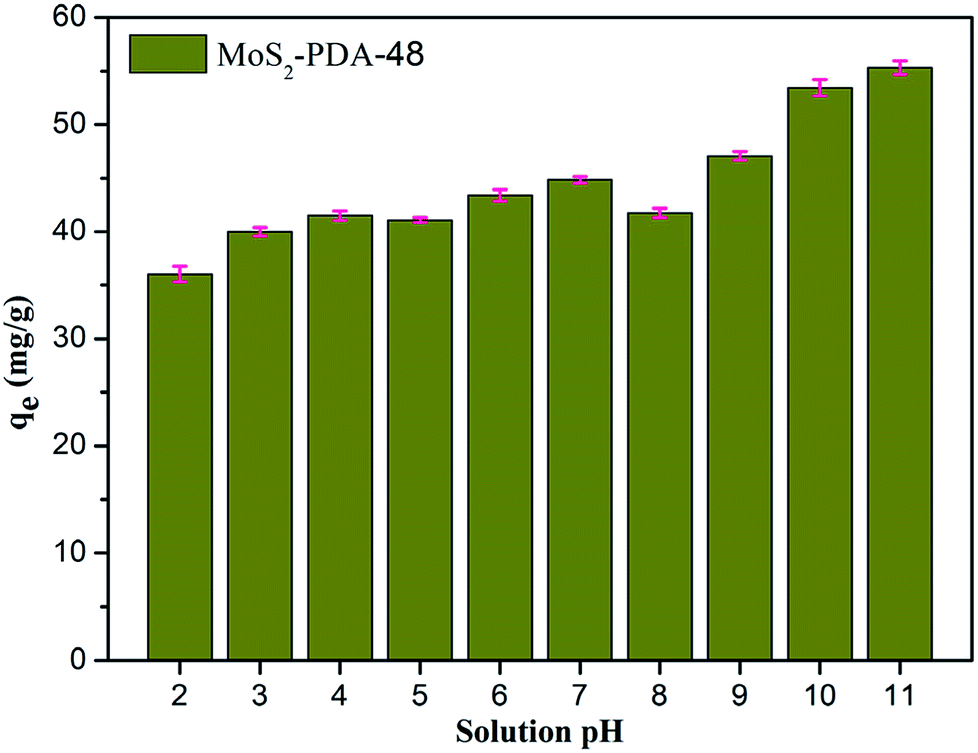
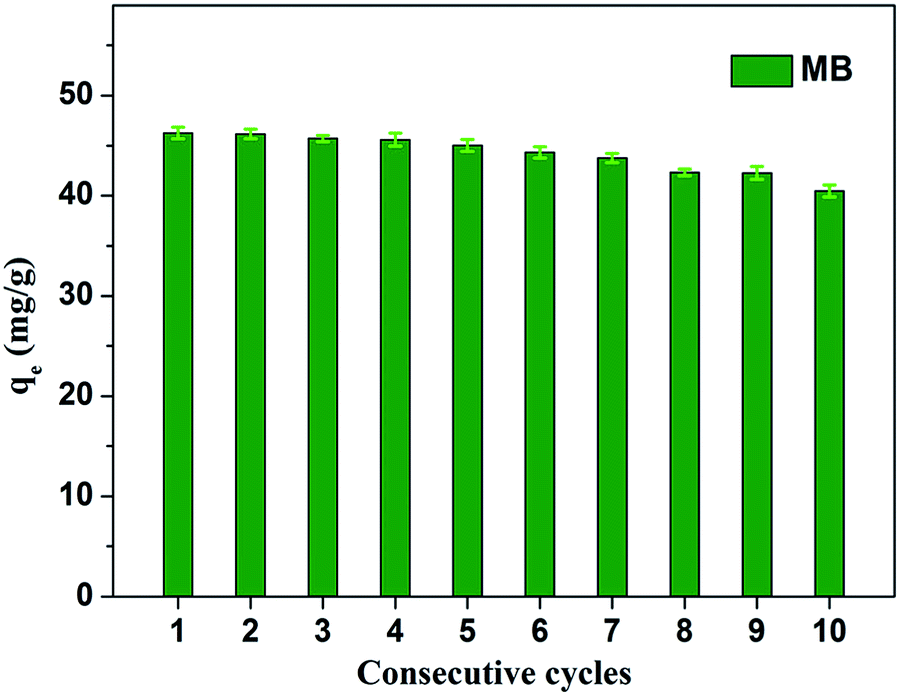
Fig. 8 shows the effect of solution pH on the adsorption of MB to MoS2–PDA-48. It can be seen that as the pH of the solution increased from 2 to 11, the adsorption capacity showed an increasing trend from 36.02 mg g−1 to 55.32 mg g−1. However, this process varied with different pH values. This difference can be presumed to be a result of the protonation of the functional groups on the nanocomposite surface as well as the π–π stacking and electrostatic interactions with the MB molecules.60 In addition, in order to demonstrate the reuse of the obtained composites, 10 consecutive cycles were repeated using the same MoS2–PDA-48 nanocomposite and fresh MB solution (Fig. 9). This showed that the adsorption capacity of the MoS2–PDA-48 nanocomposite still retained a removal rate of 87.4% towards MB and reached 40.45 mg g−1. Moreover, Table 4 lists the MB adsorptions of the relevant materials reported in the literature.61–66 In contrast, the present prepared MoS2–PDA composite materials showed a larger adsorption capacity and eco-friendly preparation process, demonstrating wide application in wastewater treatment and self-assembled core–shell composite materials.
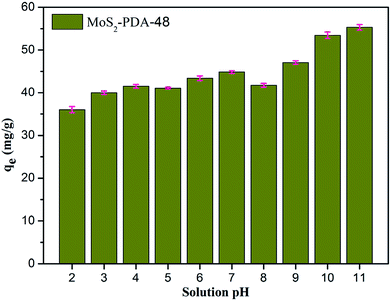 |
| | Fig. 8 The effect of solution pH on MB adsorption by MoS2–PDA-48 nanocomposites. | |
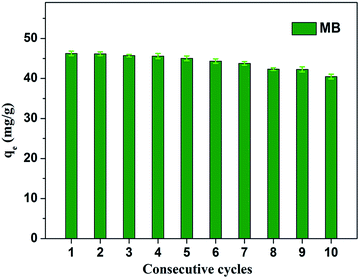 |
| | Fig. 9 Relative adsorption capacity of MoS2–PDA-48 nanocomposite towards MB for different consecutive cycles. | |
Table 4 Comparison of MB adsorption capacities in the reported literature
| Materials |
Qmax (mg g−1) |
Characteristics |
Ref. |
| Carbon nanotubes |
35 |
Simple process, environmentally friendly |
61 |
| GO/Fe3O4 nanohybrids |
32 |
Poor stability |
62 |
| PDA microspheres |
90.7 |
High-efficiency, large adsorption capacity |
63 |
| Fe3O4@MnO2 core–shell nanocomposite |
9.71 |
Weak adsorption capacity |
64 |
| PVDF/PDA membranes |
172.3 |
Good regeneration ability |
65 |
| Fe3O4@graphene |
45.27 |
Weak adsorption capacity |
66 |
| MoS2–PDA |
46.3 |
Eco-friendly preparation, good stability |
This work |
4. Conclusions
In summary, we have synthesized core–shell MoS2–PDA nanocomposites simply by using mussel-inspired chemistry. A series of characterization techniques demonstrated that the PDA was successfully coated onto the surface of the MoS2 nanosheets. The prepared MoS2–PDA composites showed effective removal capacities towards two model dyes. The adsorption process was illustrated by pseudo-first-order and pseudo-second-order kinetic models. The present research work is expected to show potential applications in wastewater treatment and self-assembled core–shell composite materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We greatly appreciate the financial supports of National Natural Science Foundation of China (No. 21473153 and 51771162), Support Program for the Top Young Talents of Hebei Province, China Postdoctoral Science Foundation (No. 2015M580214), Research Program of the College Science & Technology of Hebei Province (No. ZD2018091), and Scientific and Technological Research and Development Program of Qinhuangdao City (No. 201701B004).
Notes and references
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef PubMed
 .
. - K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson and I. V. Grigorieva, Nature, 2005, 438, 197 CrossRef PubMed .
- L. Yan, Y. B. Zheng, F. Zhao, S. Li, X. Gao, B. Xu, P. S. Weiss and Y. Zhao, Chem. Soc. Rev., 2012, 41, 97–114 RSC .
- B. K. Kim, eXPRESS Polym. Lett., 2012, 6, 772 CrossRef .
- B. H. Nguyen and V. H. Nguyen, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2016, 7, 023002 CrossRef .
- S. P. Lonkar, Y. S. Deshmukh and A. A. Abdala, Nano Res., 2015, 8, 1039–1074 CrossRef .
- F. Perreault, D. F. A. Fonfeca and M. Elimelech, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC .
- J. H. Yu, H. R. Lee, S. S. Hong, D. Kong, H. W. Lee, H. Wang, F. Xiong, S. Wang and Y. Cui, Nano Lett., 2015, 15, 1031–1035 CrossRef PubMed .
- V. W. Brar, A. R. Koltonow and J. Huang, ACS Photonics, 2017, 4, 407–411 CrossRef .
- M. K. Jana and C. N. R. Rao, Philos. Trans. R. Soc., A, 2016, 374, 20150318 CrossRef PubMed .
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766–3798 CrossRef PubMed .
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef PubMed .
- K. Kaasbjerg, K. S. Thygesen and K. W. Jacobsen, Phys. Rev. B, 2012, 85, 115317 CrossRef .
- G. Eda, T. Fujita, H. Yamaguchi, D. Voiry, M. Chen and M. Chhowalla, ACS Nano, 2012, 6, 7311–7317 CrossRef PubMed .
- D. Lembke, S. Bertolazzi and A. Kis, Acc. Chem. Res., 2015, 48, 100–110 CrossRef PubMed .
- L. Zhou, B. He, Y. Yang and Y. He, RSC Adv., 2014, 4, 32570–32578 RSC .
- G. Eda, H. Yamaguchi, D. Voiry, T. Fujita, M. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111–5116 CrossRef PubMed .
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin and A. Olivier, Adv. Funct. Mater., 2012, 22, 1385–1390 CrossRef .
- W. Qiao, S. Yan, X. Song, X. Zhang, X. He and W. Zhong, Appl. Surf. Sci., 2015, 359, 130–136 CrossRef .
- Q. Huang, M. Liu, J. Chen, Q. Wan, J. Tian and L. Huang, Appl. Surf. Sci., 2017, 419, 35–44 CrossRef .
- J. Z. Ou, A. F. Chrimes, Y. Wang, S. Tang, M. S. Strano and K. Kalantarzadeh, Nano Lett., 2014, 14, 857–863 CrossRef PubMed .
- F. K. Perkins, A. L. Friedman, E. Cobas, P. M. Campbell, G. G. Jernigan and B. T. Jonker, Nano Lett., 2013, 13, 668–673 CrossRef PubMed .
- C. Zhu, Z. Zeng, H. Li, F. Li, C. Fan and H. Zhang, J. Am. Chem. Soc., 2013, 135, 5998–6001 CrossRef PubMed .
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147 CrossRef PubMed .
- A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim and C. Y. Chim, Nano Lett., 2010, 10, 1271–1275 CrossRef PubMed .
- D. Lembke, S. Bertolazzi and A. Kis, Acc. Chem. Res., 2015, 48, 100–110 CrossRef PubMed .
- Z. Zhou, Y. Lin, P. Zhang, E. Ashalley, M. Shafa and H. Li, Mater. Lett., 2014, 131, 122–124 CrossRef .
- X. Man, L. Yu, J. Sun and S. Li, Funct. Mater. Lett., 2016, 9, 1650065 CrossRef .
- K. Maslana, Przem. Chem., 2017, 96, 689–691 Search PubMed .
- S. K. Kumar and R. Krishnamoorti, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 37–58 CrossRef PubMed .
- R. H. Gonçalves, R. Fiel, M. R. Soares, W. H. Schreiner, C. M. Silva and E. R. Leite, Chemistry, 2015, 21, 15583–15588 CrossRef PubMed .
- K. Pramoda, M. Kaur, U. Gupta and C. N. Rao, Dalton Trans., 2016, 45, 13810–13816 RSC .
- X. Wang, W. Xing, X. Feng, L. Song and Y. Hu, Polym. Rev., 2017, 57, 440–466 CrossRef .
- X. Qiao, F. Hu, D. Hou and D. Li, Mater. Lett., 2016, 169, 241–245 CrossRef .
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef PubMed .
- H. C. Yang, J. Luo, Y. Lv, P. Shen and Z. K. Xu, J. Membr. Sci., 2015, 483, 42–59 CrossRef .
- X. Zhang, M. Liu, Y. Zhang, B. Yang, Y. Ji and L. Feng, RSC Adv., 2012, 2, 12153–12155 RSC .
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef PubMed .
- M. Liu, G. Zeng, K. Wang, Q. Wan, L. Tao and X. Zhang, Nanoscale, 2016, 8, 16819–16840 RSC .
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef PubMed .
- D. E. Fullenkamp, J. G. Rivera, Y. K. Gong, K. H. Lau, L. He and R. Varshney, Biomaterials, 2012, 3, 3783–3791 CrossRef PubMed .
- C. Cheng, S. Nie, S. Li, H. Peng, H. Yang and L. Ma, J. Mater. Chem. B, 2013, 1, 265–275 RSC .
- D. Hou, H. Tao, X. Zhu and M. Li, Appl. Surf. Sci., 2017, 419, 580–585 CrossRef .
- M. L. Wu, Y. Y. Li, R. Yue, X. D. Zhang and Y. M. Huang, Sci. Rep., 2017, 7, 42773 CrossRef PubMed .
- M. Huang, X. L. Zhao, F. Li, W. Li, B. Zhang and Y. X. Zhang, J. Mater. Chem. A, 2015, 3, 12852–12857 Search PubMed .
- R. Guo, T. F. Jiao, R. R. Xing, Y. Chen, W. C. Guo, J. X. Zhou, L. X. Zhang and Q. M. Peng, Nanomaterials, 2017, 7, 317 CrossRef PubMed .
- X. N. Zhao, T. F. Jiao, R. R. Xing, H. Huang, J. Hu, Y. Qu, J. X. Zhou, L. X. Zhang and Q. M. Peng, RSC Adv., 2017, 7, 49923–49930 RSC .
- G. J. Zeng, M. Y. Liu, X. H. Liu, Q. Huang, D. Z. Xu, L. C. Mao, H. Y. Huang, F. J. Deng, X. Y. Zhang and Y. Wei, Appl. Surf. Sci., 2016, 387, 399–405 CrossRef .
- R. Guo, T. Jiao, R. Li, Y. Chen, W. Guo, L. Zhang, J. Zhou, Q. Zhang and Q. Peng, ACS Sustainable Chem. Eng., 2018, 6, 1279–1288 CrossRef .
- J. Zhou, Y. Liu, T. Jiao, R. Xing, Z. Yang, J. Fan, J. Liu, B. Li and Q. Peng, Colloids Surf., A, 2018, 538, 7–13 CrossRef .
- J. Song, R. Xing, T. Jiao, Q. Peng, C. Yuan, H. Möhwald and X. Yan, ACS Appl. Mater. Interfaces, 2018, 10, 2368–2376 Search PubMed .
- Y. Liu, C. Hou, T. Jiao, J. Song, X. Zhang, R. Xing, J. Zhou, L. Zhang and Q. Peng, Nanomaterials, 2018, 8, 35 CrossRef PubMed .
- S. Huo, P. Duan, T. Jiao, Q. Peng and M. Liu, Angew. Chem., Int. Ed., 2017, 56, 12174–12178 CrossRef PubMed .
- J. Zhou, F. Gao, T. Jiao, R. Xing, L. Zhang, Q. Zhang and Q. Peng, Colloids Surf., A, 2018, 545, 60–67 CrossRef .
- X. Luo, K. Ma, T. Jiao, R. Xing, L. Zhang, J. Zhou and B. Li, Nanoscale Res. Lett., 2017, 12, 99 CrossRef PubMed .
- K. Li, T. Jiao, R. Xing, G. Zou, J. Zhou, L. Zhang and Q. Peng, Sci. China Mater., 2018, 61, 728–736 CrossRef .
- R. Xing, T. Jiao, Y. Liu, K. Ma, Q. Zou, G. Ma and X. Yan, Polymers, 2016, 8, 181 CrossRef .
- R. Xing, K. Liu, T. Jiao, N. Zhang, K. Ma, R. Zhang, Q. Zou, G. Ma and X. Yan, Adv. Mater., 2016, 28, 3669–3676 CrossRef PubMed .
- Y. Liu, K. Ma, T. Jiao, R. Xing, G. Shen and X. Yan, Sci. Rep., 2017, 7, 42978 CrossRef PubMed .
- R. R. Xing, W. Wang, T. F. Jiao, K. Ma, Q. R. Zhang, W. Hong, H. Qiu, J. X Zhou, L. X. Zhang and Q. M. Peng, ACS Sustainable Chem. Eng., 2017, 5, 4948–4956 CrossRef .
- Y. J. Yao, F. F. Xu, M. Chen, Z. X. Xu and Z. W. Zhu, Bioresour. Technol., 2010, 101, 3040–3046 CrossRef PubMed .
- T. F. Jiao, Y. Z. Liu, Y. T. Wu, Q. R. Zhang, X. H. Yan, F. M. Gao, A. J. P. Bauer, J. Z. Liu, T. Y. Zeng and B. B. Li, Sci. Rep., 2015, 5, 12451 CrossRef PubMed .
- J. Fu, Z. Chen, M. Wang, S. Liu, J. Zhang and J. Zhang, Chem. Eng. J., 2015, 259, 53–61 CrossRef .
- W. Wang, T. F. Jiao, Q. R. Zhang, X. N. Luo, J. Hu, Y. Chen, Q. M. Peng, X. Yan and B. B. Li, RSC Adv., 2015, 5, 56279–56285 RSC .
- F. Ma, N. Zhang, X. Wei, J. Yang, Y. Wang and Z. Zhou, J. Mater. Chem. A, 2017, 5, 14430–14443 Search PubMed .
- Y. Yao, S. Miao, S. Liu, L. P. Ma, H. Sun and S. Wang, Chem. Eng. J., 2012, 184, 326–332 CrossRef .
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Ruirui Xingbc,
Jinghong Lib,
Jingxin Zhoub,
Lexin Zhang*b and
Qiuming Penga
*ab,
Ruirui Xingbc,
Jinghong Lib,
Jingxin Zhoub,
Lexin Zhang*b and
Qiuming Penga