DOI:
10.1039/C8RA02872A
(Review Article)
RSC Adv., 2018,
8, 21191-21209
Schweinfurthins A–Q: isolation, synthesis, and biochemical properties
Received
3rd April 2018
, Accepted 21st May 2018
First published on 8th June 2018
Abstract
Stilbene analogues have shown remarkable structural diversity constituting simple or tangled structures, which have attracted the synthetic as well as the medicinal chemistry communities. Schweinfurthins are a family of prenylated/geranylated/farnesylated stilbenes that are isolated from an African plant belonging to the Macaranga species. These compounds have displayed potency towards central nervous system, renal and breast cancer cell lines. Specifically, these compounds have been found to be potent and selective inhibitors of cell growth in the National Cancer Institute's 60 cell-line screen. In this review article, we described the isolation, synthesis, and biochemical properties of schweinfurthins.
1. Introduction
The role of natural products in drug discovery has been phenomenal. The search for new lead compounds displaying potency and selectivity with the fewest adverse side effects has led to the synthesis of more and more new candidates for the treatment of various diseases. Natural products have been a significant resource of drug leads in the field of medicinal chemistry, and despite an upsurge in the use of combinatorial chemistry as a part of the lead development process, they still play a pivotal role owing to their diverse biochemical properties.1–4
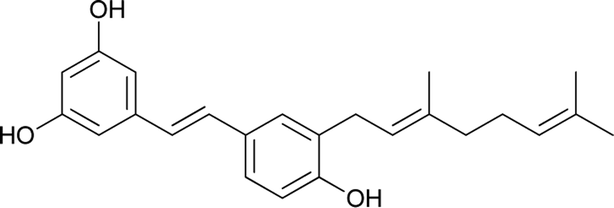
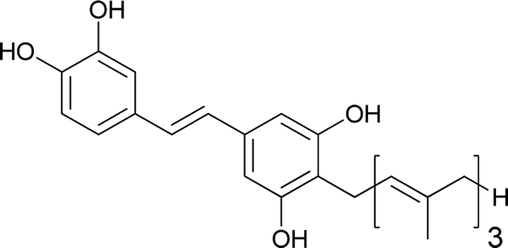
Privileged structures have been widely used as effective templates in drug discovery. Stilbenes as such are a simple class of naturally occurring compounds that have a widespread distribution in nature (Fig. 1). Although their molecular backbone is made up of only a 1,2-diphenylethylene moiety, which exists as a trans- or cis-isomer, stilbenes have shown an immense array of different structural units. The stilbene moiety has been utilised as a starting source for the synthesis of new complex molecules.5,6
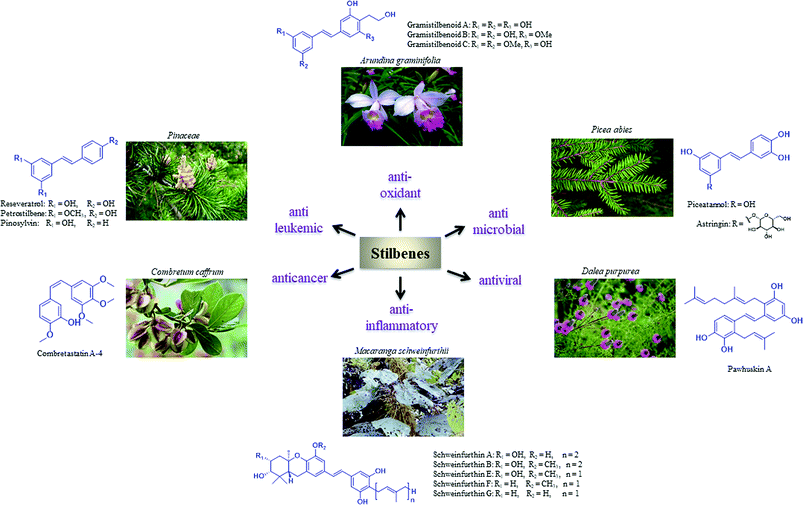 |
| | Fig. 1 Bioactive natural products with stilbene moiety. | |
Resveratrol is a major constituent of the stilbene family that has received significant attention due to its anticancer activity.7–12 However, there are numerous analogues (naturally occurring, synthetic and semisynthetic) in this family with significant properties and applications.13–20 Additionally, several stilbene-based scaffolds have been approved for clinical use. For example, diethylstilboestrol and tamoxifen are used for the treatment of prostate cancer and metastatic breast cancer, respectively.21–25
Schweinfurthins are prenylated/geranylated/farnesylated stilbenes that are isolated from numerous species of the plant genus Macaranga (Euphorbiaceae). There has been an increased interest in the schweinfurthin family because of their selective anti-proliferative activity against human cancer cells. The scarcity of natural schweinfurthins has enticed the evolution of the synthetic approach towards their total synthesis. This review provides details on the isolation, synthesis, and bioactivities of schweinfurthins. The difficulties overcome, along with the successful modification of synthetic strategies, are discussed in this review.
2. Isolation
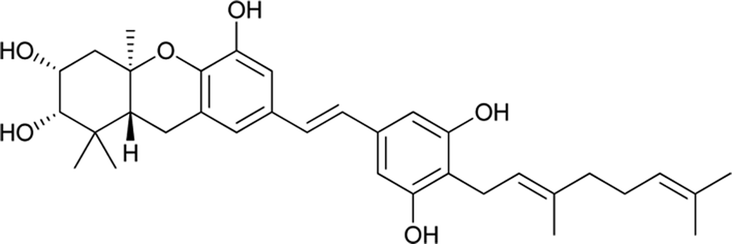
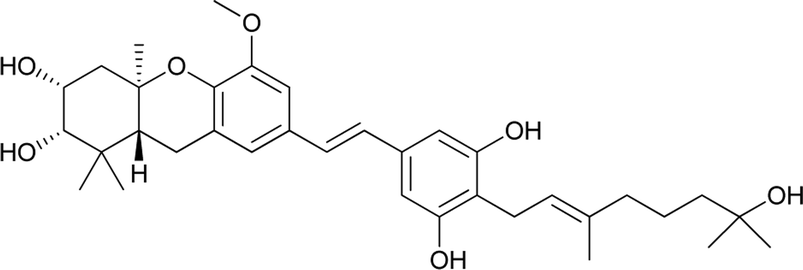
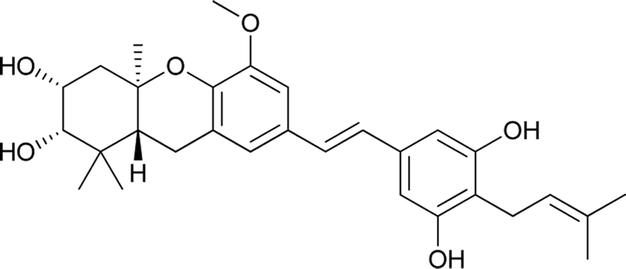
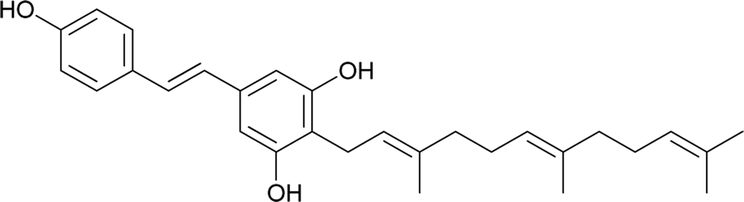
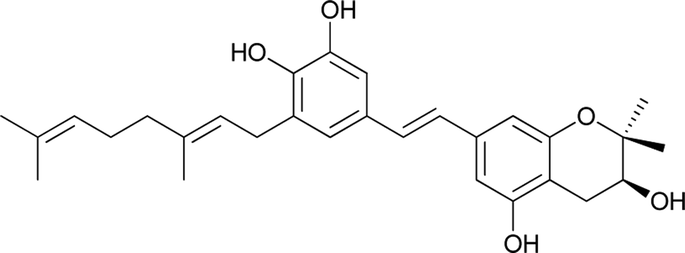
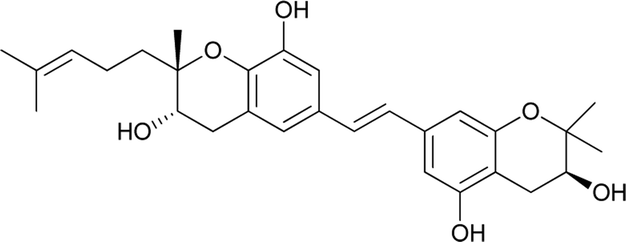
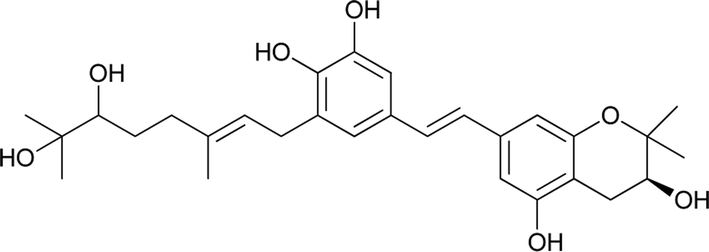
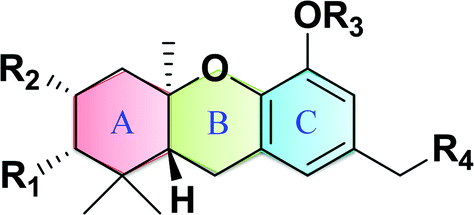
Macaranga is a genus of tropical trees that belongs to the Euphorbiaceae family and is the only genus in the subtribe Macaranginae. It is one of the largest genera of the Euphorbiaceae, comprising approximately 300 different species generally found in Africa, Australasia, Asia, and various islands in the Indian and Pacific oceans.26 Until now, 17 naturally occurring schweinfurthins (A–Q) have been isolated from the Macaranga genus (Table 1). In addition, one of the closely related congeners, vedelianin, along with two synthetic schweinfurthin analogues, is discussed.
Table 1 Structures of all schweinfurthins isolated from Macaranga species
| Comp |
Name |
Structure |
Natural source |
References |
| 1 |
Schweinfurthin A |
 |
M. schweinfurthii |
27 |
| 2 |
Schweinfurthin B |
 |
M. schweinfurthii |
27 |
| 3 |
Schweinfurthin C |
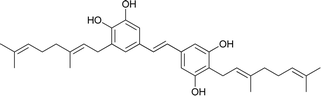 |
M. schweinfurthii |
27 |
| 4 |
Schweinfurthin D |
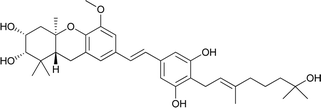 |
M. schweinfurthii |
28 |
| 5 |
Schweinfurthin E |
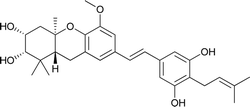 |
M. alnifolia |
29 |
| 6a |
Schweinfurthin F |
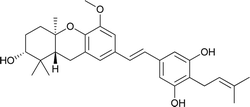 |
M. alnifolia |
29 |
| 7 |
Schweinfurthin G |
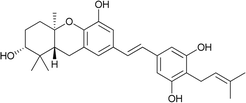 |
M. alnifolia |
29 |
| 8 |
Schweinfurthin H |
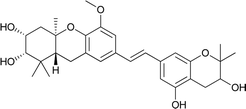 |
M. alnifolia |
29 |
| 9 |
Schweinfurthin I |
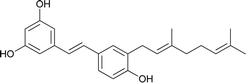 |
M. schweinfurthii |
30 |
| 10 |
Schweinfurthin J |
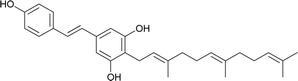 |
M. schweinfurthii |
30 |
| 11 |
Schweinfurthin K |
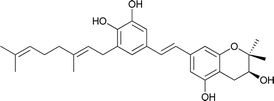 |
M. tanarius |
31 |
| 12 |
Schweinfurthin L |
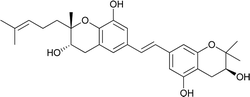 |
M. tanarius |
31 |
| 13 |
Schweinfurthin M |
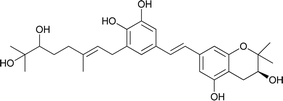 |
M. tanarius |
31 |
| 14 |
Schweinfurthin N |
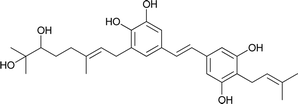 |
M. tanarius |
31 |
| 15 |
Schweinfurthin O |
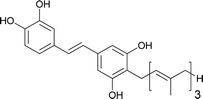 |
M. tanarius |
31 |
| 16 |
Schweinfurthin P |
 |
M. tanarius |
31 |
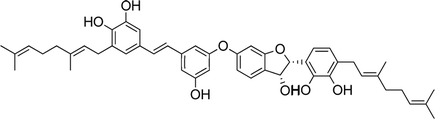
| 17 |
Schweinfurthin Q |
 |
M. tanarius |
31 |
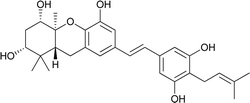
| 18 |
3-Deoxyschweinfurthin A (3dSA) |
 |
— |
— |
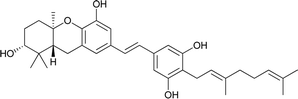
| 19 |
3-Deoxyschweinfurthin B (3dSB) |
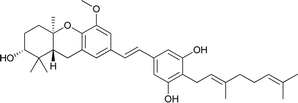 |
— |
— |
| 20 |
(+)-Vedelianin |
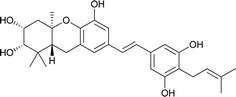 |
M. vedeliana, M. alnifolia |
29 and 32 |
In the past few years, the most common species of the Macaranga genus, M. vedeliana, M. pleiostemona, M. indica, M. tanarius and M. schweinfurthii have been thoroughly studied. A number of different classes of compounds such as prenylated stilbenes, geranylated flavonols, prenylated flavanones, chromenoflavones and diterpenes have been isolated from this species.33–36
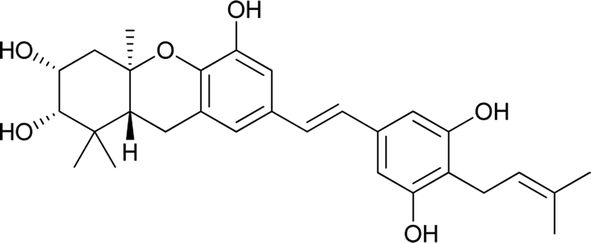
Schweinfurthins A–D were first isolated by Beutler et al. as yellowish solids from a mixed CH2Cl2–MeOH extract of M. schweinfurthii.27,28 Another unexplored species of the Macaranga genus, M. alnifolia, was highlighted by Yoder et al., who isolated four new prenylated stilbenes, schweinfurthins E–H, as pale yellow solids from an ethanolic extract of the fruit of M. alnifolia.29 Schweinfurthin H possesses a 2,2-dimethyldihydropyranol moiety, which was first observed in the schweinfurthin family. Klausmeyer et al. isolated two prenylated stilbenes, schweinfurthins I and J as yellow oils from the leaf of the M. schweinfurthii.30 Initially, Thoison et al. isolated vedelianin in 1991 from the leaves of M. vedeliana, which is closely related to the schweinfurthins.32 Later, Yoder et al. also isolated vedelianin along with schweinfurthins E-H from another Macaranga species, M. alnifolia.29
Recently, Péresse et al. isolated seven more schweinfurthins, K–Q, from M. tanarius. Among these seven newly reported natural products, only schweinfurthin Q has a hexahydroxanthene tricyclic core. Schweinfurthins K, L, and M possess a 2,2-dimethyldihydropyranol moiety. All of the compounds were isolated as orange oils from an ethanolic extract of the dried fruits of M. tanarius.31
3. Synthesis
Natural schweinfurthins possess a stilbene backbone with prenylated/geranylated/farnesylated moieties on any of the rings. Generally, the stilbene skeleton is synthesized using a Wittig or modified Wittig olefination, Heck, Suzuki, Stille or Sonogashira coupling reactions. In the case of the schweinfurthins, Horner–Wadsworth–Emmons (HWE) olefination has been utilised to selectively construct the trans-stilbene skeleton.
3.1. Synthesis of hexahydroxanthene
Hexahydroxanthene, a tricyclic moiety, is present in many schweinfurthins whose stereochemistry was not completely elucidated and hence, its synthesis was of the utmost important (Fig. 2). Previously, Mechoulam and Yagen have reported the cyclisation of geranyl olivetol under harsh reaction conditions using concentrated H2SO4 in nitromethane.37 In addition, an attempt was made to synthesize the hexahydroxanthene core from geranylated phenols by constructing A- and B-rings on the aromatic C-ring in a single step, which resulted in a poor yield, along with byproducts.38,39
 |
| | Fig. 2 Hexahydroxanthene moiety. | |
3.1.1 The Treadwell et al. approach (2002). Treadwell et al. developed a convenient route for the synthesis of hexahydroxanthene.40 The presence of phenylthio and phenylselenyl groups at the alpha position, adjacent to the terminal cation on the geranyl chain, might provide substantial stability to the emerging carbocation.41,42 Based on previous studies, 29 was viewed as a key intermediate for constructing hexahydroxanthene.43,44The synthesis commenced with vanillin (21) to give benzaldehyde analogue 23.45 The reduction of aldehyde 23 to alcohol 24 followed by protection with triethylsilyl ether afforded 25, which upon reacting with geranyl bromide in the presence of n-BuLi furnished compound 26 in 74% yield. The m-CPBA epoxidation of compound 26 yielded the desired 6,7-epoxide 27 in 53% yield along with traces of 2,3-epoxide. Compound 27 reacted smoothly with a phenyl selenide anion to afford 28, which upon treatment with 0.5 M HCl (for the deprotection of the silyl ether) and subsequently with 1 M HCl (for the deprotection of MOM) gave hydroxyselenide 29 in 66% yield. The deprotection of both groups in a single step was unsuccessful, resulting in either incomplete deprotection or lower yields along with byproducts (Scheme 1).
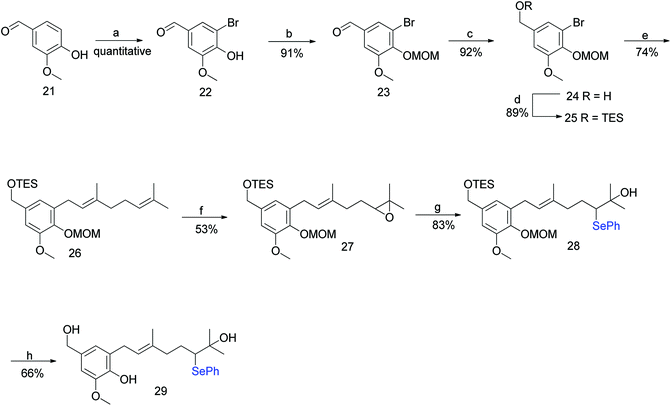 |
| | Scheme 1 Synthesis of cyclisation precursor hydroxyselenide (29). Reagents and conditions: (a) Br2, CH3CO2H, 25 °C 0.5 h; (b) MOMCl, TBAI, NaH, DMF; (c) LAH, THF, 0 °C, 11 min; (d) TESCl, imidazole, CH2Cl2; (e) n-BuLi, geranyl bromide, THF, −78 °C; (f) m-CPBA, CH2Cl2, −30 °C, 30 min; (g) NaBH4, (PhSe)2, EtOH; (h) 0.5 M or 1 M HCl, MeOH, reflux. | |
To address the deprotection issue, Treadwell et al. developed an alternative synthetic route, as shown in Scheme 2. Compound 30 obtained from vanillin was protected with TBS and selectively cleaved to obtain free phenol 32 using TBAF. Compound 32 was protected with ethyl vinyl ether (because silyl group migration from the phenolic oxygen to the adjacent ortho-carbon was observed) to give the fully protected compound 33. The geranylation of 33, followed by an acidic workup yielded compound 34, which was again protected with a silyl ether to afford 35 in 98% yield. A synthetic route similar to Scheme 1 was followed to generate the cyclisation precursor α-hydroxyselenide 29 from 35. The TFA-induced cyclisation of 29 afforded the tricyclic hexahydroxanthene core (38) in 43% yield. The formation of a single diastereomer was confirmed from NMR spectroscopy studies (Scheme 2).
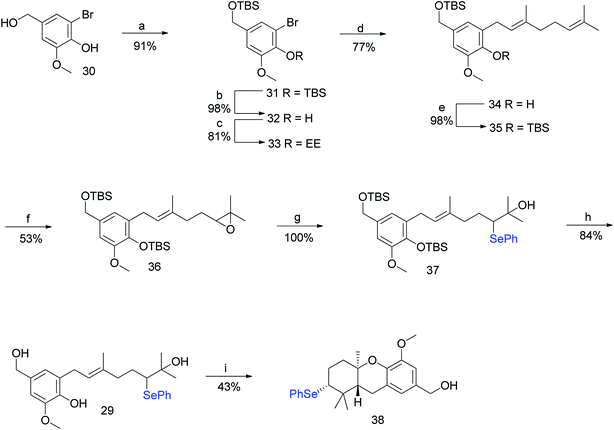 |
| | Scheme 2 Synthesis of hexahydroxanthene (38). Reagents and conditions: (a) TBSCl, CH2Cl2, 18 h; (b) TBAF, THF, 1 h; (c) pyridinium p-toluenesulphonate (PPTS), ethyl vinyl ether, CH2Cl2, 40 h; (d) n-BuLi, geranyl bromide, THF, −78 °C; (e) TBSCl, CH2Cl2, 24 h; (f) m-CPBA, CH2Cl2, −30 °C, 30 min; (g) NaBH4, (PhSe)2, EtOH, 48 h; (h) TBAF, THF, 3 days; (i) TFA, CH2Cl2, 24 h. | |
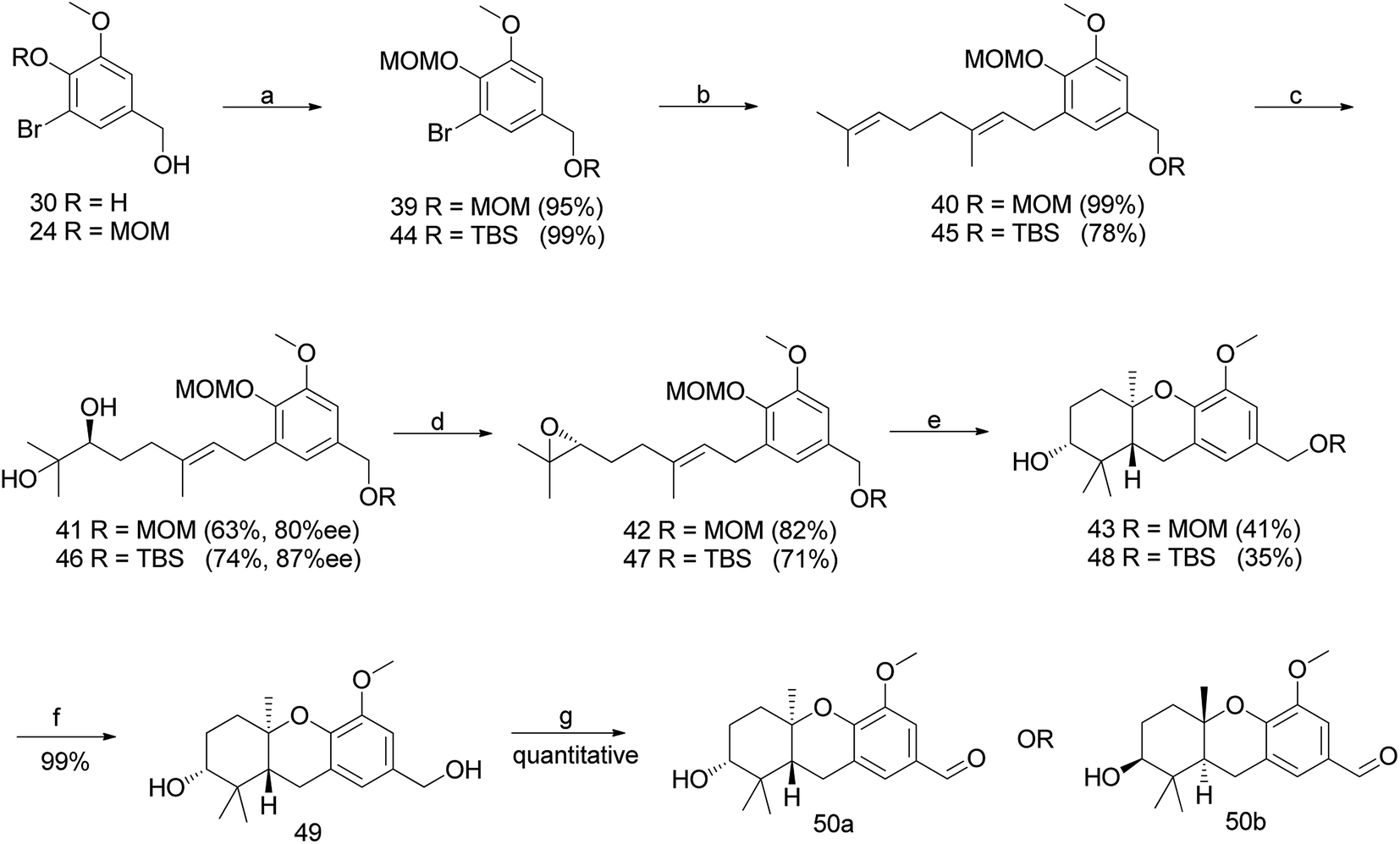
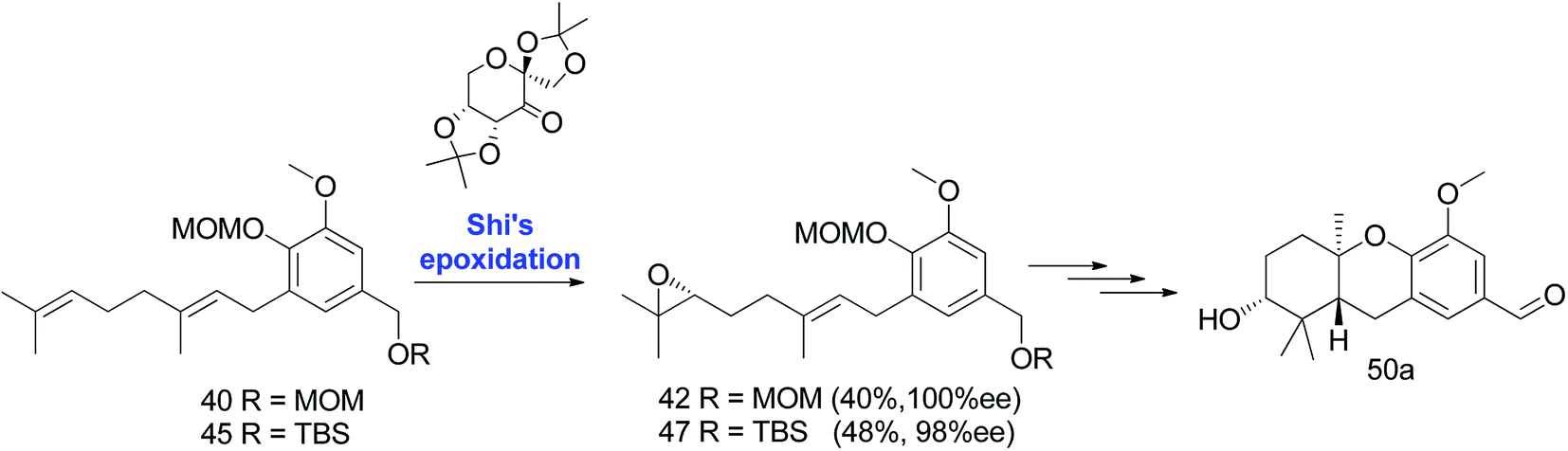
3.1.2 The Neighbors et al. approach (2008). The first-generation synthesis of hexahydroxanthene 50a, involving a cascade cyclisation and asymmetric dihydroxylation, was low in yield, with 16 steps required from vanillin (10% overall yield).46 Considering the productivity, Neighbors et al. developed efficient synthetic routes for the preparation of a geranyl epoxide ring.47
3.1.2.1 A change in protective group strategy. The sharpless dihydroxylation of a MOM-protected compound 40 gave diol 41, which was transformed to epoxide 42 using a previously developed protocol.46 Acid catalysed cyclisation using TFA afforded compound 43; however, the deprotection of an intransigent benzylic MOM group was not observed as anticipated. Hence, the reaction sequence was repeated using TBS as a protecting group to obtain compound 48. Furthermore, the deprotection of TBS using TBAF gave benzyl alcohol 49, which upon benzylic oxidation using MnO2 cleanly afforded aldehyde 50a in 9% overall yield from vanillin (enantiomer 50b was synthesized via the same route using AD-mix-β) (Scheme 3).47
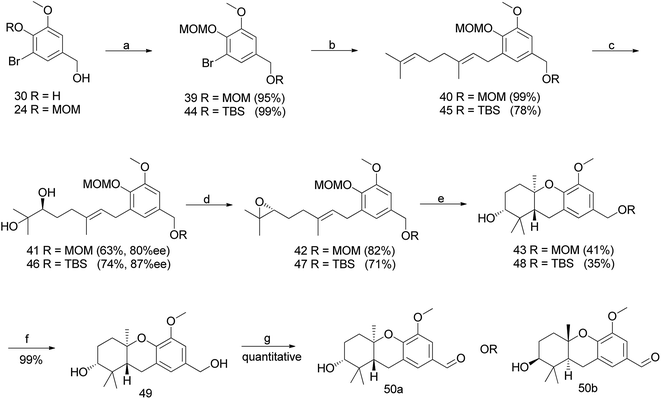 |
| | Scheme 3 Synthesis of hexahydroxanthene aldehyde (50a) via Sharpless dihydroxylation. Reagents and conditions: (a) MOMCl, DIPEA, CH2Cl2, 10 h OR TBSCl, imidazole, CH2Cl2, 16 h; (b) geranyl bromide, n-BuLi, THF, −78 °C; (c) AD-mix-α for 50a OR AD-mix-β for 50b, K2Os2O7 in H2O/t-BuOH, CH3SO2NH2, 4 h; (d) (1) MsCl, NEt3, CH2Cl2, 2 h, (2) K2CO3, MeOH, 20 h; (e) TFA, CH2Cl2, 1.5 h; (f) TBAF, THF, 1.5 h; (g) MnO2, CH2Cl2. | |
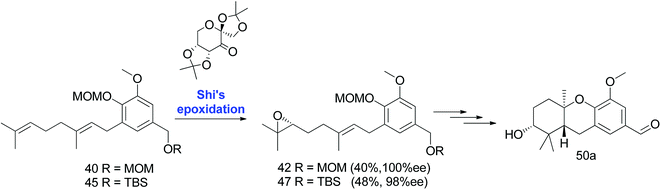 |
| | Scheme 4 Synthesis of hexahydroxanthene aldehyde (50a) via Shi's epoxidation. Reagents and conditions: Shi epoxidation diketal catalyst, buffer solution (2 M K2CO3, 0.4 mM EDTA), H2O2, CH3CN:CH2Cl2:EtOH. | |
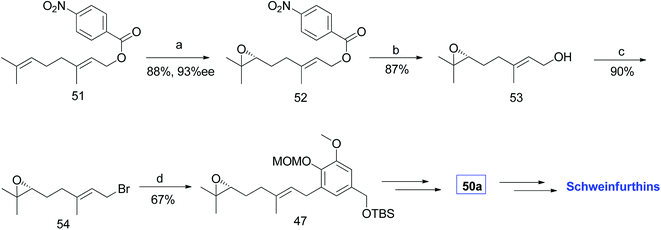 |
| | Scheme 5 Synthesis of R-(6,7)-epoxygeranyl bromide (54) via Shi's epoxidation. Reagents and conditions: (a) Shi epoxidation diketal catalyst, buffer solution (2 M K2CO3, 0.4 mM EDTA), H2O2, CH3CN:CH2Cl2:EtOH, 3 days; (b) NaOMe, TBAI, MeOH, 2.5 h; (c) (1) MsCl, NEt3, CH2Cl2, 0.5 h; (2) LiBr in THF, 2.5 h; (d) n-BuLi, 44, CuCN, THF, −78 °C. | |
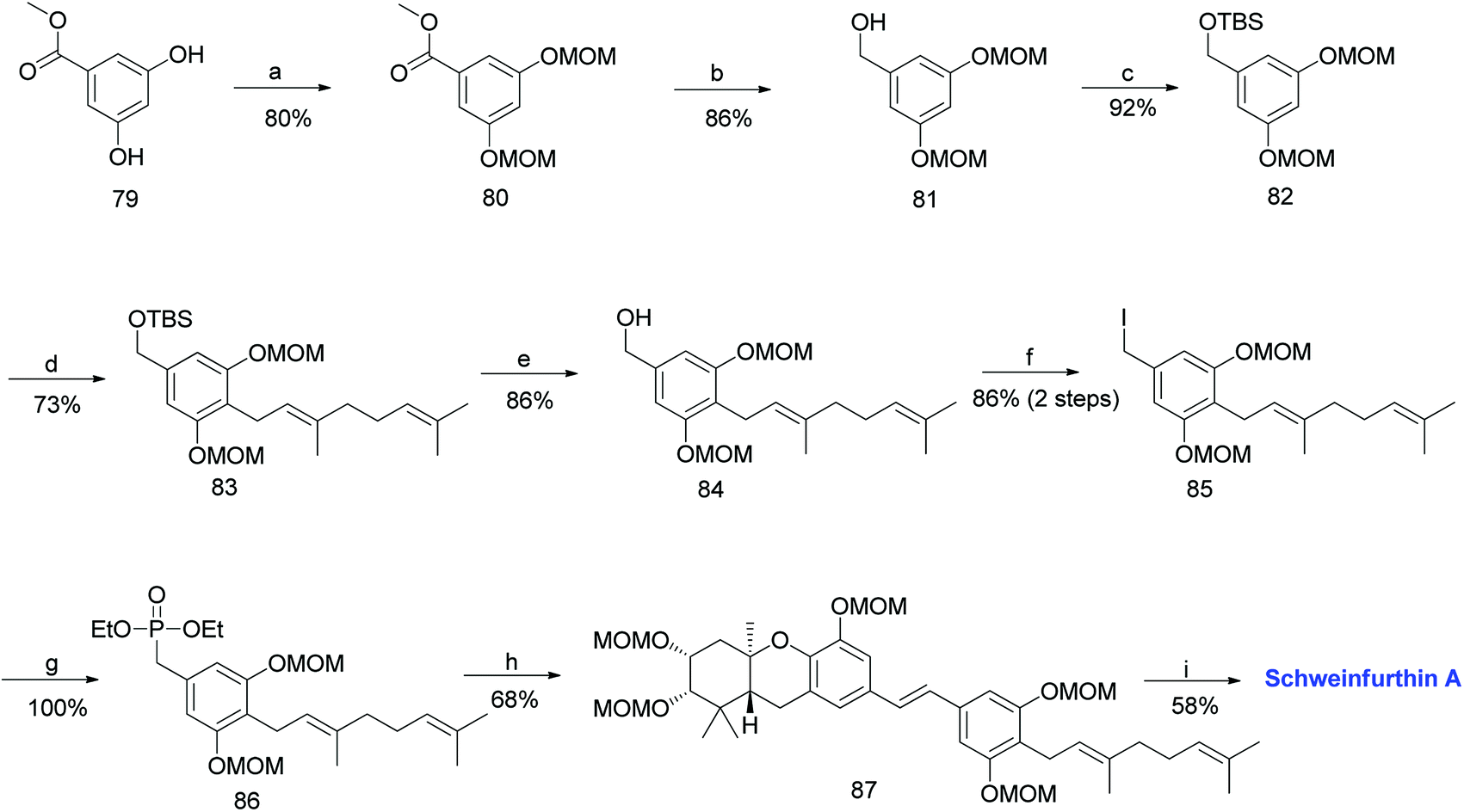
3.2. Synthesis of schweinfurthin A (2011)
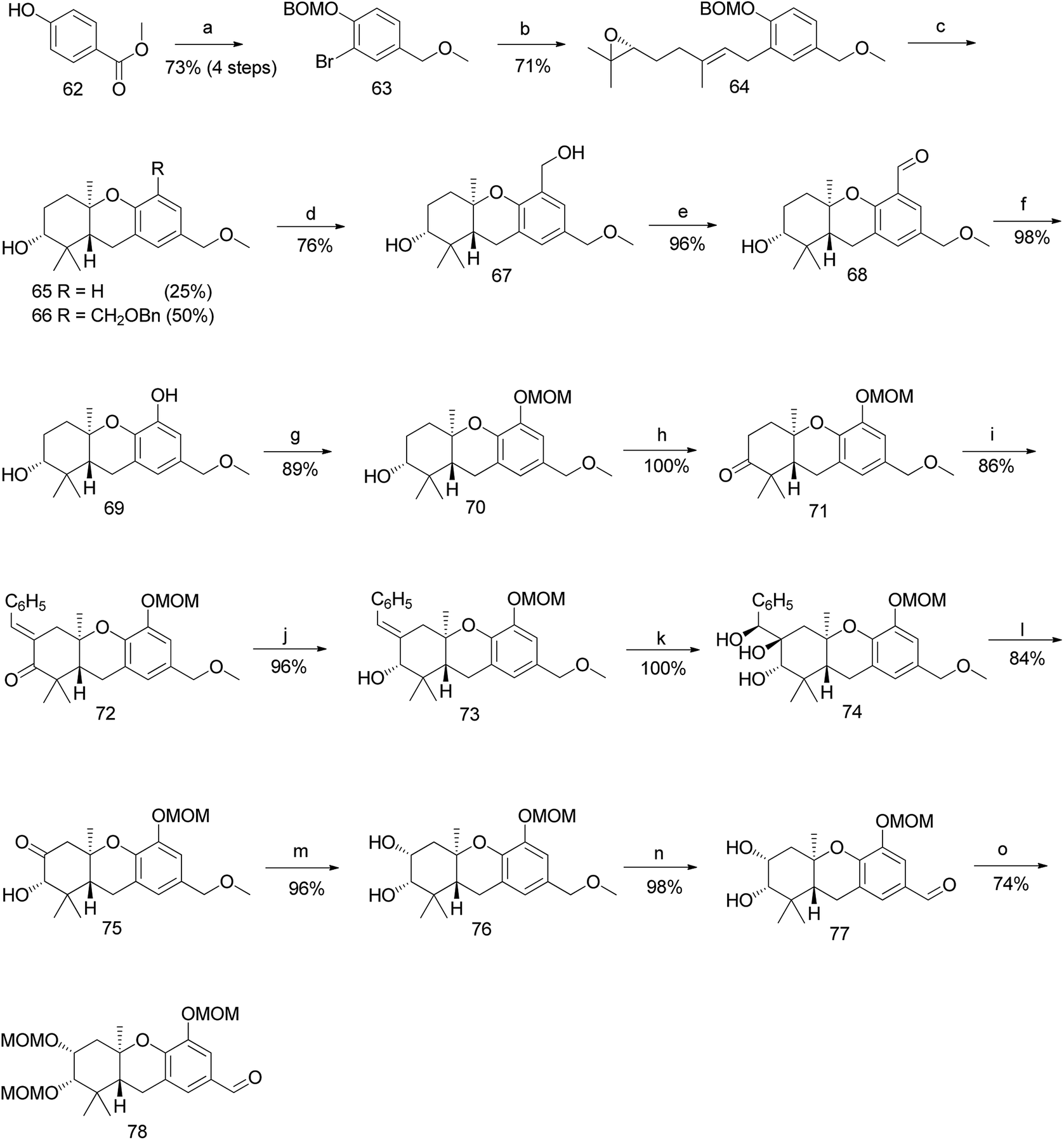
Topczewski et al. reported the total synthesis of schweinfurthin A (1), which is a potent compound in this family.50 Previous studies have reported the migration of the MOM group to the hydroxyl group of the A-ring during cascade cyclization (56). In addition, an electrophilic aromatic substitution occurs forming a new carbon–carbon bond by migration of the MOM group to an adjacent aromatic C-ring (58) (Scheme 6a).51 Hence, it was envisaged that the boron trifluoride etherate (BF3·OEt2)-mediated cyclisation52 on MOM-protected epoxide 59 may afford hexahydroxanthene 60 with the MOM group moving to the C-5 position, from which intermediate 61 with a phenolic functionality could be obtained by oxidation (Scheme 6b).
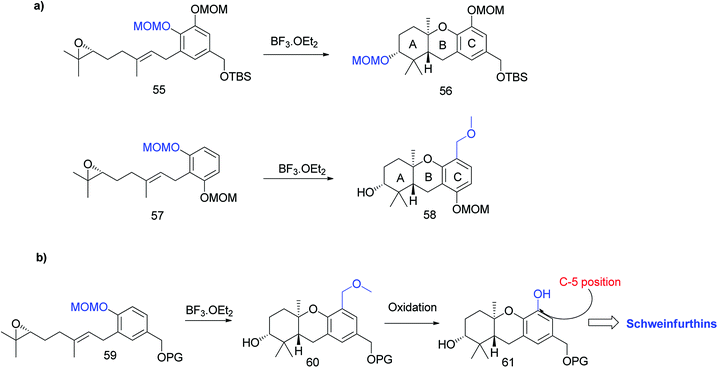 |
| | Scheme 6 Introduction of –OH on aromatic C-ring via tandem cascade cyclisation/aromatic substitution. (a) Migration of the MOM group on the A- and C-rings during the BF3·OEt2-mediated cyclisation step; (b) predicted route to introduce –OH onto the C-ring via tandem cascade cyclisation/aromatic substitution. | |
Accordingly, a compound possessing an (R)-6,7-epoxygeranyl chain (64) was synthesized from 4-hydroxybenzoate (62) in 5 steps (93% ee). The BF3·OEt2-mediated cascade cyclisation gave an inseparable mixture of 65 and 66 in 25% and 50% yield, respectively. A BOM acetal was chosen as the protecting group because it could be selectively deprotected by hydrogenolysis after being transferred to the C-5 position of the aromatic C-ring. Compounds 65 and 66 were separated by selective hydrogenolysis to afford benzylic alcohol 67 in 76% yield. Furthermore, the chemo-selective benzylic oxidation of 67 gave the corresponding aldehyde 68, which upon subsequent Baeyer–Villiger oxidation with m-CPBA and hydrolysis of formate yielded compound 69 in 98% yield. The –OH functionality was successfully introduced at the C-5 position of the aromatic C-ring, and protection with MOM gave 70 in 89% yield (Scheme 7).
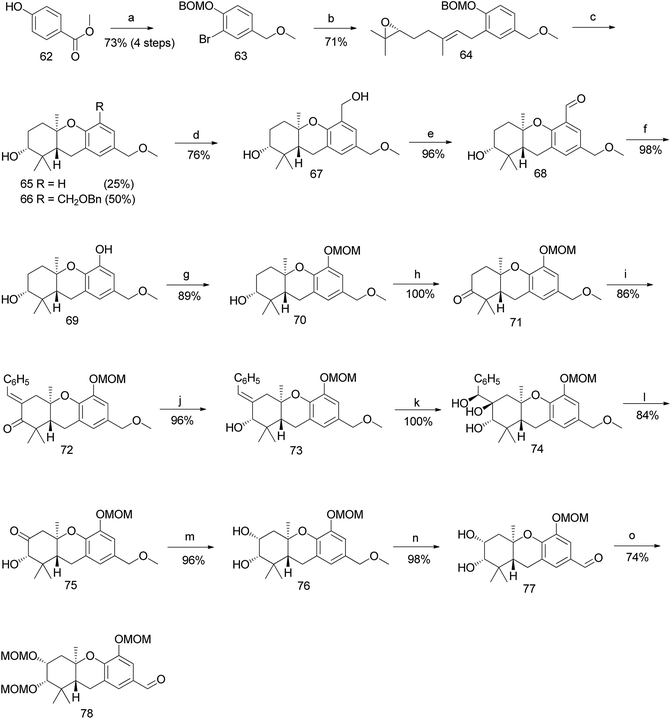 |
| | Scheme 7 Synthesis of hexahydroxanthene aldehyde (78) by Topczewski et al.50 Reagents and conditions: (a) (1) Br2, CH2Cl2; (2) BOMCl, DIPEA, CH2Cl2; (3) LiBH4, ether; (4) MeI, NaH, THF, 5 h; (b) TMEDA, n-BuLi, CuI, (R)-6,7-epoxy-geranyl bromide 54, Et2O, −8 °C; (c) BF3·OEt2, CH2Cl2, −78 °C, 8 min; (d) H2, 10% Pd/C, MeOH, 23 h; (e) MnO2·CH2Cl2, 20 h; (f) (1) m-CPBA, CH2Cl2, 2 h; (2) K2CO3, 20 h; (g) MOMCl, DIPEA, CH2Cl2; 2 h; (h) catalytic TPAP, NMO, CH2Cl2, 26 h; (i) PhCHO, KOH, EtOH, 25 min; (j) CeCl3·7H2O, NaBH4, MeOH, 2 h; (k) OsO4, t-BuOH, NMO, dioaxane:H2O, 17 h; (l) NaIO4, CH2Cl2:H2O, 24 h; (m) NaBH4, MeOH:THF, 15 min; (n) DDQ, CH2Cl2:H2O, 15 min; (o) MOMCl, DIPEA, CH2Cl2, 5 h. | |
The oxidation of hexahydroxanthene 70 under Ley's conditions gave ketone 71, which was subjected to aldol condensation with benzaldehyde to give enone 72, followed by Luche reduction to afford alcohol 73. Compound 73 was subjected to Upjohn dihydroxylation to give triol 74, which upon glycolytic cleavage in the presence of NaIO4 yielded ketone 75. The diastereoselective reduction of 75 using NaBH4 gave diol 76, and subsequent DDQ oxidation of the methyl ether on the aromatic C-ring gave the intermediate aldehyde 77. The obtained aldehyde was protected with the MOM group to give aldehyde 78 in 74% yield (Scheme 7).
The required phosphonate ester 86 was synthesized, as depicted in Scheme 8.53,59 The synthesis began with the MOM-protection of commercially available phenolic ester 79 to obtain compound 80, followed by reduction to give benzyl alcohol 81. Furthermore, compound 81 was protected with a TBS group to afford 82. The metalation of 82 with n-BuLi and TMEDA in the presence of copper cyanide (CuCN), followed by a subsequent reaction with geranyl bromide gave 83. The TBS deprotection of compound 83 using TBAF yielded benzylic alcohol 84 in 86% yield, which was sequentially converted to phosphonate ester 86 by treatment with methanesulfonyl chloride (MsCl), followed by iodination, and subjecting it to the Arbuzov reaction with P(OEt)3. The phosphonate ester 86 was condensed with MOM-protected aldehyde 78 using an HWE olefination in the presence of KHMDS as a base to give protected (E)-stilbene 87. Finally, deprotection of the MOM groups under acidic conditions afforded schweinfurthin A (1) in 58% yield (Scheme 8).
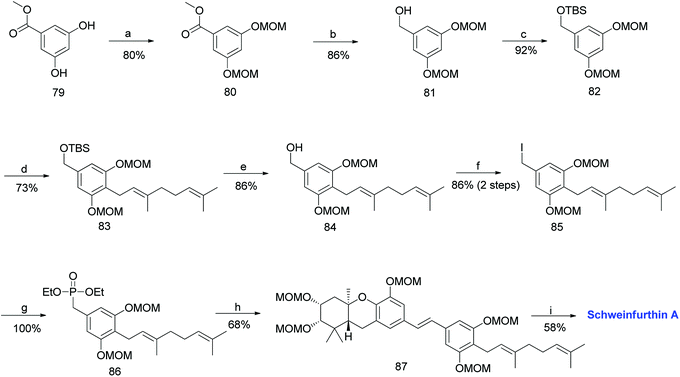 |
| | Scheme 8 Synthesis of schweinfurthin A by Topczewski et al.50 Reagents and conditions: (a) MOMCl, NaH, DMF, 4 h; (b) LAH, THF, 30 min; (c) TBSCl, imidazole, CH2Cl2, 12 h; (d) n-BuLi, TMEDA, CuCN, geranyl bromide, THF, −78 °C; (e) TBAF, THF, 3 h; (f) (1) MsCl, NEt3, CH2Cl2, 1 h; (2) NaI, acetone, 22 h; (g) P(OEt)3, reflux, 2.5 h; (h) (1) DIPEA, n-BuLi, aldehyde 78, THF, 2 h (little progress in reaction); (2) KHMDS, 2 h; (i) p-TsOH, MeOH. | |
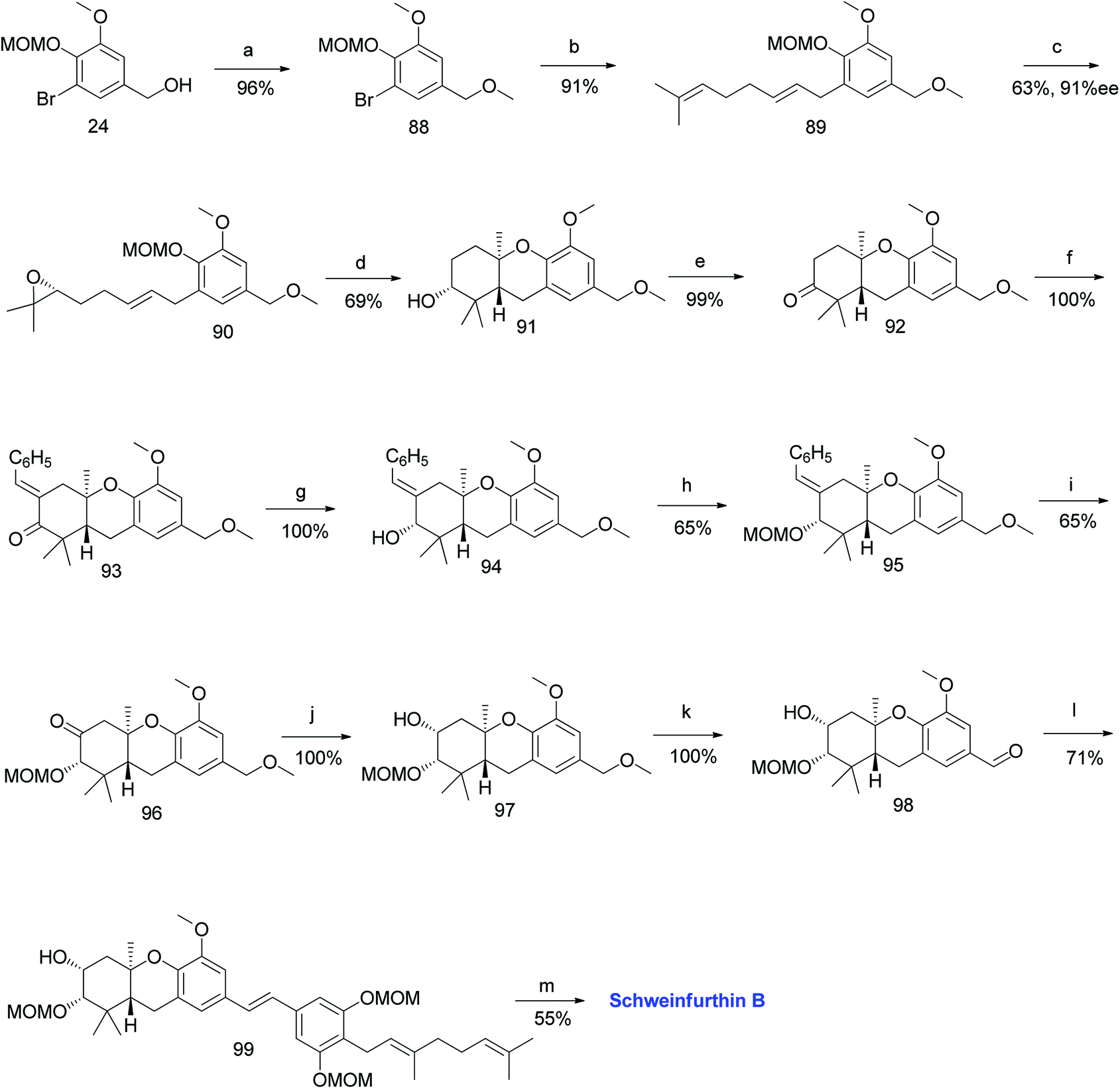
3.3. Synthesis of schweinfurthin B (2009)
Topczewski et al. reported the synthesis of schweinfurthin B (2) by eliminating the established desilylation/oxidation sequence.54 Benzyl alcohol 24 was protected with a benzyl methyl ether and converted to tricyclic compound 91 via Shi epoxidation followed by a BF3·OEt2-mediated cascade cyclisation. The oxidation of compound 91 under Ley's conditions afforded ketone 92. The aldol condensation of compound 92 with benzaldehyde gave enone 93, which upon treatment with OsO4/NaIO4 resulted in an undesirable acetal compound; however, the reduction under Luche conditions afforded the required alcohol 94. The configuration was evident from NOESY correlations. The oxidation of MOM-protected compound 95 using excess KMnO4 gave ketone 96, and subsequent NaBH4 reduction yielded secondary alcohol 97, which upon DDQ oxidation of the methyl ether gave aldehyde 98 in excellent yield. The HWE olefination of phosphonate ester 86 with aldehyde 98 yielded (E)-stilbene 99, which upon deprotection of the MOM groups gave schweinfurthin B (2) in 55% yield (Scheme 9).54
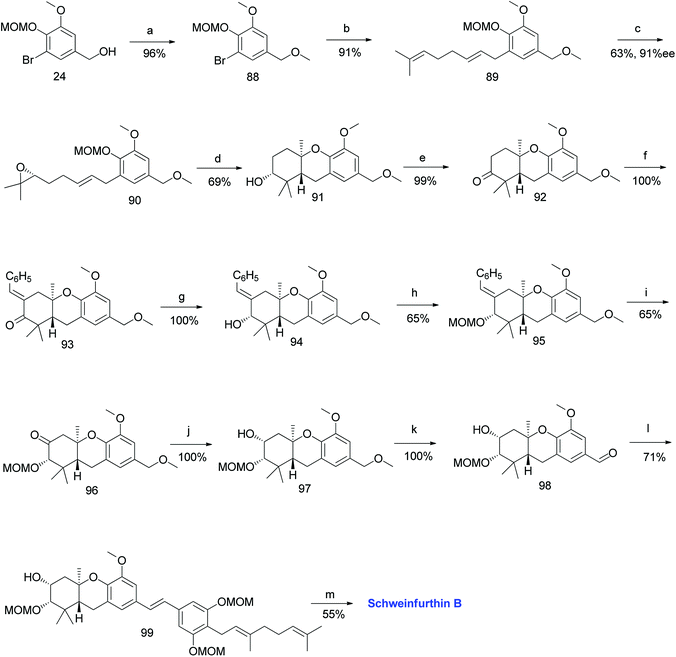 |
| | Scheme 9 Synthesis of schweinfurthin B by Topczewski et al.54 Reagents and conditions: (a) NaH, MeI, THF, 3 h; (b) n-BuLi, geranyl bromide, THF, −78 °C; (c) Shi epoxidation diketal catalyst, buffer solution (2 M K2CO3, 0.4 mM EDTA), H2O2, CH3CN:CH2Cl2:EtOH, 10 h; (d) BF3·OEt2, CH2Cl2, −78 °C, 7 min; (e) NMO, TPAP, CH2Cl2, 18.5 h; (f) benzaldehyde, KOH, EtOH, 2 h. (g) CeCl3·7H2O, NaBH4, CH3OH, 20 min; (h) MOMCl, DIPEA, CH2Cl2, 15 h; (i) KMnO4, NaHCO3, acetone, 20 h; (j) NaBH4, CH3OH, 10 min; (k) DDQ, CH2Cl2:H2O, 80 min; (l) NaH, phosphonate ester 86, 15-crown-5, THF; (m) p-TsOH, MeOH. | |
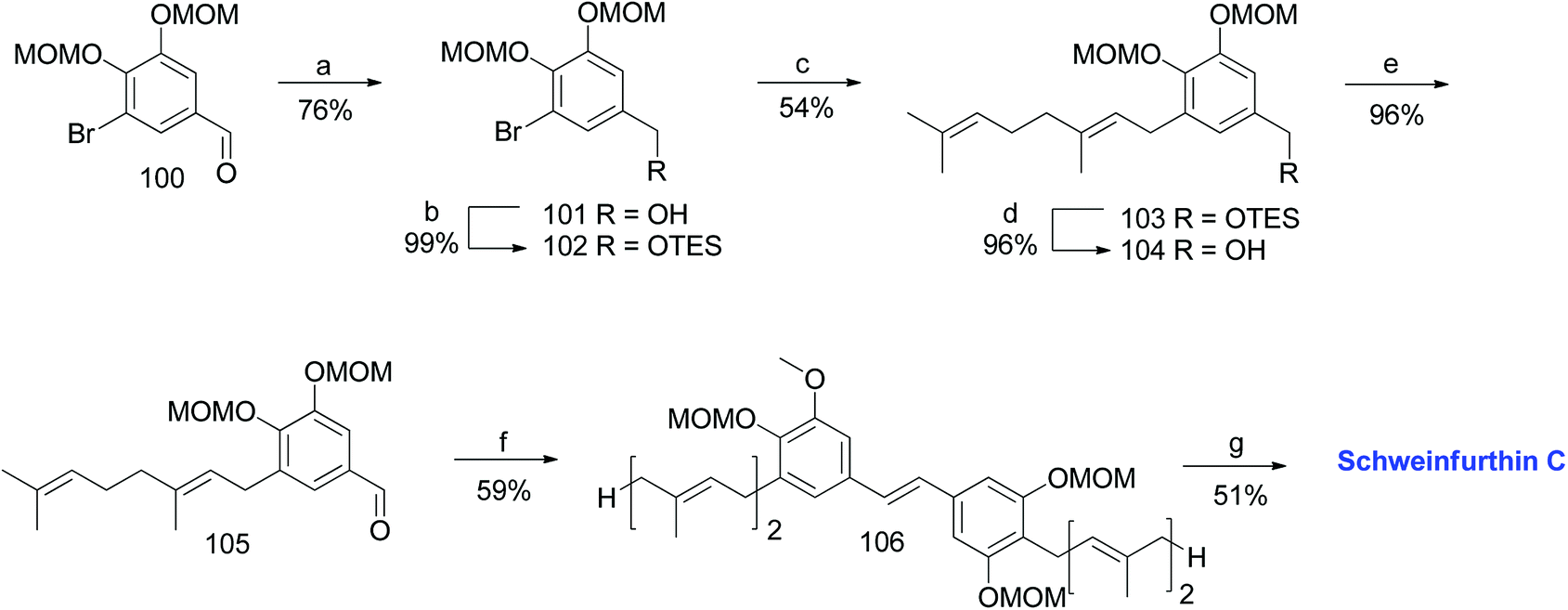
3.4. Synthesis of schweinfurthin C (1999)
The first total synthesis of schweinfurthin C (3) was reported in 1999 by Treadwell et al.53 TES-protected benzylic alcohol 102 was synthesized from the known aldehyde 100, readily prepared from vanillin.55–57 Furthermore, a geranyl moiety was installed via lithium–halogen exchange using n-BuLi and geranyl bromide to give 103, which upon deprotection followed by PDC oxidation afforded aldehyde 105 in 96% yield. The HWE olefination of phosphonate ester 86 with aldehyde 105 selectively gave (E)-stilbene 106, and deprotection of the MOM groups afforded schweinfurthin C (3) in 51% yield (Scheme 10).
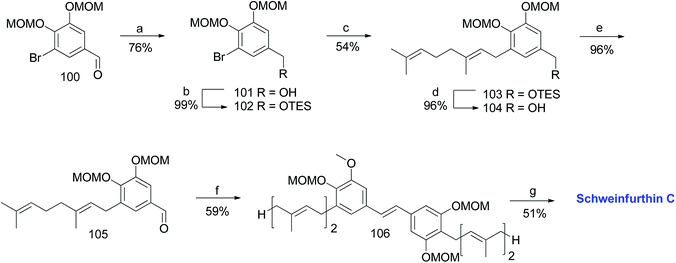 |
| | Scheme 10 Synthesis of schweinfurthin C by Treadwell et al.53 Reagents and conditions: (a) LAH, Et2O, 0 °C, 10 min; (b) TESCl, CH2Cl2, 22 h; (c) n-BuLi, geranyl bromide, Et2O, −78 °C; (d) TBAF, THF, 5 h; (e) PDC, CH2Cl2, 2.5 h; (f) phosphonate ester 86, NaH, THF, 30 min; (g) 3 M HCl, MeOH, reflux, 24 min. | |
3.5. Synthesis of schweinfurthin E (2009)
Topczewski et al. reported the synthesis of schweinfurthin E (5) using the same synthetic route as schweinfurthin B.54 A phosphonate ester 108 was synthesized using the reported method.58,59 A prenyl group was introduced by directed ortho-metalation on the known benzylic alcohol 81 to afford compound 107. Compound 107 was sequentially converted to the desired phosphonate ester 108, as depicted in Scheme 11. Phosphonate ester 108 and aldehyde 98 were condensed using HWE olefination to give protected (E)-stilbene 109, which was then deprotected under acidic conditions to obtain schweinfurthin E (5) in 81% yield (Scheme 11).
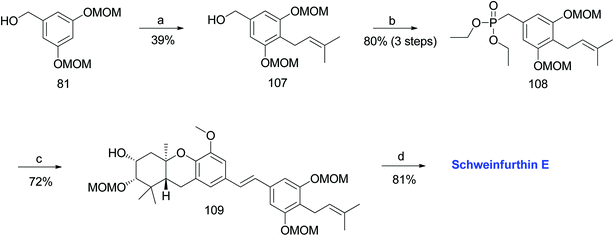 |
| | Scheme 11 Synthesis of schweinfurthin E by Topczewski et al.54 Reagents and conditions: (a) n-BuLi, TMEDA, CuBr.DMS, prenyl bromide, −20 °C, 4 h, THF; (b) (1) NEt3, MsCl, CH2Cl2, 4.5 h; (2) NaI, acetone, 13.5 h; (3) P(OEt)3, 100 °C, 4 h; (c) NaH, aldehyde 98, 15-crown-5, THF; (d) p-TsOH, MeOH. | |
3.6. Synthesis of schweinfurthin F (2007)
Mente et al. reported the synthesis of two enantiomers of schweinfurthin F in 2007.58 Having established the synthetic route for aldehydes 50a,b and phosphonate ester 108, HWE olefination followed by the acid-catalysed removal of the MOM groups afforded both the enantiomers schweinfurthin F [(R,R,R)-enantiomer 6a (69% yield) and (S,S,S)-enantiomer 6b (53% yield)]. Optical rotations and bioassay results indicated the formation of the 6a enantiomer, which is the natural form of schweinfurthin F (Scheme 12).
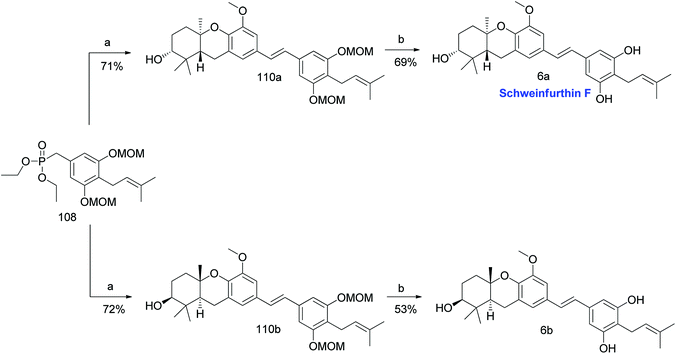 |
| | Scheme 12 Synthesis of schweinfurthin F by Mente et al.58 Reagents and conditions: (a) 50a or 50b, NaH, 15-crown-5, THF, 0 °C to rt, 24 h; (b) CSA, MeOH, rt for 16 h, 55 °C for 3.5 h. | |
3.7. Synthesis of schweinfurthin G (2008)
Mente et al. reported the synthesis of schweinfurthin G (7) via an efficient cascade cyclisation using BF3·Et2O as a Lewis acid.52 Initially, several Lewis acids were examined to cyclize epoxide 47, such as Ti(OiPr)4, TiCl4, SnCl4, MeAlCl2, In(OTf)3 and BF3·Et2O. The best result was obtained with the use of BF3·Et2O, and the cascade cyclisation was convenient and productive at larger scales. Using this approach, hexahydroxanthene was obtained in excellent yield (∼75%) compared with the protic acid-catalysed cyclisation (30–40%).60–64
The MOM-protected aldehyde 100 was sequentially converted to epoxide 55 via Shi epoxidation. The cascade cyclisation was achieved by reacting with BF3·Et2O to give hexahydroxanthenes 113 and 56 in 52% and 30% yield, respectively. The formation of the A-ring MOM ether 56 was observed due to the reaction of the CH3OCH2+ cation with the nucleophilic oxygen of the C-2 hydroxyl group during cyclisation. The deprotection of TBS using TBAF gave compound 114, which upon benzylic oxidation using MnO2 furnished aldehyde 115 in excellent yield. The HWE olefination of phosphonate ester 108 with aldehyde 115 yielded (E)-stilbene 116, which upon MOM-deprotection gave schweinfurthin G (7) in 57% yield (Scheme 13).
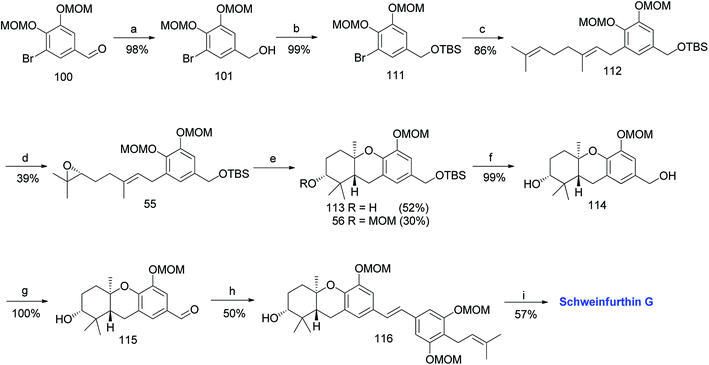 |
| | Scheme 13 Synthesis of schweinfurthin G by Mente et al.52 Reagents and conditions: (a) NaBH4, THF; (b) TBSCl, imidazole, CH2Cl2, 15 h; (c) n-BuLi, geranyl bromide, Et2O, −78 °C, 16 h; (d) Shi epoxidation diketal catalyst, buffer solution (2 M K2CO3, 0.4 mM EDTA), H2O2, CH3CN:CH2Cl2:EtOH, 22 h; (e) BF3·Et2O, CH2Cl2, 3.5 min; (f) TBAF, THF; (g) MnO2, CH2Cl2, 16.5 h; (h) NaH, phosphonate ester 108, 15-crown-5, THF; (i) p-TsOH, MeOH. | |
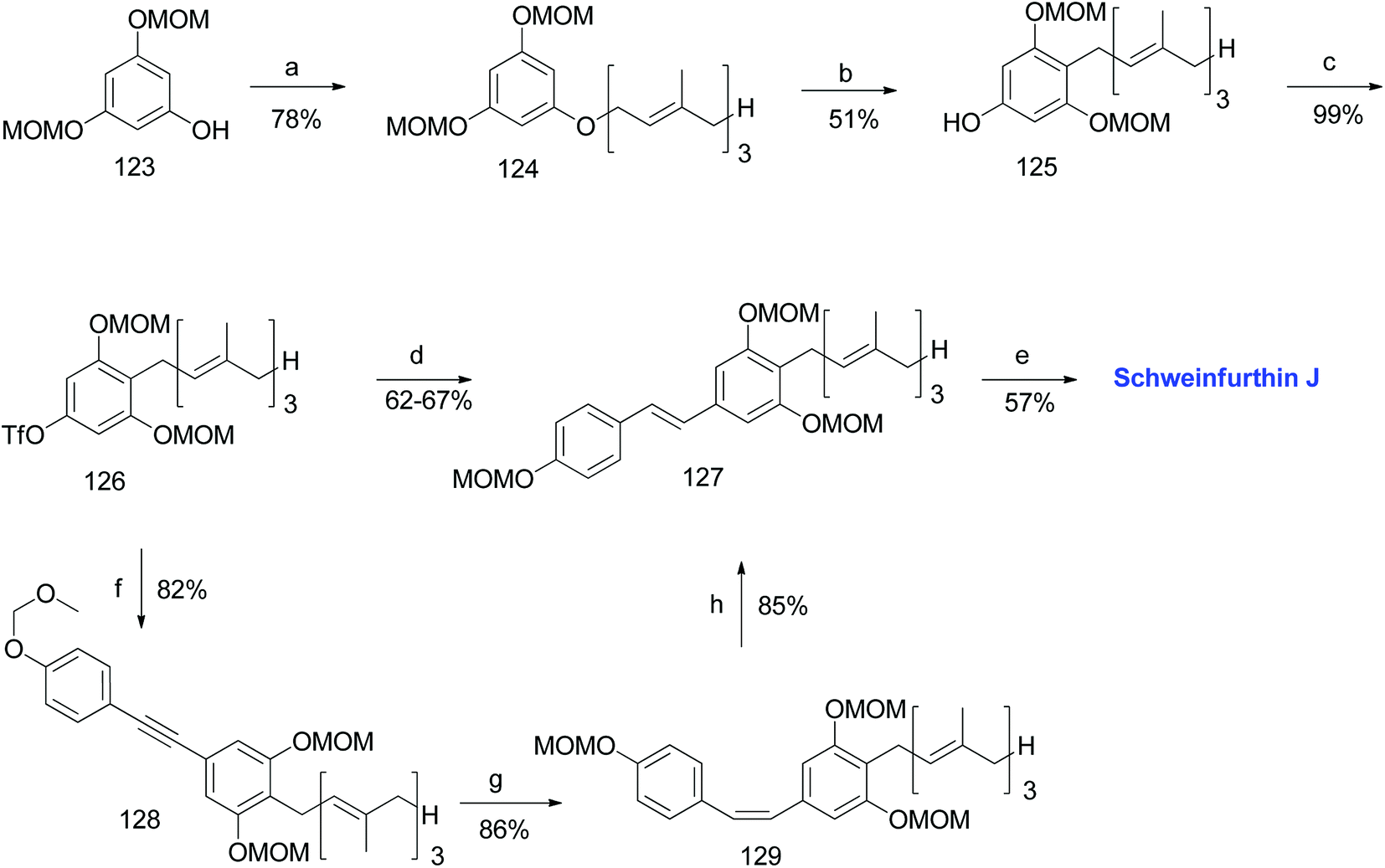
3.8. Synthesis of schweinfurthin J (2012)
Argade et al. reported the synthesis of farnesylated stilbene schweinfurthin J (10).65 The required coupling partners for Heck (118), Stille (120), Suzuki (122), and Sonogashira (119) coupling reactions were synthesized from aldehyde 117, as outlined in Scheme 14. A standard Wittig reaction with MOM-protected aldehyde 117 gave alkene 118 in 65% yield. The treatment of aldehyde 117 with the Bestmann–Ohira reagent gave 119, and followed by radical hydrostannation yielded stannane 120. Additionally, Takai olefination of aldehyde 117 afforded vinyl iodide 121, which was subsequently converted to pinacolborane 122 in 80% yield (Scheme 14).
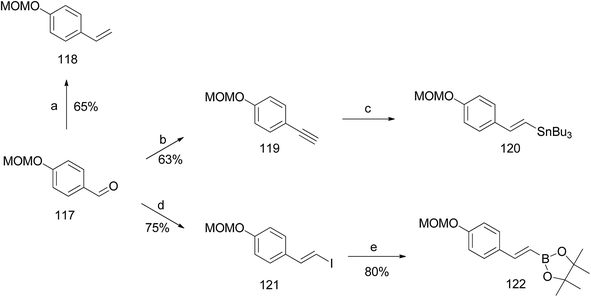 |
| | Scheme 14 Synthesis of intermediates 118–120 and 122. Reagents and conditions: (a) n-BuLi, MePPh3I, THF, 24 h; (b) Bestmann–Ohira reagent, K2CO3, MeOH, 24 h; (c) n-Bu3SnH, AIBN, 80 °C, 12 h; (d) CrCl2, CHI3, dioxane:THF, 0 °C, 2 h; (e) t-BuLi, boronate, THF, −78 °C, 2 h. | |
The phenolic compound 123 was alkylated using farnesyl bromide to give 124, followed by Claisen rearrangement to give p-farnesylated compound 125. The free phenolic group was converted to a triflyloxy-leaving group, resulting in the formation of intermediate 126 in modest yield, which underwent Heck, Stille, or Suzuki coupling reactions with the respective coupling partners to yield protected (E)-stilbene 127 (62–67% yield). Finally, the acid-catalysed deprotection of 127 afforded schweinfurthin J (10) in 57% yield. Alternatively, the Sonogashira coupling of 119 and triflate 126 yielded alkyne 128, which upon reduction gave cis-stilbene 129 using Pd(OAc)2 in the presence of KOH in dimethylformamide (DMF). The isomerization of cis-stilbene 129 to trans-stilbene 127 (85% yield) was achieved using Pd(MeCN)2Cl2 as a catalyst (Scheme 15).
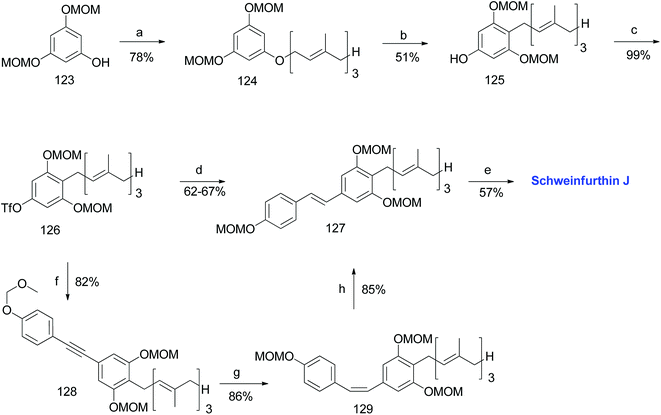 |
| | Scheme 15 Synthesis of schweinfurthin J by Argade et al.65 Reagents and conditions: (a) K2CO3, farnesyl bromide, DMF, 12 h; (b) DMA, 200 °C, 3 h; (c) Tf2O, pyridine, CH2Cl2, −20 °C, 2 h; (d) (1) Heck coupling: 118, Pd(OAc)2, Ph3P, Et3N, MeCN, 85 °C, 24 h, 63%; (2) Stille coupling: 120, Pd(PPh3)4, LiCl, DMF, 120 °C, 8 h, 67%; (3) Suzuki coupling: 122, Pd(PPh3)4, Na2CO3, dioxane:H2O, 90 °C, 8 h, 66%; (e) CSA, MeOH; 12 h; (f) Sonogashira coupling: 119, PdCl2(PPh3)2, CuI, TBAI, DMF:Et3N, 70 °C, 1.5 h; (g) Pd(OAc)2, KOH, DMF; (h) Pd(MeCN)2Cl2, CH2Cl2, 25 °C, 12 h. | |
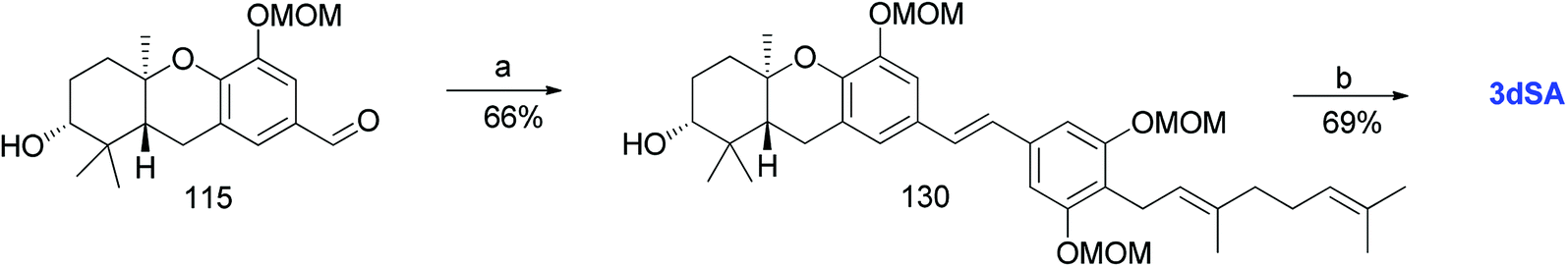
3.9. Synthesis of 3-deoxyschweinfurthin A (3dSA) (2008)
Mente et al. reported the synthesis of 3dSA (18) from aldehyde 115 and phosphonate ester 86, similar to that of schweinfurthin G (Scheme 16).52
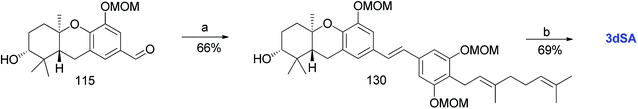 |
| | Scheme 16 Synthesis of 3-deoxyschweinfurthin A (3dSA) by Mente et al.52 Reagents and conditions: (a) NaH, phosphonate ester 86, 15-crown-5, THF; (b) p-TsOH, MeOH. | |
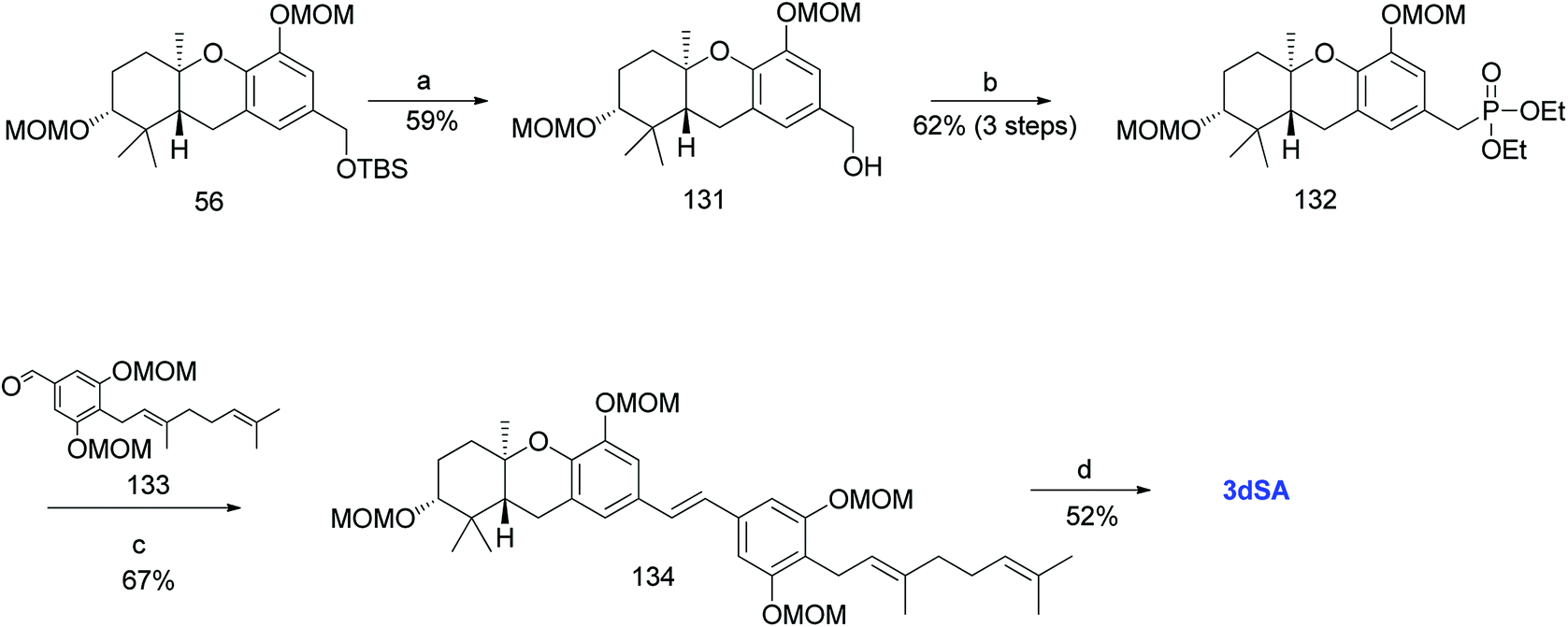
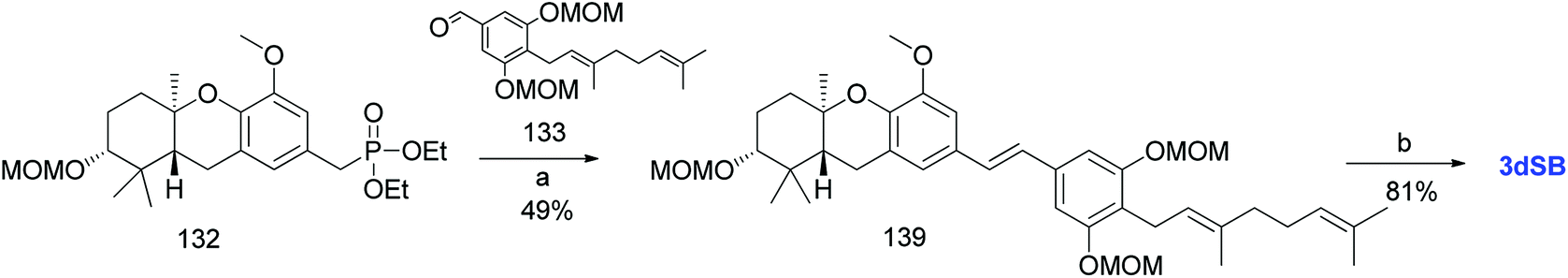
Additionally, Mente et al. utilised the byproduct compound 56 and transformed it into the phosphonate ester 132. The MOM-protected geranyl aldehyde 133 was synthesized by according to a reported procedure66 and was condensed with phosphonate ester 132 using an HWE reaction to give (E)-stilbene 134. Finally, deprotection of the MOM groups under acidic conditions gave 3dSA (18) in 52% yield (Scheme 17).52
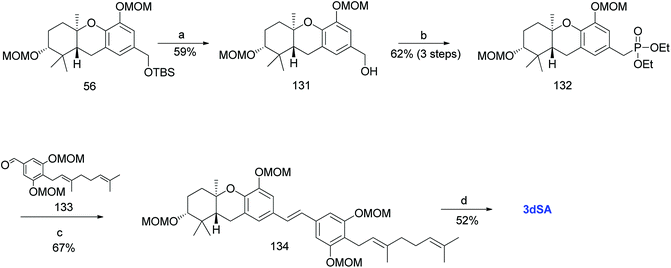 |
| | Scheme 17 Synthesis of 3-deoxyschweinfurthin A (3dSA) by Mente et al.52 Reagents and conditions: (a) TBAF, THF, 4 h; (b) (1) MsCl, NEt3, CH2Cl2; (2) NaI, acetone, 15 h; (3) P(OEt)3, 80 °C, 7 h; (c) NaH, geranyl aldehyde 133, 15-crown-5, THF, 16 h; (d) p-TsOH, MeOH. | |
3.10. Synthesis of 3-deoxyschweinfurthin B (3dSB)
3.10.1 The Neighbors et al. approach (2005). Neighbors et al. reported the first synthesis of 3dSB (19) while targeting the synthesis of a natural product, schweinfurthin B.46 The regioselective dihydroxylation on the terminal olefin of compound 35 via Sharpless dihydroxylation gave the desired diol 135 in 68% yield with 83% ee. The (S) configuration was assigned to the new asymmetric centre in diol 135, which was confirmed by treating it with the (S)- and (R)-enantiomers of O-methylmandelic acid under standard mixed anhydride coupling conditions, followed by spectroscopic studies. The diol 135 was transformed to non-racemic epoxide 136, as depicted in Scheme 18, in 75% yield, followed by deprotection of TBS using TBAF to afford 137. Furthermore, acid-catalysed cyclisation gave the expected tricyclic core 49 in 38% yield, which upon benzylic oxidation using MnO2 gave aldehyde 50a. The HWE olefination reaction between phosphonate ester 86 and aldehyde 50a yielded protected (E)-stilbene 138 and deprotection with camphorsulfonic acid (CSA) afforded 3dSB (19) in 77% yield (Scheme 18).
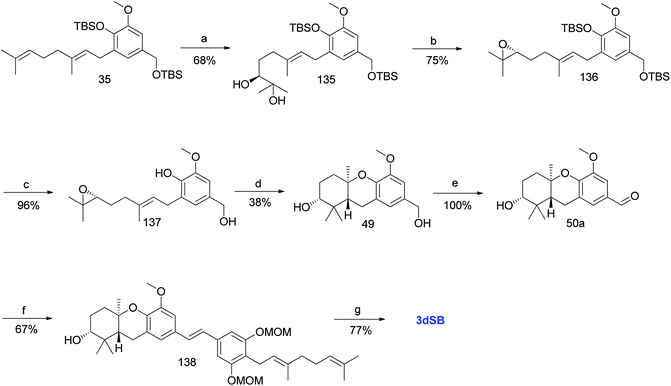 |
| | Scheme 18 Synthesis of 3-deoxyschweinfurthin B (3dSB) by Neighbors et al.46 Reagents and conditions: (a) AD-mix-α in H2O/t-BuOH, CH3SO2NH2, 6 °C, 15 h; (b) (1) MsCl, NEt3, CH2Cl2, 2 h; (2) K2CO3, MeOH, 20 h; (c) TBAF, THF, 1.5 h; (d) TFA, CH2Cl2, 2 h; (e) MnO2, CH2Cl2; (f) phosphonate ester 86, NaH, THF, 18 h; (g) CSA, MeOH, 5 h. | |
3.10.2 Mente et al. approach (2008). Mente et al. synthesized 3dSB (19) using an alternative route similar to that of 3dSA (Scheme 18).52 Phosphonate ester 132 and MOM-protected geranyl aldehyde 133 were condensed using an HWE olefination to give (E)-stilbene 139, which was deprotected under acidic conditions to yield 3dSB (19) in 81% yield (Scheme 19).
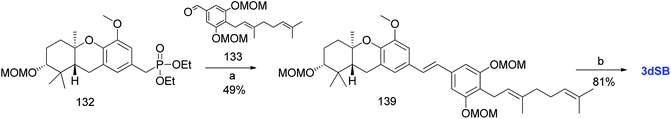 |
| | Scheme 19 Synthesis of 3-deoxyschweinfurthin B (3dSB) by Mente et al.52 Reagents and conditions: (a) NaH, geranyl aldehyde, 133, 15-crown-5, THF, 16 h; (b) p-TsOH, MeOH. | |
3.11. Synthesis of (+)-vedelianin (2011)
Topczewski et al. reported the synthesis of (+)-vedelianin (20) using a synthetic route similar to that of schweinfurthin A. A MOM-protected aldehyde 78 was synthesized from 101 via Shi epoxidation and a BF3·OEt2-mediated cyclisation. The HWE olefination of phosphonate ester 108 with aldehyde 78 selectively yielded (E)-stilbene 141, which upon MOM-deprotection afforded (+)-vedelianin (20) (Scheme 20).67
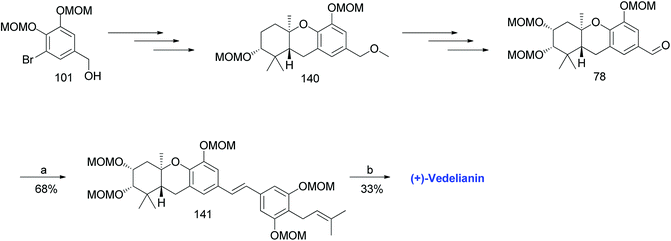 |
| | Scheme 20 Synthesis of (+)-vedelianin (2011) by Topczewski et al.67 Reagents and conditions: (a) KHMDS, phosphonate ester 108, THF, 30 min; (b) p-TsOH, MeOH, 48 h. | |
4. Biological activities
Schweinfurthins have shown a wide range of bioactivities, such as anticancer, antimicrobial, cytotoxicity and radical scavenging effects. However, the focus has always been on their anticancer properties, since they have displayed excellent inhibition of the growth of human cancer cell lines in the National Cancer Institute's 60-cell line screen (NCI-60).
4.1. Anticancer activity
The cytotoxicity of schweinfurthins towards cancer cell lines was first reported by Beutler et al. when they isolated three novel geranyl stilbenes, schweinfurthins A (1), B (2), and C (3).27 Schweinfurthins A (1) and B (2) were evaluated in an in vitro experiment using the lung cancer-derived A549 and glioblastoma multiforme (GBM)-derived SF295 cell lines. A 1 hour treatment led to negligible cytotoxicity, whereas evaluation at 48 hours showed modest cytotoxicity for each compound (Table 2). However, schweinfurthin C was found to be biologically inactive.
Table 2 Cytotoxicity of schweinfurthins A (1) and B (2) on SF295 and A549 cell lines
| Compound |
1, IC50 (μM) |
2, IC50 (μM) |
| Protocol |
A549 |
SF295 |
A549 |
SF295 |
| 1 h |
78.3 |
85.6 |
>170 |
>170 |
| 1 h washout |
36.4 |
0.109 |
142.1 |
5.1 |
| 48 h |
6.7 |
<0.00002 |
12.6 |
0.016 |
The SF295 cell line was found to be the most sensitive for compound 1, with a growth inhibition (GI50) of 11 nM and total growth inhibition (TGI) of 52 nM.27 Schweinfurthin A is a potent member of this family that inhibits the SF295 line at a low nanomolar concentration and displays 1000-fold selectivity compared to that of the A549 cell line; however, its mechanism of action has not been fully elucidated.
GBM is the most common, deadly brain tumor in adults, which is highly infiltrative, resistant to chemotherapy and incurable. Previous studies have shown that neurofibromatosis type 1 (NF1), an autosomal dominant genetic disorder, plays a role in GBM tumorigenesis.68,69 Turbyville et al. studied the mode of action of schweinfurthin A and observed that it inhibited SF295 and KR158 cell lines in a dose-dependent manner with no effect on A549, indicating that schweinfurthin A acts via a cytotoxic mechanism rather than a cytostatic one. Further findings have indicated that schweinfurthin A does not act at the level of Ras inhibition, but selectively inhibits proliferation and Rho signalling in the glioma and NF1 tumor cells in an NF1-GRD-dependent manner.70
Beutler et al. performed a 2 day cytotoxicity assay on schweinfurthin D (4) using two cell lines, SF295 and A549. Schweinfurthin D (4) (IC50 3.44 nM) was found to be equipotent to schweinfurthin B (2) (IC50 5.33 nM) for the SF295 line whereas, for the A549 cell line, the IC50 for compounds 4 and 2 were 4.82 μM and 9.77 μM, respectively.28
Mente et al. tested both the enantiomers of schweinfurthin F (6a and 6b) for growth inhibition in RPMI-8226 human-derived myeloma cells using a 3H-thymidine incorporation assay of DNA synthesis.58 The enantiomers 6a and 6b exhibited an IC50 of 0.58 μM and 3.40 μM, respectively (Table 3). In a glioma-derived SNB 75 cell line, a large divergence of bioactivity was observed, where compound 6a displayed a GI50 of 10 nM and its enantiomer 6b had a GI50 of 1.7 μM. The activity pattern was reversed in the malignant glioblastoma-derived human cell line U251, where 6b showed very high activity with a GI50 of <10 nM, while 6a had a GI50 of 13 μM (Table 3).58
Table 3 Cytotoxicity of schweinfurthin F (6a) and its enantiomer (6b) on RPMI-8226, SNB 75, and U251 cell lines
| Cell line |
Compounds |
| 6a |
6b |
| RPMI-8226 |
IC50 = 0.58 μM |
IC50 = 3.40 μM |
| SNB 75 |
GI50 = 10 nM |
GI50 = 1.7 μM |
| U251 |
GI50 = 13 μM |
GI50 < 10 nM |
Yoder et al. evaluated the cytotoxicity of schweinfurthins E–H and a congener vedelianin against the A2780 ovarian cancer cell line (Table 4).29 Vedelianin (20) was found to be the most potent compound with an IC50 of 0.13 μM. In a comparison of their cytotoxicity, vedelianin (20) was found to be twice as potent as schweinfurthin E (5) (IC50 0.26 μM) and three times as potent as schweinfurthin G (7) (IC50 0.39 μM).
Table 4 Cytotoxicity of schweinfurthins E–H and vedelianin on A2780 cells
| Compound |
IC50 (μM) |
| 5 |
0.26 |
| 6a |
5.0 |
| 7 |
0.39 |
| 8 |
4.5 |
| 20 |
0.13 |
Péresse et al. evaluated the cytotoxicity of schweinfurthins G, K–Q, and vedelianin against GBM-derived U87 and lung cancer-derived A549 cells (Table 5).31 All of the compounds showed good to moderate cytotoxicity on the U87 cell line, except for schweinfurthin P (16). As expected, schweinfurthins G (7), Q (17), and vedelianin (20) containing the hexahydroxanthene moiety, were found to be highly cytotoxic with an EC50 of 0.04, 0.05, and 0.03 μM, respectively on the U87 cell line. However, on the A549 cell line, only schweinfurthins G (7), K (11), and L (12) and vedelianin (20) were found to be active, exhibiting cytotoxicity in the sub-micromolar range.31
Table 5 Cytotoxicity of schweinfurthins G, K–Q, and vedelianin on the GBM (U87) and lung cancer (A549) cell lines
| Compound |
U87, EC50 (μM) |
A549, EC50 (μM) |
| 11 |
0.25 ± 0.08 |
0.73 ± 0.01 |
| 12 |
0.45 ± 0.03 |
0.24 ± 0.12 |
| 13 |
0.50 ± 0.04 |
>10 |
| 14 |
0.10 ± 0.01 |
>10 |
| 15 |
0.025 ± 0.006 |
>10 |
| 16 |
>10 |
>10 |
| 17 |
0.050 ± 0.003 |
>10 |
| 7 |
0.045 ± 0.005 |
0.80 ± 0.04 |
| 20 |
0.030 ± 0.009 |
0.190 ± 0.002 |
| Monomethyl auristatin E |
0.00020 ± 0.00003 |
0.00050 ± 0.00005 |
The schweinfurthin family has been found to strongly inhibit the growth of human cancer cell lines when evaluated in the NCI 60-cell line screen. The mean GI50 values of the schweinfurthins are shown in Table 6. Schweinfurthin A (1), one of the potent compounds in this family, displayed a mean GI50 of 0.36 μM, whereas schweinfurthin B (2) was found to have a mean GI50 of 0.81 μM.27 Schweinfurthin E (5) was found to be slightly more potent than compounds 1 and 2, with a mean GI50 of 0.19 μM.29 The two enantiomers of schweinfurthin F, 6a and 6b were found to have a mean GI50 of 0.41 and 0.13 μM, respectively (Table 6).58
Table 6 Growth inhibition activity of schweinfurthins in the NCI 60-cell line screen
| Compounds |
Mean GI50 (μM) |
References |
| 1 |
0.36 |
27 |
| 2 |
0.81 |
27 |
| 5 |
0.19 |
29 |
| 6a |
0.41 |
58 |
| 6b |
0.13 |
58 |
| 9 |
10 |
30 |
| 10 |
2.8 |
30 |
| 19 |
0.21 |
46 |
| 20 |
0.08 |
58 and 67 |
Schweinfurthin I (9) was observed to be the least potent compound with a mean GI50 of 10 μM, whereas schweinfurthin J (10) exhibited moderate growth inhibitory activity with a mean GI50 of 2.8 μM.30 The synthetic analogue of schweinfurthin B, 3dSB (19) was found to have a mean GI50 of 0.21 μM.46 Vedelianin (20) was the most potent compound, which exhibited a mean GI50 of 0.08 μM (Table 6).58,67 The results suggest that the presence of dihydroxy groups on the A-ring of the tricyclic hexahydroxanthene core [schweinfurthins A (1), B (2), and E (5) and vedelianin (20)] may be critical for inhibitory activity. Additionally, the mean GI50 values of schweinfurthin E (5) and vedelianin (20) indicated that a shorter prenyl side chain could enhance the inhibitory activity (Table 6).
4.2. Free radical scavenging activity
A high level of free radicals in living systems causes adverse effects that can lead to tissue damage, neural disorders, skin irritation, inflammation, cell death or various diseases, including cancer.71 The harmful effects caused by free radicals are counteracted by antioxidants, either from natural sources in the form of food or synthetic antioxidants.72–74 Petrova et al. evaluated the free radical scavenging activity of schweinfurthins A (1) and B (2) against DPPH radicals, but they were found to be less active than other extracted lignans (Table 7).75
Table 7 Free radical scavenging activity of schweinfurthins A (1) and B (2)
| Compound |
DPPHa free radical scavenging activity, % inhibitionb |
| DPPH solution 0.02% in EtOH. Mean value of three measurements ± S.D. Solution 0.21 mg ml−1 in ethanol. |
| 1 |
25.4 ± 0.7 |
| 2 |
15.6 ± 0.4 |
| Caffeic acidc |
65.5 ± 0.1 |
4.3. Antimicrobial activity
Natural products have particularly shown a protective role against microbial invasion. Petrova et al. tested schweinfurthins A (1) and B (2) for antibacterial activity against Staphylococcus aureus, and both 1 and 2 demonstrated antimicrobial activity, as shown in Table 8. However, they were not active against Candida albicans and Escherichia coli.75
Table 8 Antimicrobial activity of schweinfurthins A (1) and B (2) against S. aureus (at 400 μg in the cup)a
| Compound |
Zone of inhibition (mm) |
| Tests were done in triplicate, values are mean ± S.D. 100 μg in the cup. |
| 1 |
19 ± 1 |
| 2 |
19 ± 1 |
| Streptomycinb |
30 ± 1 |
4.4. Pleiotropic effects of 3-deoxyschweinfurthin on isoprenoid homeostasis
Holstein et al. studied the pleiotropic effects of a synthetic schweinfurthin, 3dSB (18), on isoprenoid homeostasis to determine whether 3dSB-induced cytotoxicity could be prevented by co-incubation with an isoprenoid species. MTT assays to assess cell metabolic activity were performed by treating human multiple myeloma cell lines (RPMI-8226 and U266) with 3dSB and/or lovastatin in the presence or absence of mevalonate, farnesyl pyrophosphate (FPP) or geranylgeranyl pyrophosphate (GGPP). Cytotoxicity assays demonstrated that 3dSB had a synergistic interaction with lovastatin but not with other isoprenoid biosynthetic pathway (IBP) inhibitors in diverse human cancer cell lines. 3dSB enhanced the lovastatin-induced decrease in protein prenylation. Additionally, intracellular FPP and GGPP levels were decreased in both established cell lines and primary cells. Holstein et al. reported that 3dSB alters isoprenoid levels and controls IBP and sterol homeostasis by disrupting multiple aspects of the regulatory elements.76
5. Conclusion
Schweinfurthins display multiple biological and pharmacological activities. In particular, schweinfurthins possessing hexahydroxanthene have exhibited potent growth inhibitory activity in NCI 60-cell cancer screening. The synthesis of hexahydroxanthene has smoothed the path towards exploring these natural products and carrying out diverse structure–activity relationship studies. Further investment in optimization by preparing new synthetic schweinfurthin analogues that are metabolically stable, potent and specific may enhance the drug discovery process. The Macaranga species will continue to be investigated in the future due to the anticancer activity of the schweinfurthins and their congeners. In the present review, we have mainly focused on the synthesis and biological activities of the schweinfurthins. We expect that the information provided in this review might be helpful for the synthesis of schweinfurthin analogues and may offer a platform upon which to develop useful therapeutic candidates.
Conflicts of interest
There are no conflicts to declare.
Abbreviations
| TBAI | Tetrabutylammonium iodide |
| m-CPBA | m-Chloroperoxybenzoic acid |
| MOM | Methoxymethyl |
| TBS | tert-Butyldimethylsilyl |
| TBAF | Tetrabutylammonium fluoride |
| TFA | Trifluoroacetic acid |
| NMR | Nuclear magnetic resonance |
| TMEDA | Tetramethylethylenediamine |
| BOM | Benzyloxymethyl |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| KHMDS | Potassium bis(trimethylsilyl)amide |
| NOESY | Nuclear Overhauser effect spectroscopy |
| PDC | Pyridinium dichromate |
| NCI60 | National Cancer Institute; 60 human tumor cell line |
| GI50 | Concentration of compound required to inhibit growth by 50% |
| TGI | Concentration of compound required for the total inhibition of growth |
| IC50 | Concentration of compound required to inhibit by 50% |
| EC50 | Concentration of a compound expected to produce an effect in 50% of test organisms |
| DPPH | 2,2-diphenyl-1-picrylhydrazyl |
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF), grant funded by the Korea government (MSIT) (No. 2012M3A9C1053532 and 2015M3A6A4065734).
References
- M. S. Butler, J. Nat. Prod., 2004, 67, 2141 CrossRef PubMed.
- M. S. Butler, Nat. Prod. Rep., 2005, 22, 162 RSC.
- B. Shen, Cell, 2015, 163, 1297 CrossRef PubMed.
- M. Dong, B. Pfeiffer and K. Altmann, Drug Discovery Today, 2017, 22, 585 CrossRef PubMed.
- T. Shen, X. Wang and H. Lou, Nat. Prod. Rep., 2009, 26, 916 RSC.
- Z. I. Chrząścik, Crit. Rev. Anal. Chem., 2009, 39, 70 CrossRef.
- S. Fulda, Drug Discovery Today, 2010, 15, 757 CrossRef PubMed.
- L. Carter, J. D'Orazio and K. J. Pearson, Endocr.-Relat. Cancer, 2014, 21, R209 CrossRef PubMed.
- B. Aggarwal, A. Bhardwaj, R. Aggarwal, N. Seeram, S. Shishodia and Y. Takada, Anticancer Res., 2004, 24, 2783 Search PubMed.
- Z. A. Bishayee, Cancer Prev. Res., 2009, 2, 409 CrossRef PubMed.
- M. Jang, L. Cai, G. Udeani, K. Slowing, C. Thomas, C. Beecher, H. Fong, N. Farnsworth, A. Kinghorn, R. Mehta, R. Moon and J. M. Pezzuto, Science, 1997, 275, 218 CrossRef PubMed.
- L. Stivala, M. Savio, F. Carafoli, P. Perucca, L. Bianchi, G. Maga, L. Forti, U. Pagnoni, A. Albini, E. Prosperi and V. Vannini, J. Biol. Chem., 2001, 276, 22586 CrossRef PubMed.
- H. Piotrowska, M. Kucinska and M. Murias, Mutat. Res., 2012, 750, 60 Search PubMed.
- M. Nalli, G. Ortar, A. Moriello, E. Morera, V. Marzo and L. Petrocellis, Bioorg. Med. Chem. Lett., 2016, 26, 899 CrossRef PubMed.
- R. Mikstacka, T. Stefański and J. Różański, Cell. Mol. Biol. Lett., 2013, 18, 368 Search PubMed.
- J. Sirerol, M. Rodríguez, S. Mena, M. Asensi and A. Ortega, Oxid. Med. Cell. Longevity, 2016, 3128951 Search PubMed.
- R. Mikstacka, A. Rimando, K. Szalaty, K. Stasik and W. Dubowska, Xenobiotica, 2006, 36, 269 CrossRef PubMed.
- G. Belofsky, A. French, D. Wallace and S. Dodson, J. Nat. Prod., 2004, 67, 26 CrossRef PubMed.
- M. Liu, Y. Han, D. Xing, Y. Shi, L. Xu, L. Du and Y. Ding, J. Asian Nat. Prod. Res., 2004, 6, 229 CrossRef PubMed.
- Q. Hu, B. Zhou, Y. Ye, Z. Jiang, X. Huang, Y. Li, G. Du, G. Yang and X. M. Gao, J. Nat. Prod., 2013, 76, 1854 CrossRef PubMed.
- J. Ingle, D. Ahmann, S. Green, J. Edmonson, H. Bisel, L. Kvols, W. Nichols, E. Creagan, R. Hahn, J. Rubin and S. Frytak, N. Engl. J. Med., 1981, 304, 16 CrossRef PubMed.
- Z. A. Herbst, H. Ulfelder and D. Poskanzer, N. Engl. J. Med., 1971, 284, 878 CrossRef PubMed.
- V. Jordan, Br. J. Pharmacol., 2006, 147, S269 CrossRef PubMed.
- Z.
A. Howell, J. Cuzick, M. Baum, A. Buzdar, M. Dowsett, J. Forbes, G. Hoctin-Boes, J. Houghton, G. Locker and J. Tobias, Lancet, 2005, 365, 60 CrossRef.
- E. Bommel, T. Hendriksz, A. Huiskes and A. G. Zeegers, Ann. Intern. Med., 2006, 144, 101 CrossRef PubMed.
- G. Webster, Ann. Mo. Bot. Gard., 1994, 81, 33 CrossRef.
- J. Beutler, R. Shoemaker, T. Johnson and M. Boyd, J. Nat. Prod., 1998, 61, 1509 CrossRef PubMed.
- J. Beutler, J. Jato, G. Cragg and M. Boyd, Nat. Prod. Lett., 2000, 14, 399 CrossRef.
- B. Yoder, S. Cao, A. Norris, J. Miller, F. Ratovoson, J. Razafitsalama, R. Andriantsiferana, V. Rasamison and D. Kingston, J. Nat. Prod., 2007, 70, 342 CrossRef PubMed.
- P. Klausmeyer, Q. Van, J. Jato, T. McCloud and J. Beutler, J. Nat. Prod., 2010, 73, 479 CrossRef PubMed.
- T. Péresse, G. Jézéquel, P. Allard, V. Pham, D. Huong, F. Blanchard, J. Bignon, H. Lévaique, J. Wolfender, M. Litaudon and F. Roussi, J. Nat. Prod., 2017, 80, 2684 CrossRef PubMed.
- O. Thoison, E. Hnawia, F. Gueritte-voegelein and T. Sevenet, Phytochemistry, 1992, 31, 1439 CrossRef.
- E. Hnawia, O. Thoison, F. Guéritte-Voegelein, D. Bourret and T. Sévenet, Phytochemistry, 1990, 29, 2367 CrossRef.
- B. Schutz, A. Wright, T. Rali and O. Sticher, Phytochemistry, 1995, 40, 1273 CrossRef.
- S. Sultana and M. Ilyas, Phytochemistry, 1986, 25, 953 CrossRef.
- W. Hui, K. Ng, N. Fukamiya, M. Koreeda and K. Nakanishi, Phytochemistry, 1971, 10, 1617 CrossRef.
- R. Mechoulam and B. Yagen, Tetrahedron Lett., 1969, 10, 5349 CrossRef.
- G. Manners, L. Jurd and K. Stevens, Tetrahedron, 1972, 28, 2949 CrossRef.
- G. Trammell, Tetrahedron Lett., 1978, 19, 1525 CrossRef.
- E. Treadwell, J. Neighbors and D. Wiemer, Org. Lett., 2002, 4, 3639 CrossRef PubMed.
- B. Branchaud and H. Blanchette, Tetrahedron Lett., 2002, 43, 351 CrossRef.
- Z. A. Toshimitsu, C. Hirosawa and K. Tamao, Synlett, 1996, 465 CrossRef.
- T. Kametani, K. Suzuki, H. Kurobe and H. Nemoto, J. Chem. Soc., Chem. Commun., 1979, 1128 RSC.
- T. Kametani, H. Kurobe, H. Nemoto and K. Fukumoto, J. Chem. Soc., Perkin Trans. 1, 1982, 1085 RSC.
- D. Boger and I. Jacobson, J. Org. Chem., 1991, 56, 2115 CrossRef.
- J. Neighbors, J. Beutler and D. Wiemer, J. Org. Chem., 2005, 70, 925 CrossRef PubMed.
- J. Neighbors, N. Mente, K. Boss, D. Zehnder and D. Wiemer, Tetrahedron Lett., 2008, 49, 516 CrossRef.
- Z. Wang and Y. Shi, J. Org. Chem., 1998, 63, 3099 CrossRef.
- L. Shu and Y. Shi, Tetrahedron Lett., 1999, 40, 8721 CrossRef.
- J. Topczewski, J. Kodet and D. Wiemer, J. Org. Chem., 2011, 76, 909 CrossRef PubMed.
- J. Topczewski, M. Callahan, J. Neighbors and D. Wiemer, J. Am. Chem. Soc., 2009, 131, 14630 CrossRef PubMed.
- N. Mente, J. Neighbors and D. Wiemer, J. Org. Chem., 2008, 73, 7963 CrossRef PubMed.
- E. Treadwell, S. Cermak and D. Wiemer, J. Org. Chem., 1999, 64, 8718 CrossRef.
- J. Topczewski, J. Neighbors and D. Wiemer, J. Org. Chem., 2009, 74, 6965 CrossRef PubMed.
- J. Stafford and N. Valvano, J. Org. Chem., 1994, 59, 4346 CrossRef.
- P. Manchand, P. Belica and H. S. Wong, Synth. Commun., 1990, 20, 2659 CrossRef.
- R. Lange, J. Org. Chem., 1962, 27, 2037 CrossRef.
- N. Mente, A. Wiemer, J. Neighbors, J. Beutler, R. Hohl and D. Wiemer, Bioorg. Med. Chem. Lett., 2007, 17, 911 CrossRef PubMed.
- J. Neighbors, M. Salnikova and D. Wiemer, Tetrahedron Lett., 2005, 46, 1321 CrossRef.
- N. Yee and R. Coates, J. Org. Chem., 1992, 57, 4598 CrossRef.
- E. Corey and K. Liu, J. Am. Chem. Soc., 1997, 119, 9929 CrossRef.
- E. Corey and M. Sodeoka, Tetrahedron Lett., 1991, 32, 7005 CrossRef.
- J. Zhang and E. Corey, Org. Lett., 2001, 3, 3215 CrossRef PubMed.
- S. Taylor, M. Ivanovic, L. Simons and M. Davis, Tetrahedron: Asymmetry, 2003, 14, 743 CrossRef.
- M. Singh and N. P. Argade, Synthesis, 2012, 44, 2895 CrossRef.
- B. Wang, Y. Zhang, J. Chen, W. Zhang, J. Song, B. Del, L. Brown and G. Miller, PCT Int. Appl., CODEN: PIXXD2 WO 2003009807 A220030206 CAN 138:153318 AN 2003:97274, 2003.
- J. Topczewski and D. Wiemer, Tetrahedron Lett., 2011, 52, 1628 CrossRef PubMed.
- K. Reilly, D. Loisel, R. Bronson, M. McLaughlin and T. Jacks, Nat. Genet., 2000, 26, 109 CrossRef PubMed.
- Y. Zhu, F. Guignard, D. Zhao, L. Liu, D. Burns, R. Mason, A. Messing and L. Parada, Cancer Cell, 2005, 8, 119 CrossRef PubMed.
- T. Turbyville, D. Gürsel, R. Tuskan, J. Walrath, C. Lipschultz, S. Lockett, D. Wiemer, J. Beutler and K. Reilly, Mol. Cancer Ther., 2010, 9, 1234 CrossRef PubMed.
- I. Gulcin, Chem.-Biol. Interact., 2009, 179, 71 CrossRef PubMed.
- S. Kedare and R. Singh, J. Food Sci. Technol., 2011, 48, 412 CrossRef PubMed.
- E. Koksal and I. Gulcin, Turk. J. Agric. For., 2008, 32, 65 Search PubMed.
- T. Kulisic, A. Radonic, V. Katalinic and M. Milos, Food Chem., 2004, 85, 633 CrossRef.
- Z. A. Petrova, M. Popova, C. Kuzmanova, I. Tsvetkova, H. Naydenski, E. Muli and V. Bankova, Fitoterapia, 2010, 81, 509 CrossRef PubMed.
- S. Holstein, C. Kuder, H. Tong and R. Hohl, Lipids, 2011, 46, 907 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jyotirling R. Malia,
Aneesh Sivaramana,
Yongseok Choi
a,
Jyotirling R. Malia,
Aneesh Sivaramana,
Yongseok Choi