
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient donor–acceptor host materials for green organic light-emitting devices: non-doped blue-emissive materials with dual charge transport properties†
Jayaraman Jayabharathi *,
Palanisamy Sujatha,
Venugopal Thanikachalam and
Pavadai Nethaji
*,
Palanisamy Sujatha,
Venugopal Thanikachalam and
Pavadai Nethaji
Department of Chemistry, Annamalai University, Annamalainagar 608 002, Tamilnadu, India. E-mail: jtchalam2005@yahoo.co.in; Tel: +91 9443940735
First published on 31st May 2018
Abstract
Comparative optical, electroluminescence and theoretical studies were performed for (E)-4′-(1-(4-(2-(1-(4-morpholinophenyl)-1H-phenanthro[9,10-d]imidazol-2-yl)vinyl)phenyl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-[1,1′-biphenyl]-4-amine (SMPI-TPA) and (E)-4-(4-(2-(4-(2-(4-(9H-carbazol-9-yl)phenyl)-1H-phenanthro[9,10-d]imidazol-1-yl)styryl)-1H-phenanthro[9,10-d]imidazol-1-yl)phenyl)morpholine (SMPI-Cz). These compounds show excellent thermal properties, dual charge transport properties and form thin films under thermal evaporation. Blue OLEDs (CIE: 0.16, 0.08) based on SMPI-TPA show efficient device performance (ηex 6.1%; ηc 5.3 cd A−1; ηp 5.2 lm W−1) at low turn-on voltages. Both SMPI-TPA and SMPI-Cz were utilised as hosts for green OLEDs. The devices with SMPI-Cz (30 nm):5 wt% Ir(ppy)3 exhibit maximum luminance of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 725 cd m−2, and ηc and ηp values of 61.4 cd A−1 and 63.8 lm W−1, respectively. In comparison, devices with SMPI-TPA (30 nm):5 wt% Ir(ppy)3 exhibit high ηc and ηp values of 65.2 cd A−1 and 67.1 lm W−1, respectively. Maximum ηex values of 19.6% and 23.4% were obtained from SMPI-TPA:Ir(ppy)3 and SMPI-Cz:Ir(ppy)3, respectively. These device performances indicate that the phenanthroimidazole unit is a tunable building unit for efficient carrier injection and it may also be employed as a host for green OLEDs.
725 cd m−2, and ηc and ηp values of 61.4 cd A−1 and 63.8 lm W−1, respectively. In comparison, devices with SMPI-TPA (30 nm):5 wt% Ir(ppy)3 exhibit high ηc and ηp values of 65.2 cd A−1 and 67.1 lm W−1, respectively. Maximum ηex values of 19.6% and 23.4% were obtained from SMPI-TPA:Ir(ppy)3 and SMPI-Cz:Ir(ppy)3, respectively. These device performances indicate that the phenanthroimidazole unit is a tunable building unit for efficient carrier injection and it may also be employed as a host for green OLEDs.
1. Introduction
Efficient green or red OLEDs with pure color CIE coordinates have been reported1–4 and blue emitters with less power consumption in organic optoelectronics have also been broadly studied.5,6 However, there is need for long-lifetime blue emitters with pure colour CIE coordinates due to wide band gaps (Eg), which require limited π-conjugation length.7–9 Simultaneous carrier injection into a blue-emissive layer becomes very difficult due to its wide Eg, resulting in a decrease in device efficiency.10,11 Therefore, for OLED applications, highly efficient and low-cost blue OLEDs are of urgent demand. The external quantum efficiency (ηex) can be deduced from the following equation: ηex = ηIQE × ηout = ηrec × ηPL × ηS × ηout, where ηIQE is the internal quantum efficiency, ηout (∼1/2n2) is the light out-coupling efficiency (n = 1.5, ηout ∼ 20%), ηrec is the electron–hole recombination efficiency (100%), ηPL is the photoluminescence efficiency and ηS is the utilization efficiency. The two key parameters ηPL and ηS required for high ηex can be tuned by altering the emitter molecular design.12A blue-emissive material with high triplet energy (ET) may be employed as a host for green OLEDs.13–16 However, the high triplet energy enables green emitters to harvest the triplet energy of a blue emitter; also, efficient non-doped blue emitters are not suitable as hosts for phosphorescent OLEDs due to their low ET as well as poor carrier transport properties.17 Moreover, efficient hosts for green emitter exhibit low efficiency when they are used as emissive materials in blue OLEDs.18–20 Hence, efforts are still required to achieve efficient OLEDs based on blue emissive materials.
Therefore, to develop dual-functional emissive materials, i.e., emitters for blue OLEDs and hosts for green OLEDs, synthesis of molecules with donor (D)/acceptor (A) (electron/hole transport moieties) configuration has gained interest. To achieve deep blue emission, the donor–acceptor (D–A) molecule should have relatively weak charge transfer properties since a strong D–A geometry can induce red-shifted emission. In addition, the singlet–triplet splitting should be small to ensure that the triplet-excited state energy is high enough to excite the green phosphorescent dopant.
With our continuous interest to synthesize efficient n-type imidazole derivatives as OLED emitters,21–23 herein we report the synthesis of (E)-4′-(1-(4-(2-(1-(4-morpholinophenyl)-1H-phenanthro[9,10-d]imidazol-2-yl)vinyl)phenyl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-[1,1′-biphenyl]-4-amine (SMPI-TPA) and (E)-4-(4-(2-(4-(2-(4-(9H-carbazol-9-yl)phenyl)-1H-phenanthro[9,10-d]imidazol-1-yl)styryl)-1H-phenanthro[9,10-d]imidazol-1-yl)phenyl)morpholine (SMPI-Cz) derivatives and utilize them as blue emitters in non-doped devices and hosts for green OLEDs. These derivatives exhibit higher stability with balanced injection property, leading to excellent device performances.
2. Materials and measurements
The structure of emissive materials was confirmed by 1H and 13C NMR spectroscopies and mass spectrometry, recorded using a Bruker spectrometer (400 MHz) and Agilent (LCMS VL SD), respectively. Redox potentials were measured using a potentiostat CHI 630A electrochemical analyzer. The Lambda 35 PerkinElmer instrument spectrophotometer with integrated sphere (RSA-PE-20) was used to measure absorbance in both solution and film states. Emissive properties (PL) were analyzed via PerkinElmer LS55 fluorescence spectrometer measurements. Thermal characteristics such as decomposition (Td) and glass transition (Tg) temperatures were analyzed using a PerkinElmer thermal analysis system and NETZSCH-DSC-204, respectively, with heating rate of 10 °C min−1 under N2 atmosphere. Lifetime measurements of SMPI-TPA and SMPI-Cz were recorded using a time-correlated single-photon counting spectrometer (TCSPC: Horiba Fluorocube-01-NL lifetime system). The Φ (PL quantum yield) was measured in dichloromethane using 0.5 M H2SO4:quinine (0.54) as reference.2.1. Theoretical calculations
Using the Gaussian 09 software package,24 the electron density on frontier molecular orbitals of SMPI-TPA and SMPI-Cz was identified.2.2. Devices fabrication
OLEDs with SMPI-TPA and SMPI-Cz emitters were fabricated by vacuum deposition at 5 × 10−6 Torr on a precleaned ITO substrate with 20 Ω per square resistance (rate −0.1 nm s−1). LiF and Al were also evaporated thermally on the organic layer and the thickness was measured using a quartz crystal thickness monitor. A series of fabricated devices with multilayer configuration are denoted as follows: ITO/NPB (4,4′-bis[N-(1-naphthyl)-N-phenylamino]biphenyl) (50 nm)/TCTA (tris(4-carbazoyl-9-ylphenyl)amine) (5 nm)/SMPI-TPA (I) or SMPI-Cz (II) (30 nm)/TPBI (2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole)) (15 nm)/LiF (1 nm)/Al (100 nm); ITO/NPB (50 nm)/SMPI-TPA (30 nm):5 wt% Ir(PPy)3 or (III) or SMPI-Cz (30 nm):5 wt% Ir(PPy)3 (IV)/BCP (2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline) (15 nm)/Alq3 (50 nm)/LiF (1 nm)/Al (100 nm). Further, single-carrier devices were also been fabricated. The hole-only device has the configuration of ITO/NPB (50 nm)/SMPI-TPA or SMPI-Cz (30 nm)/NPB (50 nm)/Al (200 nm), while the electron-only device has the configuration of ITO/TPBI (20 nm) SMPI-TPA or SMPI-Cz (30 nm)/TPBI (20 nm)/LiF (1 nm)/Al (200 nm). Electrical measurements were recorded using a Keithley 2400 sourcemeter.:HCl mixture, and the obtained white product was used after purification. Anal. calcd C33H28N4O: C, 79.87; H, 5.62; N, 11.18. Found: C, 79.82; H, 5.60; N, 11.15. 1H NMR (400 MHz, CDCl3): δ 2.72 (s, 4H), 3.36 (s, 4H), 4.12 (s, 2H), 6.43 (s, 1H), 6.93 (d, J = 16.2 Hz, 1H), 7.06 (d, J = 15.4 Hz, 1H), 7.23 (s, 2H), 7.36 (d, J = 8.6 Hz, 3H), 7.59–7.68 (m, 5H), 7.92 (s, 2H), 8.03 (d, J = 8.6 Hz, 2H), 8.62 (d, J = 7.2 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 42.28, 57.36, 112.61, 120.18, 120.76, 121.121, 121.43, 122.28, 122.67, 123.73, 124.08, 124.65, 125.12, 125.56, 125.92, 126.28, 126.32, 126.49, 127.52, 127.63, 127.82, 128.27, 128.56, 130.63, 132.53, 133.06, 145.27. MS: m/z 496.65 [M+]; calcd 496.60.3. Results and discussion
As depicted in Scheme S1,† the blue emitters used as hosts for green OLEDs, namely, (E)-4′-(1-(4-(2-(1-(4-morpholinophenyl)-1H-phenanthro[9,10-d]imidazol-2-yl)vinyl)phenyl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-[1,1′-biphenyl]-4-amine (SMPI-TPA) and (E)-4-(4-(2-(4-(2-(4-(9H-carbazol-9-yl)phenyl)-1H-phenanthro[9,10-d] imidazol-1-yl)styryl)-1H-phenanthro[9,10-d]imidazol-1-yl)phenyl)morpholine (SMPI-Cz) were prepared from phenanthrenequinone (0.416 g, 2 mmol), 4′-(diphenylamino)biphenyl-4-carbaldehyde (SMPI-TPA)/4-(9H-carbazol-9-yl)benzaldehyde (SMPI-Cz), 4-((E)-2-(1-(4-morpholinophenyl)-1H-phenanthro[9,10-d]imidazol-2-yl)vinyl)benzenamine and ammonium acetate in acetic acid (120 °C; 12 h; N2 stream). The formed SMPI-TPA and SMPI-Cz were analysed by various spectroscopic methods, from which their molecular structures were confirmed.3.1. Potential energy surface (PES) scan studies and thermal properties
The potential energy surface scan of N24–C25–C31–C30 (SMPI-Cz)/N24–C25–C31–C26 (SMPI-TPA) was performed using DFT/B3LYP/6-31G (d,p). During calculation, all the geometrical parameters were simultaneously relaxed, while their respective torsional angles were varied in steps of 0°, 20°, 40°, 60°…360°. The potential energy surface diagrams (Fig. 1a and b) reveal that the minimum energy conformation corresponds to that in which the 4-morpholinophenyl ring attached to the imidazole nitrogen atom (N23) is tilted to an angle of 98.21° (SMPI-Cz)/101.1° (SMPI-TPA) and the phenyl ring with carbazole core (SMPI-Cz) and another phenyl ring with triphenylamine core (SMPI-TPA) are attached to the imidazole carbon atom (C25) at an angle of 91.56° (SMPI-Cz)/110.1° (SMPI-TPA) (Fig. 1c). Owing to rigid molecular back bone and non-coplanar geometry, these compounds (SMPI-TPA and SMPI-Cz) exhibit suitable thermal stability [high decomposition (Td5)/high glass transition (Tg) temperature: 526/163 °C-SMPI-TPA and 518/172 °C-SMPI-Cz] (Fig. 1d; Table 1). The high Tg and Td5 values improve the lifetime of the OLEDs by forming thin films upon vacuum evaporation.21 | ||
| Fig. 1 Potential energy scan diagram of (a) SMPI-TPA and (b) SMPI-Cz; (c) dihedral angle of SMPI-Cz: (1) C(7)–N(23)–C(49)–C(48); (2) C(24)–N(25)–C(31)–C(30) and SMPI-TPA: (1) C(7)–N(23)–C(61)–C(66); (2) N(24)–C(25)–C(31)–C(26); (d) DSC and TGA of SMPI-TPA and SMPI-Cz. | ||
| Emitters | SMPI-TPA (I) | SMPI-Cz (II) | SMPI-TPA:Ir(PPy)3 (III) | SMPI-Cz:Ir(PPy)3 (IV) |
|---|---|---|---|---|
| a Measured in dilute toluene solution (×10−5 mol L−1) at room temperature.b Measured neat film by coating.c Absolute PL quantum yield evaluated using an integrating sphere.d Measured by cyclic voltammetry calculated by comparing with ferrocene (Fc). | ||||
| Optical and thermal properties | ||||
| λab (nm) (asol/bfilm) | 250, 358/252, 360 | 248, 355/250, 357 | — | — |
| λem (nm) (asol/bfilm) | 441/439 | 412/401 | — | — |
| Tg/Td (°C) | 163/526 | 172/518 | — | — |
| PLQYc (asol/bfilm) | 0.81/0.56 | 0.69/0.71 | — | — |
| HOMO/LUMOd (eV) | −5.41/−2.15 | −5.39/−2.17 | — | — |
|
||||
| Device efficiency | ||||
| L (cd m−2) | 12680 |
12468 |
1094 | 20725 |
| Von (V) | 3.1 | 3.2 | 2.8 | 2.9 |
| ηex (%) | 6.1 | 5.9 | 19.6 | 23.4 |
| ηc (cd A−1) | 5.3 | 5.1 | 65.2 | 61.4 |
| ηp (lm W−1) | 5.2 | 4.6 | 67.1 | 63.8 |
3.2. HOMO–LUMO
The HOMO of both SMPI-TPA and SMPI-Cz is localized on the fragment attached to phenanthrimidazole nitrogen, while the LUMO is distributed on the phenanthrimidazole with TPA (SMPI-TPA) and Cz (SMPI-Cz) fragments (Fig. 2). The HOMO and LUMO of SMPI-TPA and SMPI-Cz display adequate separation in electron density features, which enhances the hole- and electron-transport functions and also reduces the singlet–triplet splitting.22 Moreover, SMPI-TPA and SMPI-Cz exhibit redox waves, supporting their carrier transport abilities. The HOMO energies of −5.41 eV (SMPI-TPA) and −5.39 eV (SMPI-Cz) are determined from their respective oxidation onset potentials of 0.61 V (SMPI-TPA) and 0.59 V (SMPI-Cz) [EHOMO = −(Eox + 4.8 eV)].23 The LUMO energies of −2.15 eV (SMPI-TPA) and −2.17 eV (SMPI-Cz) are calculated using the equation ELUMO = EHOMO − 1239/λonset. The charge-transporting properties of SMPI-TPA and SMPI-Cz were investigated by fabricating single-carrier devices. The hole-only device has the configuration of ITO/NPB (50 nm)/SMPI-TPA or SMPI-Cz (30 nm)/NPB (50 nm)/Al (200 nm) and the electron-only device has the configuration of ITO/TPBI (20 nm) SMPI-TPA or SMPI-Cz (30 nm)/TPBI(20 nm)/LiF (1 nm)/Al (200 nm). NPB on the cathode side of the hole-only device and TPBI (1,3,5-tri(phenyl-2-benzimidazolyl)benzene) on the anode side of the electron-only device were used to block electron injection from Al and hole injection from ITO, respectively. Fig. 3 shows that both devices can significantly conduct current, indicating that SMPI-TPA and SMPI-Cz are capable of transporting both holes and electrons and they exhibit a bipolar transporting nature. This is beneficial for balancing the holes and electrons in the emitting layer based on SMPI-TPA and SMPI-Cz.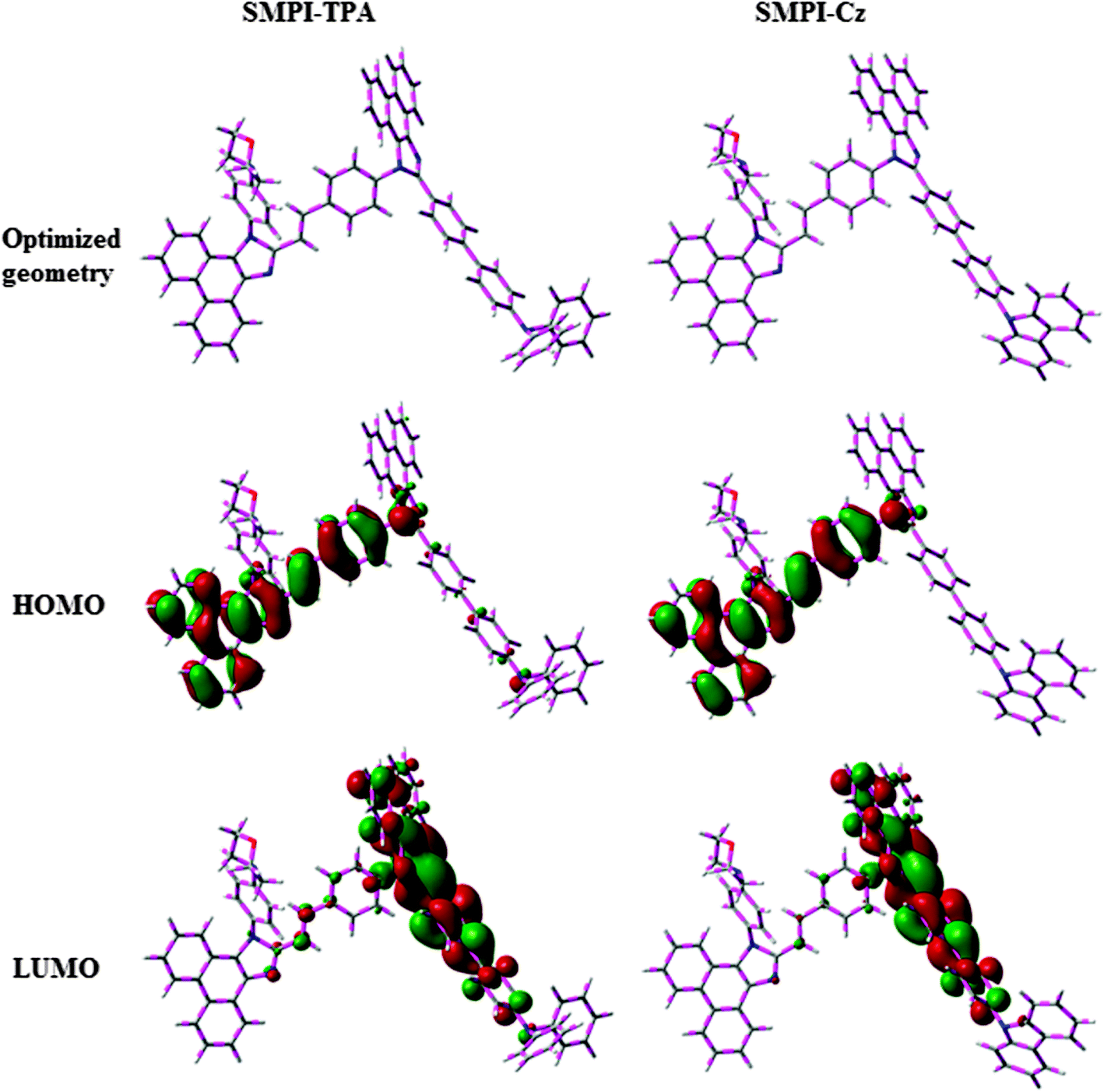 | ||
| Fig. 2 Optimised geometry, and HOMO and LUMO contour plots of SMPI-TPA and SMPI-Cz. | ||
 | ||
| Fig. 3 Normalized (a) absorption, emission and (b) EL spectra of SMPI-TPA, SMPI-Cz, SMPI-TPA:5 wt% Ir (PPy)3:SMPI-TPA film and SMPI-Cz:Ir (PPy)3:SMPI-Cz film and (c) hole-only and electron-only devices. | ||
3.3. Photophysical properties
Electronic spectral studies of SMPI-TPA and SMPI-Cz were carried out in dichloromethane (Fig. 3). The results indicated that λabs at around 248 nm originated from the aryl group attached to the nitrogen of phenanthrimidazole plane, while λabs at around 355 nm was attributed to π → π* electronic transition of the styryl phenanthrimidazole ring. The origin of absorption of SMPI-TPA and SMPI-Cz was studied by computational methods. The outcomes of computational and experimental methods are similar [HOMO–LUMO+1 transition; Table S1(a) & (b)†]. These SMPI-TPA and SMPI-Cz derivatives show blue emission at 441 and 412 nm, respectively, and the measured PLQY values for SMPI-TPA and SMPI-Cz in solution/film are of 0.81/0.56 and 0.69/0.71, respectively. The emission maximum of SMPI-Cz (412 nm) is blue-shifted when compared with SMPI-TPA (441 nm) due to the additional phenyl ring in SMPI-TPA, which induces an elongated conjugation. Such high fluorescence efficiencies of SMPI-TPA and SMPI-Cz confirmed that they could be efficient deep blue emitters. The calculated triplet energy levels of 2.32 eV (SMPI-TPA) and 2.40 eV (SMPI-Cz) are sufficiently high for the excitation of green dopants. The theoretically estimated singlet-to-triplet energy gaps (ΔEST) of SMPI-TPA and SMPI-Cz are 0.36 eV and 0.30 eV, respectively, which are unfavourable for TADF emission. A small ΔEST value is required for efficient energy transfer from host T* (triplet excited state) to green phosphorescent emitters. The observed intense blue emission and high Tg of SMPI-TPA and SMPI-Cz confirmed their suitability as blue emitters. The device performances of the blue emitters were analysed by fabricating non-doped OLEDs with a configuration of ITO/NPB (50 nm)/TCTA (5 nm)/SMPI-TPA (I)/SMPI-Cz (II) (30 nm)/TPBI (15 nm)/LiF (1 nm)/Al (100 nm). It is clear from Fig. 4 and Table 1 that the as-fabricated novel SMPI-TPA- and SMPI-Cz-based blue OLEDs exhibit maximum brightness at low voltage. The blue EL and PL spectra of SMPI-TPA and SMPI-Cz in the solid state are similar (Fig. 3), and the hole injection barrier between SMPI-TPA and HTL (hole transport layer) is very small. Thus, effective electron–hole radiative recombination occurs in SMPI-TPA and SMPI-Cz layers. The small injection barrier of 0.28 (SMPI-TPA) and 0.32 eV (SMPI-Cz) for charge carriers accounts for the observed low turn-on voltages. The SMPI-TPA film exhibits smooth surface (roughness of 0.24 nm) and remains unchanged even after annealing (100 °C: 10 h) (Fig. 4: inset). The value of ηex is calculated using the formula ηex = ηout × ηrc × ηγ × ΦPL,23 where ηout is the light out-coupling efficiency (20%), ηrc is the product of the charge recombination efficiency (100%), ηγ is the efficiency of radiative exciton formation (25%) and ΦPL is the photoluminescence quantum yield of emitters SMPI-TPA (0.56) and SMPI-Cz (0.71). Thus, the calculated ηex values of SMPI-TPA- and SMPI-Cz-based devices are 2.8 and 3.6%, respectively. However, the obtained external quantum efficiencies (ηex) of SMPI-TPA- and SMPI-Cz-based devices are 6.1 and 5.9%, respectively, and the current efficiencies (ηc) of SMPI-TPA- and SMPI-Cz-based devices are 5.3 and 5.1 cd A−1, respectively. The harvested ηex exceeds the theoretical limit since delayed fluorescence is not expected and the obtained high ηex is not in accordance with TADF.25–30 Time-resolved lifetime studies revealed monoexponential decay for SMPI-TPA and SMPI-Cz (Fig. S1†) with lifetime values in nanoscale, indicating the absence of delayed component (TADF) in emission. Both SMPI-TPA and SMPI-Cz do not possess delayed lifetime component. Only prompt species for SMPI-TPA (1.88 ns) and SMPI-Cz (1.92 ns) confirmed that SMPI-TPA and SMPI-Cz are different from TADF materials in luminous mechanism.31 The as-fabricated device structure ITO/NPB (50 nm)/TCTA (5 nm)/SMPI-TPA (I) or SMPI-Cz (II) (30 nm)/TPBI (15 nm)/LiF (1 nm)/Al (100 nm) shows that the TCTA layer plays a key role in achieving higher efficiency32 and the NPB layer was used as the hole-transporting layer (HTL) to be deposited in proximity to the SMPI-TPA and SMPI-Cz emissive layers. Due to the relatively small energy gap of NPB (Eg = −3.00 eV) in comparison to the energy gaps of the emissive layers SMPI-TPA (Eg = −3.26 eV) and SMPI-Cz (Eg = −3.22 eV), the excitons generated in the emissive layer are more likely to leak into HTL, leading to loss of excitons. However, herein, we used TCTA with Eg of −3.40 eV as a buffer layer to confine the excitons within the emissive layer, resulting in higher efficiency.33 Further, the EL spectra of the SMPI-TPA- and SMPI-Cz-based devices are identical to the corresponding PL emission of the thin film, implying balanced carrier transport and efficient confinement of excitons33 (Fig. 3). In the present study, some factors account for the efficiency of the devices: (i) bipolar carrier transporting properties of SMPI-TPA and SMPI-Cz, which contribute to better balance of carrier transport and wider distribution of recombination region within the emission layer (Fig. 3) and (ii) suitable HOMO and LUMO energies of SMPI-TPA and SMPI-Cz (Fig. 5); the hole injection barrier at TCTA:EML interface is 0.42 eV (SMPI-TPA):0.44 eV (SMPI-Cz) and electron injection barrier at TPBI:EML interface is 0.55 eV (SMPI-TPA):0.53 eV (SMPI-Cz). This reveals that there is only a small barrier for carrier injection, leading to high exciton formation even under high current density, resulting in higher device performances.32,33 The PL spectra of SMPI-TPA or SMPI-Cz were recorded in THF/water mixture with different water fractions (fw) to understand whether these materials show aggregation-induced emission. When a small amount of water (fw = 10–90 vol%) was added to the THF solution of both SMPI-TPA and SMPI-Cz, the PL intensity remained unchanged, which shows that both SMPI-TPA and SMPI-Cz materials are AIE-inactive (Fig. S1†).
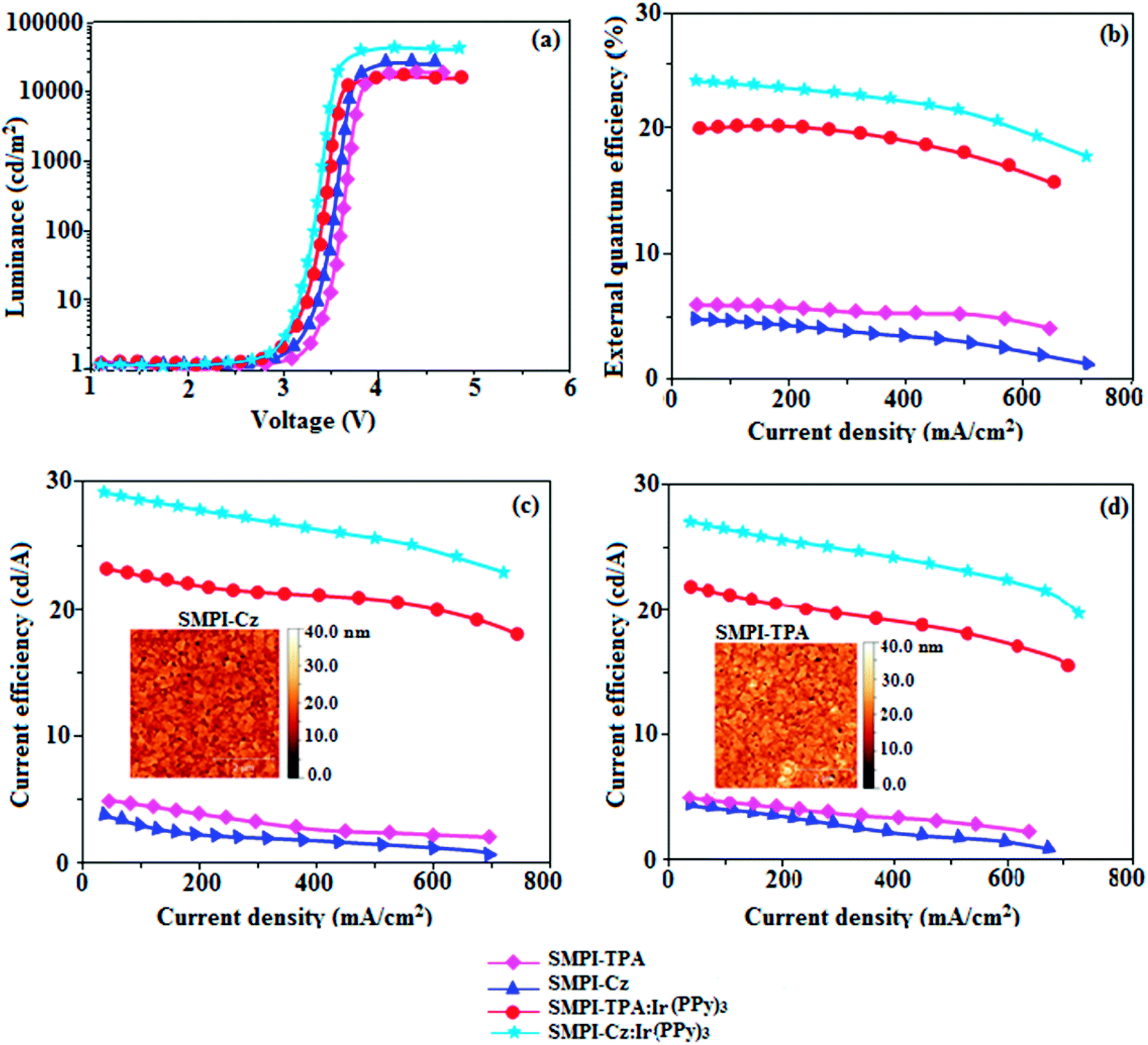 | ||
| Fig. 4 Electroluminescence performances: (a) luminance versus voltage; (b) external quantum efficiency versus current density; (c) current efficiency versus current density and (d) power efficiency versus current density of devices I–IV based on SMPI-TPA (I), SMPI-Cz (II), SMPI-TPA:Ir(PPy)3 (III) and SMPI-Cz:Ir(PPy)3 (IV), [inset: AFM images of SMPI-TPA film and SMPI-Cz film]. | ||
The small singlet–triplet splitting and good carrier transport properties allow both SMPI-TPA and SMPI-Cz to be used as hosts to fabricate red, green and yellow phosphorescence-emitting layers for PhOLEDs. The calculated PLQY of Ir(ppy)3-doped SMPI-TPA and Ir(ppy)3-doped SMPI-Cz was 0.61 and 0.79, respectively. In addition to high ηex and ηc, SMPI-TPA- and SMPI-Cz-based devices show high ηp (power efficiency) of 5.2 and 4.6 lm W−1, respectively. The blue emitters exhibit CIE coordinates of (0.16, 0.08 – SMPI-TPA) and (0.15, 0.07 – SMPI-Cz). The device based on SMPI-TPA shows maximum luminance of 12680 cd m−2, and maximum current and power efficiencies of 5.3 cd A−1 and 5.2 lm W−1, respectively, at 3.1 V. These SMPI-TPA and SMPI-Cz compounds were also utilised as hosts for green dopants in the fabricated devices (Fig. 4) with a configuration of ITO/NPB (50 nm)/SMPI-TPA (30 nm):5 wt% Ir(PPy)3 or (III) or SMPI-Cz (30 nm):5 wt% Ir(PPy)3 (IV)/BCP (2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline) (15 nm)/Alq3 (50 nm)/LiF (1 nm)/Al (100 nm). The energy level diagram of the as-fabricated device and molecular structures of materials used in the devices are shown in Fig. 5. Devices with SMPI-Cz (30 nm):5 wt% Ir(ppy)3 exhibit maximum luminance of 20725 cd m−2, and ηc and ηp of 61.4 cd A−1 and 63.8 lm W−1, respectively, whereas SMPI-TPA:Ir(ppy)3-based devices exhibit high ηc and ηp values of 65.2 cd A−1 and 67.1 lm W−1, respectively. The maximum ηex values of 19.6% and 23.4% were exhibited by SMPI-TPA:Ir(ppy)3 and SMPI-Cz:Ir(ppy)3, respectively. The lower efficiency from SMPI-TPA:Ir(ppy)3-based devices could be attributed to the lower triplet energy (ET = 2.32 eV) of SMPI-TPA, which may cause back energy transfer from the guest triplet states to the host, resulting in loss of efficiency. The device performances reveal that SMPI-TPA and SMPI-Cz are universal hosts for green emitters. The EL spectra of devices III and IV were consistent with their corresponding PL spectra (Fig. 3), and no new peaks were observed under different operation voltages (2.8–10 V: Fig. S1†), implying that no excitons were wasted for host emission and effective exothermic energy transfer occurs from the host to the dopant in the emissive layer, which results in higher efficiency.34,35 In the present study, some factors account for the higher efficiency of the devices: (i) high triplet energies of 2.32 eV (SMPI-TPA) and 2.40 eV (SMPI-Cz) efficiently suppress the energy return from the dopant to the host, resulting in higher efficiency and (ii) good thermal stability restrains the strong bimolecular interaction of the phosphorescent emitters to reduce the triplet–triplet annihilation.34 These results indicate that the introduction of a bipolar molecule is a practical strategy for achieving highly efficient OLEDs both as a blue emitter and as a host for green emission.
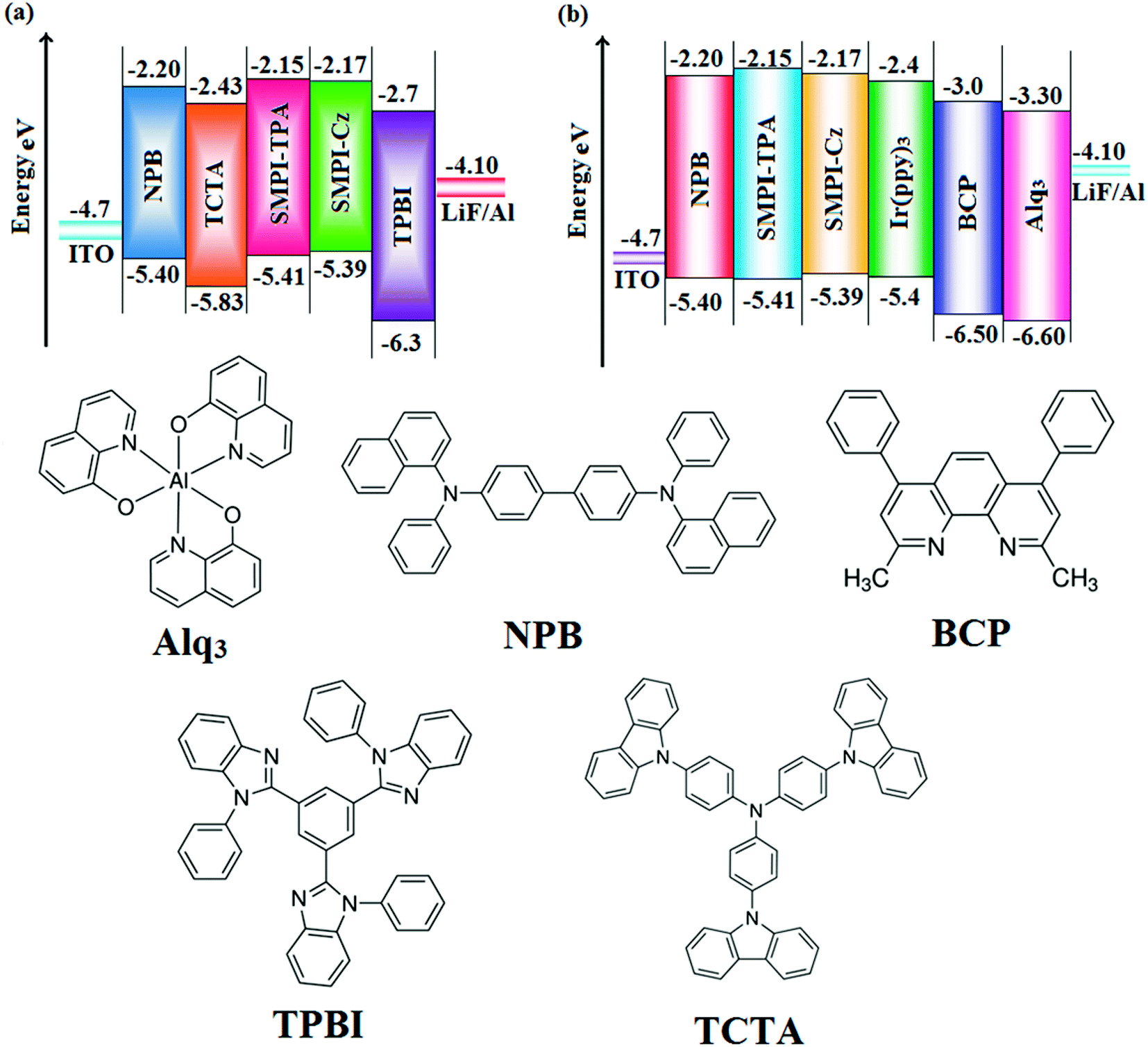 | ||
| Fig. 5 Energy-level diagram of (a) non-doped and (b) doped devices and the materials used for the fabrication of devices. | ||
4. Conclusion
In this study, we have reported the newly fabricated efficient deep blue-emissive materials with balanced transport and high thermal properties. Non-doped blue emitters with SMPI-TPA and SMPI-Cz exhibit higher electroluminescent efficiencies. Devices with SMPI-TPA show maximum luminance of 12680 cd m−2 and maximum current (ηc) and power (ηp) efficiencies of 5.3 cd A−1 and 5.2 lm W−1, respectively, at 3.1 V. The device with configuration of SMPI-Cz (30 nm):5 wt% Ir(ppy)3 exhibits maximum luminance of 20725 cd m−2, whereas the ηc and ηp exhibited by the SMPI-TPA:Ir(ppy)3 device are 65.2 cd A−1 and 67.1 lm W−1, respectively. The maximum ηex of 19.6% and 23.4% were exhibited by SMPI-TPA:Ir(ppy)3- and SMPI-Cz:Ir(ppy)3-based devices, respectively. The lower efficiency from SMPI-TPA:Ir(ppy)3-based devices could be attributed to the lower triplet energy (ET = 2.32 eV) of SMPI-TPA, which may cause back energy transfer from the guest triplet states to the host, resulting in loss of efficiency. The device performance indicates that introduction of a bipolar molecule is a practical strategy for achieving efficient OLEDs both as a blue emitter and as a host for green emission.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr J. Jayabharathi thank the DST (Department of Science and Technology – EMR/2014/000094, F. No. SR/S1/1C-73/2010, F. No. SR/S1/1C-07/2007), DRDO (Defence Research and Development Organization – 213/MAT/10-11), CSIR (Council of Scientific and Industrial Research – No. 01/(2707)/13EMR-II), UGC (University Grant Commission – 36-21/2008, F.·No. 30-71/2004(SR))and DST-Nano Mission (SR/NM/NS-1001/2016) for financial support.References
- J. P. J. Markham, E. B. Namdas, T. D. Anthopoulos, I. D. W. Samuel, G. R. Richards and P. L. Burn, Appl. Phys. Lett., 2004, 85, 1463–1465 CrossRef.
- H. J. Bolink, E. Coronado, S. G. Santamaria, M. Sessolo, N. Evans, C. Klein, E. Baranoff, K. Kalyanasundaram, M. Graetzel and M. K. Nazeeruddin, Chem. Commun., 2007, 3276–3278 RSC.
- W. Y. Hung, L. C. Chi, W. J. Chen, Y. M. Chen, S. H. Chou and K. T. Wong, J. Mater. Chem., 2010, 20, 10113–10119 RSC.
- E. B. Namdas, T. D. Anthopoulos, D. W. Samuel, M. J. Frampton, S. C. Lo and P. L. Burn, Appl. Phys. Lett., 2005, 86, 161104–161113 CrossRef.
- (a) C. Chein, C. Chen, F. Hsu, C. Shu, P. Chou and C. Lai, Adv. Funct. Mater., 2009, 19, 560–566 CrossRef; (b) C. H. Chang, M. C. Kuo, W. C. Lin, Y. T. Chen, K. T. Wong, S. H. Chou, E. Mondal, R. C. Kwong, S. Xia, T. Nakagawa and C. Adachi, J. Mater. Chem., 2012, 22, 3832–3838 RSC.
- D. H. Kim, N. S. Cho, H. Y. Oh, J. H. Yang, W. S. Jeon, J. S. Park, M. C. Suh and J. H. Kwon, Adv. Mater., 2011, 23, 2721–2726 CrossRef PubMed.
- (a) H. Fukagawa, T. Shimizu, H. Hanashima, Y. Osada, M. Suzuki and H. Fujikake, Adv. Mater., 2012, 24, 5099–5103 CrossRef PubMed; (b) Y. Cho and J. Lee, Low Driving Voltage, Adv. Mater., 2011, 23, 4568–4572 CrossRef PubMed.
- (a) M. T. Lee, H. H. Chen, C. H. Liao, C. H. Tsai and C. H. Chen, Appl. Phys. Lett., 2004, 85, 3301–3303 CrossRef; (b) M. T. Lee, C. H. Liao, C. H. Tsai and C. H. Chen, Highly Efficient, Deep-Blue Doped Organic Light-Emitting Devices, Adv. Mater., 2005, 17, 2493–2497 CrossRef.
- (a) A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402–428 CrossRef; (b) H. Sasabe, N. Toyota, H. Nakanishi, T. Ishizaka, Y. J. Pu and J. Kido, Adv. Mater., 2012, 24, 3212–3217 CrossRef PubMed; (c) H. Huang, Y. X. Wang, S. Q. Zhuang, X. Yang, L. Wang and C. L. Yang, J. Phys. Chem. C, 2012, 116, 19458–19466 CrossRef.
- J. Hu, Y. Pu, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, Adv. Mater., 2014, 24, 2064–2071 Search PubMed.
- T. C. Chao, Y. T. Lin, C. Y. Yang, T. S. Hung, H. C. Chou, C. C. Wu and K. T. Wong, Adv. Mater., 2005, 17, 992–996 CrossRef.
- J. R. Sheats, H. Antoniadis, M. Hueschen, W. Leonard, J. Miller, R. Moon, D. Roitman and A. Stocking, Science, 1996, 273, 884–888 Search PubMed.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750–753 CrossRef PubMed.
- D. Kenneth, S. Jianmin, S. Nigel, F. Eric and C. M. David, Inorg. Chem., 2005, 44, 4445–4447 CrossRef PubMed.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef PubMed.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC.
- (a) W. Y. Lai, Q. Y. He, R. Zhu, Q. Q. Chen and W. Huang, Adv. Funct. Mater., 2008, 18, 265–276 CrossRef; (b) L. Wang, Y. Jiang, J. Luo, Y. Zhou, J. H. Zhou, J. Wang, J. Pei and Y. Cao, Adv. Mater., 2009, 21, 4854–4858 CrossRef PubMed; (c) L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef PubMed; (d) C. G. Zhen, Y. F. Dai, W. J. Zeng, Z. Ma, Z. K. Chen and J. Kieffer, Adv. Funct. Mater., 2011, 21, 699–707 CrossRef; (e) K. Chen, H. R. Zhao, Z. K. Fan, G. Yin, Q. M. Chen, Y. W. Quan, S. H. Li and S. H. Ye, Org. Lett., 2015, 17, 1413–1416 CrossRef PubMed.
- (a) K. Wang, F. C. Zhao, C. G. Wang, S. Y. Chen, D. Chen, H. Y. Zhang, Y. Liu, D. G. Ma and Y. Wang, Adv. Funct. Mater., 2013, 23, 2672–2680 CrossRef; (b) K. Wang, S. P. Wang, J. B. Wei, S. Y. Chen, D. Liu, Y. Liu and Y. Wang, J. Mater. Chem. C, 2014, 2, 6817–6826 RSC.
- Y. Kawamura, S. Yanagida and S. R. Forrest, J. Appl. Phys., 2002, 92, 87–93 CrossRef.
- F. Shen, H. Xia, C. Zhang, D. Lin, L. He and Y. Ma, J. Phys. Chem. B, 2004, 108, 1014–1019 CrossRef.
- C. Fan, Y. H. Chen, Z. Q. Jiang, C. L. Yang, C. Zhong, J. G. Qin and D. G. Ma, J. Mater. Chem., 2010, 20, 3232–3237 RSC.
- (a) L. Duan, J. Qiao, Y. D. Su and Y. Qiu, Adv. Mater., 2011, 23, 1137–1144 CrossRef PubMed; (b) C. J. Zheng, J. Wang, J. Ye, M. F. Lo, X. K. Liu, M. K. Fung, X. H. Zhang and C. S. Lee, Adv. Mater., 2013, 25, 2205–2211 CrossRef PubMed; (c) D. D. Zhang, L. Duan, Y. Li, H. Y. Li, Z. Y. Bin, D. Q. Zhang, J. Qiao, G. D. Dong, L. D. Wang and Y. Qiu, Adv. Funct. Mater., 2014, 24, 3551–3561 CrossRef.
- Y. Yuan, J. X. Chen, F. Lu, Q. X. Tong, Q. D. Yang, H. W. Mo, T. W. Ng, F. L. Wong, Z. Q. Guo, J. Ye, Z. Chen, X. H. Zhang and C. S. Lee, Chem. Mater., 2013, 25, 4957–4965 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09 (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- V. Jankus, C. J. Chiang, F. Dias and A. P. Monkman, Adv. Mater., 2013, 25, 1455–1459 CrossRef PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef PubMed.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef PubMed.
- J. W. Sun, J. H. Lee, C. K. Moon, K. H. Kim, H. Shin and J. J. Kim, Adv. Mater., 2014, 26, 5684–5688 CrossRef PubMed.
- D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050–5055 CrossRef PubMed.
- X. K. Liu, Z. Chen, C. J. Zheng, M. Chen, W. Liu, X. H. Zhang and C. S. Lee, Adv. Mater., 2015, 27, 2025–2030 CrossRef PubMed.
- C. Li, S. Wang, W. Chen, J. Wei, G. Yang, K. Ye, Y. Liu and Y. Wang, Chem. Commun., 2015, 51, 10632–10635 RSC.
- Z. Wang, Y. Feng, H. Li, Z. Gao, X. Zhang, P. Lu, P. Chen, Y. Ma and S. Liu, Phys. Chem. Chem. Phys., 2014, 16, 10837–10843 RSC.
- B. Liu, J. Zhao, C. Luo, F. Lu, S. Tao and Q. Tong, J. Mater. Chem. C, 2016, 4, 2003–2010 RSC.
- S. Zhang, Q. L. Xu, Y. M. Jing, X. Liu, G. Z. Lu, X. Liang, Y. X. Zheng and J. L. Zuo, RSC Adv., 2015, 5, 27235–27241 RSC.
- Z. Chen, X. K. Liu, C. J. Zheng, J. Ye, X. Y. Li, F. Li, X. M. Ou and X. H. Zhang, J. Mater. Chem. C, 2015, 2, 4283–4289 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02840k |
| This journal is © The Royal Society of Chemistry 2018 |