
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe digestibility of hydrothermally-treated bovine serum albumin glycated by glyoxal
Guoying Su a,
Lin Liabc,
Di Zhaod,
Bing Li*ac and
Xia Zhang*ac
a,
Lin Liabc,
Di Zhaod,
Bing Li*ac and
Xia Zhang*ac
aSchool of Food Science and Engineering, South China University of Technology, 381 Wushan Road, Guangzhou, 510640, China. E-mail: lcbingli@scut.edu.cn; Tel: +86 20 87113252
bSchool of Chemical Engineering and Energy Technology, Dongguan University of Technology, College Road 1, Dongguan, 523808, China
cGuangdong Province Key Laboratory for Green Processing of Natural Products and Product Safety, 381 Wushan Road, Guangzhou, 510640, China
dKey Laboratory of Meat Processing and Quality Control, MOE, Jiangsu Collaborative Innovation Center of Meat Production and Processing, Quality and Safety Control, Key Laboratory of Meat Products Processing, MOA, Nanjing Agricultural University, Nanjing, 210095, P. R. China
First published on 22nd October 2018
Abstract
The digestion of dietary advanced glycation end products (AGEs) largely determines their absorption in humans. To help elucidate the health effects of dietary AGEs, changes in the digestive behavior of bovine serum albumin (BSA, dietary protein) caused by glycation derived from glyoxal (GO, an important precursor of AGEs) in a simulated food heating system have been investigated. The hydrothermal aggregation of BSA was suppressed by GO derived glycation, generating glycated aggregates of loose and branched structures, according to dynamic light scattering (DLS), circular dichroism (CD) spectroscopy, free sulfhydryl group, transmission electron microscopy (TEM) and small angle X-ray scattering (SAXS) results. Analysis of protein digests showed that glycation reduced the gastric and gastrointestinal digestibility of BSA and the bioavailability of all seven detected amino acids. A comparative analysis of the distribution of CML and lysine in glycated BSA digests with different molecular weights showed that carboxymethylation directly blocked the action of proteases on Lys residues.
Introduction
Dietary advanced glycation end products (AGEs) have been demonstrated to be involved in the pathology of various diseases, such as diabetes, cardiovascular disease, and cataracts.1–3 The effects of dietary AGEs on human health are determined by their absorbability by the small intestine. Only 10–30% of dietary AGEs are estimated to be absorbed and enter circulation.4,5As important precursors of AGEs, α-dicarbonyl compounds are widely generated during the Maillard reaction (this process is referred to as glycation) and caramelization reactions during thermal processing and the storage of sugar-rich foods.6,7 3-Deoxyglucosone (3-DG), 3-deoxygalactosone (3-DGal), glyoxal (GO), methylglyoxal (MGO) and butanedione (BU) are the main α-dicarbonyl compounds involved in glyoxidation,7–9 and the oxidation of lipids can also produce GO, MGO, and BU.10 α-Dicarbonyl compounds are found in commonly consumed foods, such as soft drinks, honey, milk, wine and baked food.11–14 Heat treatment increases the content of these compounds in foods. For example, the GO content was reported to increase from less than 0.3 mg L−1 to approximately 2.5 mg L−1 after the ultra-high temperature (UHT) treatment of milk.15
After being generated during food processing or storage, α-dicarbonyl compounds will react with Lys and Arg residues of proteins to generate AGEs.16 GO has been proposed as the precursor to N(ε)-carboxymethyllysine (CML), GO hydroimidazolone (G-H) and glyoxal lysine dimer (GOLD), whereas MGO is the precursor to N(ε)-carboxyethyllysine (CEL), MGO hydroimidazolone (MG-H1) and methylglyoxal lysine dimer (MOLD).17,18
As dietary AGEs and glycated proteins are digested in the gastrointestinal tract, they are enzymatically hydrolyzed into absorbable or unabsorbable fractions.19 Therefore, the digestion of glycated proteins is closely related to their absorption in the small intestine. The decreased digestibility of glycated proteins has been mostly attributed to the reduction of lysine and arginine residues, which are crucial cleavage sites for trypsin.20,21 Controversially, conformational changes caused by glycation are usually neglected as a cause of decreased digestibility. During α-dicarbonyl compound-derived glycation, the active α-dicarbonyl compounds not only act on the lysine and arginine sites, but also cause changes to the conformation and aggregation behavior of proteins. These changes may influence contact between proteins and enzymes excreted in the digestive system, as well as the digestibility of proteins. However, limited information has been reported on the digestibility of dietary proteins glycated by α-dicarbonyl compounds.
Bovine serum albumin (BSA) is the dominant form of dietary protein and is wildly used in food systems as a nutrient, antioxidant, stabilizer, etc.;22–25 it is also used as a molecular model protein for food systems, due to its well characterized structural and physico-chemical properties.26 To study the changes in the digestive behavior of dietary protein caused by glycation derived from α-dicarbonyl compounds in food systems, BSA, a well-studied dietary protein, and GO, one of the major α-dicarbonyls, were selected to establish the glycation system. CML, a typical GO-derived AGE existing in cooked meat and dairy foods at high levels,27 and seven kinds of amino acids were chosen as the indicators to be detected after simulated gastric and intestinal digestion to evaluate the digestibility of both glycated and non-glycated BSA aggregates. Moreover, conformational changes in the glycated aggregates were measured to elucidate the mechanism underlying changes in digestibility brought about by glycation.
Experimental
Materials
CML (98%) was obtained from TCR (Canada). BSA and all chemicals used for simulated gastrointestinal digestion, namely pepsin (>2500 units per mg protein), trypsin (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mg protein), chymotrypsin (>40 units per mg protein) and bile salts, were purchased from Sigma-Aldrich (Steinheim, Germany). GO was obtained from Aladdin (Shanghai China).
000 units per mg protein), chymotrypsin (>40 units per mg protein) and bile salts, were purchased from Sigma-Aldrich (Steinheim, Germany). GO was obtained from Aladdin (Shanghai China).
Glycation of BSA
BSA (5 mg ml−1) was incubated at 98 °C in 100 mM phosphate buffer (pH = 7.0) with GO (1 mM, 10 mM or 50 mM) for 2 h to simulate heat treatment during food processing. All samples were dialyzed using dialysis bags with a molecular weight cut-off of 3000 Da for 3 days at 4 °C before digestion to ensure any excess GO and other small molecules were removed.Particle size analysis via dynamic light scattering (DLS)
The particle sizes of the heated samples were determined using a Zetasizer Nano system (Malvern Instruments Inc., Worcester, UK). Measurements were carried out at 25 °C with a scattering angle of 173°, and the viscosity of the solvent was presumed to be the same as water.Circular dichroism (CD) spectroscopy
Far-UV CD spectra of the protein samples (0.2 mg mL−1) were recorded in cells (1 mm path length) over a wavelength range of 190 to 260 nm at 25 °C using Chirascan apparatus (Applied Photophysics, UK). Each CD spectrum was calculated from the average of three scans using a band width of 0.5 nm, a step interval of 0.5 nm, and a scanning speed of 60 nm min−1.Transmission electron microscopy (TEM) analysis
Aliquots of the protein samples (5 μl, 10 μg ml−1) were withdrawn and applied to carbon-stabilized grids for 30 s. Excess sample was removed by applying ashless filter paper to the edges of the grids, and then the grids were stained with 0.5% phosphotungstic acid (PTA) in distilled deionized water for another 30 s. After removing the excess stain, the grids were left to air dry for 30 min before being examined and photographed with a transmission electron microscope (TECNAI G212, FEI, Netherlands) at an accelerating voltage of 200 kV.Small-angle X-ray scattering (SAXS) analysis
SAXS (PW3810, PANalytical, Netherlands) analysis was conducted as follows. The X-ray exciter was set to 40 kV and 50 mA, a Gobel mirror and collimator were used to provide monochromatic light (λ = 0.1542 nm), and samples (10 mg ml−1) were injected into vacuum capillaries, 1 mm in diameter, that were sealed at each end. Samples were kept at 26 °C throughout testing. Two-dimensional images of each sample were recorded with an imaging plate and then translated into a one-dimensional scattering result.Simulated gastrointestinal digestion
The in vitro digestion procedure followed the method of Martínez and Pinto.20,28 For the simulated gastric digestion, 4 mg of sample was dissolved in 1.33 ml of simulated gastric fluid (SGF; 0.15 M NaCl; pH 2.5, adjusted with 1 M HCl). Then, a 0.48% (w/v) solution of porcine pepsin in SGF was added to each sample at a simulated physiological ratio of enzyme to substrate (1:20, w/w) to obtain 165 units per mg of protein. Digestion was performed at 37 °C for 5 min, 10 min, 15 min, 30 min, 60 min and 120 min. 40 mM NH4HCO3 was added to inactivate the pepsin in each sample. For the following intestinal digestion,11 the pH was adjusted to 6.5 with 0.1 M NaOH and bile salts (4 mM sodium taurocholate; 4 mM sodium glycodeoxycholate; 26.1 mM bis–tris buffer, pH 6.5). Chymotrypsin and trypsin were added at 0.4 U per mg of test protein and 34.5 U per mg of test protein, respectively. Digestion was performed at 37 °C for 5 min, 10 min, 15 min, 30 min, 60 min, 120 min, and 240 min.
Both glycated and unglycated samples digested after 5 min, 10 min, 30 min, 120 min and 240 min were analyzed for their gastric and duodenal digestive behavior via the determination of the free amino group liberation trend using the o-phthaldialdehyde (OPA) method. Peptide mixtures after the whole simulated gastrointestinal procedure were used for the following FPLC separation.
OPA assay
The level of primary amino groups was measured using a modified OPA method to investigate the extent of glycation during incubation and the dynamic hydrolytic process during simulated gastrointestinal digestion.29 The OPA reagent was prepared as follows: 40 mg of OPA in 25 mL of 10 mM sodium tetraborate was mixed with 2.5 mL of 20% (w/w) SDS and 100 μL of β-mercaptoethanol. Then, the mixed solution was diluted with distilled water to a final volume of 50 mL as the working solution. During the assay, 100 μL of glycated protein or digest was mixed with 2 mL of working solution and incubated at 40 °C for 3 min. The absorbance at 340 nm was measured using a Varian Cary 50 instrument (Varian, USA), which is controlled by Cary Win UV software, version 2.0.The loss of amino groups during glycation was expressed as a relative amount, assuming that 100% was equal to the amino group content in the control proteins before heat treatment. Increased absorbance during digestion was calculated as the difference between the absorbance values before and after gastric or intestinal digestion.
Free sulfhydryl group (–SH) measurements
The –SH content was detected using the method of Veraverbeke et al..30 A 290 mL aliquot of either glycated or control sample solution was mixed with 30 μL of 1 mM DTNB aqueous solution and 1 mL of sample buffer (containing 5 mM phosphate buffer, 2.0% SDS and 3.0 M urea; pH 6.5). This mixture was incubated in the dark at room temperature for 30 min before testing. 250 μL of reaction solution was transferred to a microtiter plate, and the UV absorbance at 412 nm was measured using an Infinite M200 PRO microplate reader (Tecan, Grödig, Austria). The absorbances of DTNB and the sample were both recorded and subtracted. The content of –SH was determined as a relative percentage, assuming that the absorbance of control β-Lg before heat treatment represented 100%. This assay was repeated in triplicate for each sample.Fast protein liquid chromatography (FPLC) analysis of the digestion products
Analytical separations of the peptide mixtures were performed using an FPLC system (AKTA, GE, USA). Superdex peptide HR 10/30 (Pharmacia Bio-tech, Uppsala, Sweden) was used for the analytical runs. Each run was performed for 70 min with phosphate-buffered saline (PBS, 0.30 M, 0.15 M NaCl, pH 7.0) as the mobile phase at a flow rate of 0.5 ml min−1. All samples were membrane-filtered (0.45 μm) before analysis. The injection volume was 500 μl. Detection was performed at 280 nm, and the eluate was collected every 5 min during the run, starting at 10 min. BSA (66.446 kDa), cytochrome (12.384 kDa), bacitracin (6.511 kDa), lysozyme (1.450 kDa), Gly-Gly-Try-Arg (451 Da), and Gly-Gly-Gly (189 Da) were used for mass calibration.Relative amounts of CML and selected amino acids in peptide mixtures after gastrointestinal digestion
Each fraction collected from FPLC separation was hydrolyzed in 6 M HCl at 110 °C for 24 h and was then vacuum dried (40 °C).31 After drying, each fraction was dissolved in 0.5 ml of aqueous methanol (10%) and filtered through a membrane (0.46 μm) before HPLC-MS analysis.The HPLC-MS method was the same as that of Han, with some modifications:32 CML was analyzed via HPLC (Waters 1525, Waters, USA) in tandem with a single quadrupole mass spectrometer (Waters Micromass ZQ, Waters, USA). An Atlantis C18 (Waters, USA) column was selected for the HPLC separations, with aqueous methanol (10%) as the mobile phase at a flow rate of 0.5 ml min−1. The measurements were collected under the following conditions: a capillary voltage of 3.0 kV; a cone voltage of 20 V; a source temperature of 100 °C; and a desolvation temperature of 300 °C. Electrospray ionization (ESI) in positive mode and single ion recording (SIR) were applied to the +205.1 (CML), +166.2 (Phe), +182.2 (Tyr), +132.2 (Leu), +118.1 (Val), +120.1 (Thr), +116.1 (Pro), and +133.1 (Asn) channels. The CML/Lys content in digested fractions was expressed in relative amounts, considering the ion suppression from the salts of the SEC mobile phase on the MS signal, and assuming that 100% was equal to the sum of CML or Lys in all fractions.
Statistical analysis
Triplicate analyses (n = 3) were performed for each dependent variable. Analysis of variance (ANOVA) with Duncan's multiple range test was performed using IBM SPSS Statistics 19.0 software (SPSS Inc., Chicago, IL, USA). Differences were considered significant at P < 0.05. The values shown in the tables are means of values from triplicate analyses (standard deviations).Results and discussion
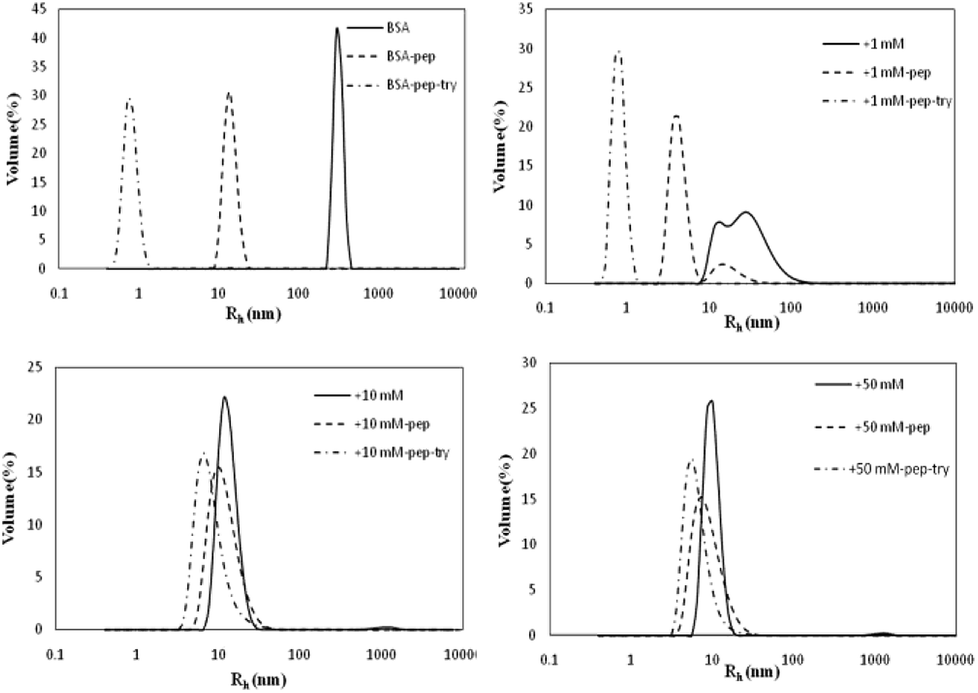
Influence of glycation on BSA hydrothermal aggregation
The Rh values of the non-glycated aggregates after heating for 2 h were approximately 100 nm, much larger than those of the glycated samples. As illustrated in Fig. 1, the Rh values of BSA glycated with 1 mM GO were distributed over a wide range (between 10 and 100 nm) after 120 min of heating at 98 °C. In contrast, more intense glycation with a higher concentration of GO (1 mM and 50 mM) significantly suppressed the radius of BSA to approximately 10 nm. According to Philippe Rondeau,33 hydrothermal treatment induces the partial opening of the native protein conformation. Consequently, some specific regions, such as hydrophobic sites or free –SH groups, become more exposed to new intermolecular interactions and so contribute to the formation of aggregates. The DLS data indicates that GO-derived glycation may involve interactions with the opened specific regions, resulting in the reduction of BSA particle size. A similar effect of glycation was found in the studies of Pinto and Liu,34,35 in which the decreased size of glycated aggregates was attributed to glycation-induced increases in steric hindrance and electrostatic interactions.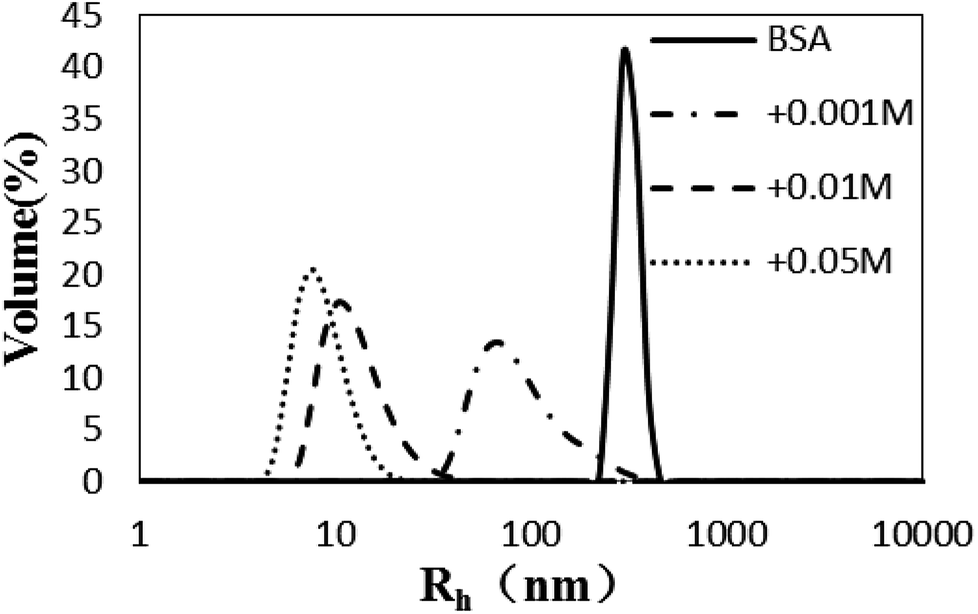 | ||
| Fig. 1 The size distribution (by volume) measured via dynamic light scattering of glyoxal-derived BSA after hydrothermal incubation for 2 h. | ||
Fig. 2 presents representative TEM micrographs of non-glycated and glycated BSA after heating for 2 h at 98 °C. A majority of the aggregates were large and compact spherical granules, 500–1000 nm in diameter, and only a minority was small in size. Aggregates glycated with 1 mM GO (Fig. 2B) were much smaller spherical particles (approximately 50–500 nm in diameter) than the aggregates formed from non-glycated BSA. Increased glycation mainly results in smaller branched chains of globular aggregates, as illustrated in Fig. 2C. Analogous TEM micrographs were obtained from BSA samples co-incubated with 5 mM glyoxylic acid after 83 weeks at 37 °C.36 Although more research is needed to elucidate the underlying mechanism, we speculate that the formation of branched chain species might be due to steric hindrance and crosslinks formed during incubation with GO, which is consistent with Rondeau's study of BSA aggregation at its denaturation temperature (58 °C).33
 | ||
| Fig. 2 (A) A representative TEM micrograph of non-glycated BSA after incubation for 2 h at 98 °C. (B) A representative TEM micrograph of a BSA sample with 10 mM glyoxal after incubation for 2 h at 98 °C. (C and D) Representative TEM micrographs of a BSA sample with 50 mM glyoxal after incubation for 2 h at 98 °C. | ||
To further confirm the conformation changes of the glycated BSA aggregates, SAXS was used to explore the changes in the chain structures of the protein aggregates brought on by glycation. A Kratky plot (Fig. 3A), which is prepared from the original intensity data, conveys conformational information.37 Typical bell-shaped curves, which are observed for non-glycated BSA and mildly glycated (BSA-1 mM) samples, indicate the particles in these samples are compact and spherical,38 which is consistent with what was observed in the TEM micrographs. In contrast, these bell-shaped curves become less intense and broader (BSA-10 mM) and eventually transform into flat-topped curves (BSA-5 mM) when the concentration of GO is further increased. This change indicates the collapse of the spherical structures and the formation of random coils or other looser structures without cores, which is consistent with what is found in the TEM micrographs.
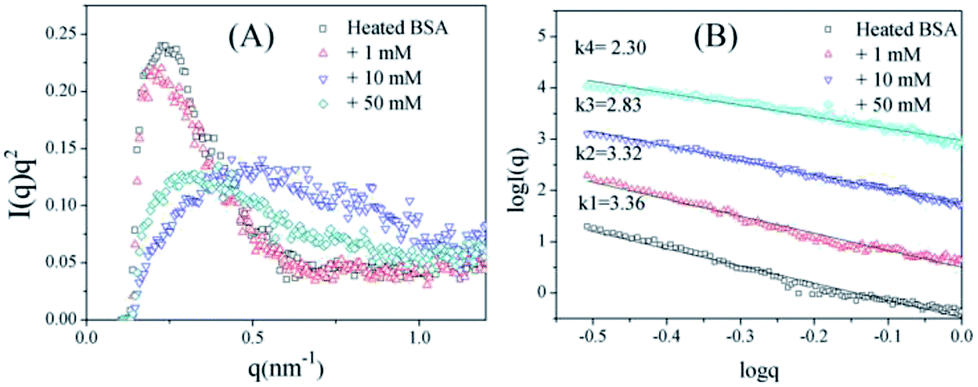 | ||
| Fig. 3 Kratky plots (A) and the typical curves (B) of non-glycated and glycated samples after incubation for 2 h at 98 °C. | ||
Fig. 3B shows typical curves obtained from the SAXS measurements, and the intensity of small-angle scattering (I(q)) follows the equation:
| I(q) ∝ q−k |
I(q) vs. logq. For mass fractals, one can show that Dm = k and 1 < k < 3 in three-dimensional space, and that Dm increases as the compactness of the scattering particles increases. In contrast, we have Dm = 6 − k (3 < k < 4) for the surface fractals, which suggests that the structure of the scattering particles is originally compact, and that Dm increases as the roughness of the surface decreases.39,40 For the BSA samples, the slope (k) decreases as the concentration of GO increases. Fig. 3B shows that the k value of BSA is 3.36, which is the surface fractal dimension, and the particles have a compact structure. The k values of BSA glycated with 10 mM and 50 mM GO are 2.83 and 2.30, respectively, which are the mass fractal dimensions. The shift in the k value caused by GO glycation is consistent with the phenomenon shown in Fig. 3A.
These results clearly indicate that BSA glycated under these conditions no longer has the compact and spherical structure of unmodified BSA aggregates. The initial stage of thermal aggregation involves unfolding, which exposes all residues to the external environment. During this process, GO can react with lysine and arginine residues in Maillard reactions to generate stem grafting-type and crosslinking-type AGEs. Steric hindrance increases dramatically during this procedure, inhibiting subsequent hydrophobic aggregation to a large extent, and the generation of intermolecular AGEs may involve combining different aggregates to form branched structures. Glycated aggregates, which have smaller and looser structures, are theoretically better substrates for protease, but we cannot neglect the increased steric hindrance of the glycated structures, which may hinder interactions between the aggregates and protease.
The CD profile of unheated BSA shows a negative peak at 208 nm (Fig. 4), indicating the predominant α-helix structure of BSA. Heat treatment was shown to increase the negative peak, which could thus imply the elevation of a disordered structure.41 In addition, glycation further enlarges the negative peak, and a blue-shift occurred for 50 mM GO-glycated BSA from 208 nm to 206 nm. These results indicated that glycation was shown to lead to an additional reduction in the α-helix structure which is combined with the elevation of an unordered structure.41
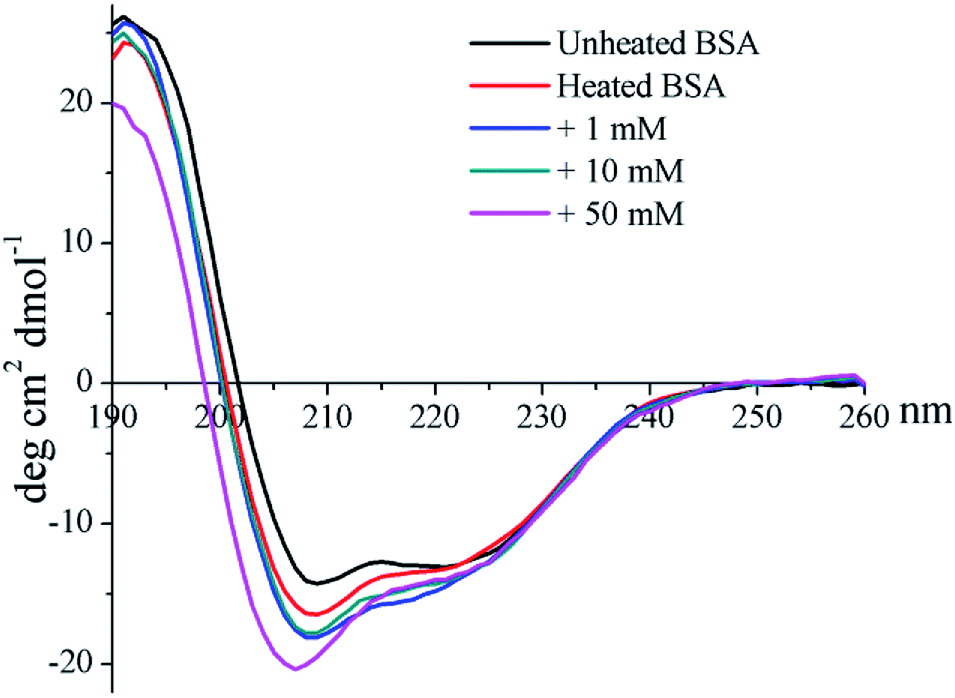 | ||
| Fig. 4 Far-UV CD spectra of non-glycated and glycated BSA after incubation for 2 h at 98 °C. | ||
The formation of intermolecular disulfide bonds was investigated via measuring the loss of –SH groups. The free –SH group at Cys 34 of BSA can take part in the formation of intermolecular disulfide bonds. As shown in Fig. 5, the amounts of free –SH in the control, 1 mM GO-glycated, 10 mM GO-glycated and 50 mM GO-glycated BSA were shown to decease by 67%, 74%, 80% and 89% after 1 h of incubation (P < 0.05). These data suggest a faster loss of sulfhydryl groups in the glycated samples. Analogous to amino and guanidyl groups, free sulfhydryl groups were also reported to participate in glycation based on nucleophilic reactions, giving rise to the corresponding Maillard reaction products, such as S-(carboxymethyl)-cystine.42,43 Therefore, the formation of intermolecular disulfide bonds during incubation is speculated to be blocked once glycation at Cys34 has occurred.
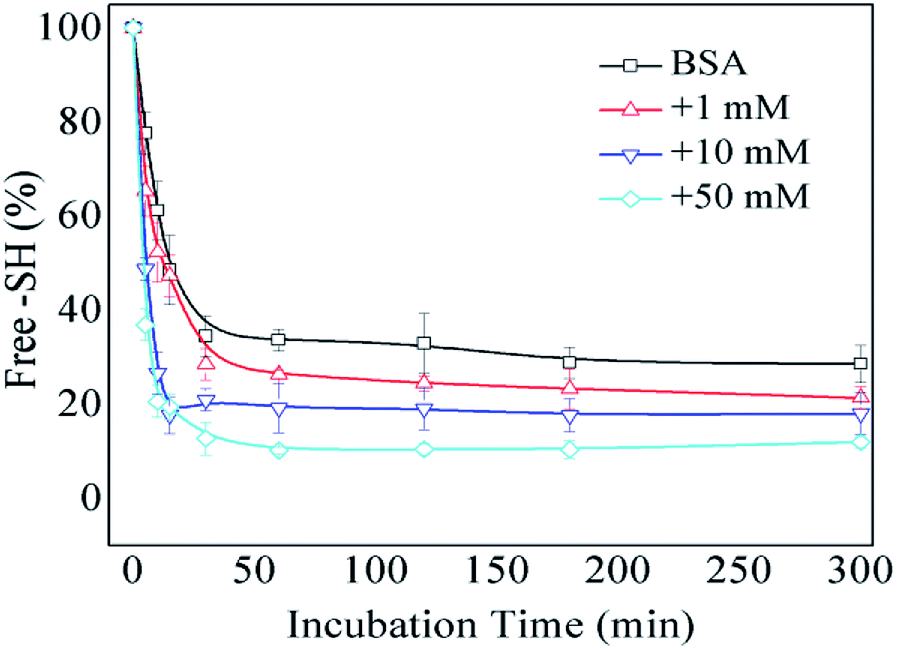 | ||
| Fig. 5 The free –SH group content of non-glycated and glycated BSA after incubation for 2 h at 98 °C. | ||
Influence of BSA modification on gastric and intestinal digestibility
The trend in the liberation of dynamic free amino groups during simulated digestion reflects the digestive behavior of protein samples. In Fig. 6A, the initial (before 15 min) gastric digestion rate of non-glycated BSA is less than those of the glycated samples (P < 0.05) and the particle size of non-glycated BSA (100 nm < Rh < 1000 nm) is larger than those of the glycated samples (8 nm < Rh < 100 nm) (P < 0.05). According to Joseph S. Fruton,44 hydrophobic amino acid residues (mainly Phe, Trp, and Tyr) are the main cleavage sites for pepsin and they remain complete during glycation. Therefore, the phenomenon may be explained through the larger and more compact structure of the non-glycated aggregates, which inhibits interactions between pepsin and hydrophobic amino residues (Phe, Tyr, and Trp). The final increased absorption of the non-glycated sample is similar to the sample glycated with 1 mM GO (P > 0.05) and is greater than those of the samples co-incubated with 10 mM and 50 mM GO (P < 0.05), indicating that the non-glycated samples show better gastric digestibility. This is because the spherical structures of the non-glycated sample will gradually collapse during gastric digestion, exposing the hydrophobic cleavage sites to pepsin. In contrast, covalent stem grafting from glycation hindered pepsin attack. After gastric digestion, most aggregates lose their overall structure and are hydrolyzed into polypeptides for subsequent intestinal digestion. In intestinal digestion, as shown in Fig. 6B, the non-glycated sample is much more digestible than the glycated samples. This is consistent with the Rh results after pepsin and trypsin digestion shown in Fig. 7. | ||
| Fig. 6 Trends in the liberation of the free amino groups of non-glycated and glycated BSA samples after heating for 2 h during simulated gastric (A) and intestinal (B) digestion. | ||
 | ||
| Fig. 7 Particle size distributions of simulated gastric and intestinal digestion products of non-glycated and glycated BSA. | ||
As shown in Fig. 7, the BSA peak after pepsin and trypsin digestion is at Rh < 1 nm, while the peaks of the samples glycated with 10 M and 50 M GO are at 8 nm < Rh < 50 nm, similar to the peaks of their own glycated and pepsin-digested products. Trypsin always cleaves proteins at Lys and Arg residues, which are preferentially blocked during the Maillard reaction.20,21 These findings indicate that reducing the number of lysine and arginine residues that are blocked during glycation accounts for this result, since these two residues are the cleavage sites for trypsin.
As shown in Fig. 8, FPLC detects the accumulation of digests in different MW ranges. The fractions collected in the first 20 min contained digests with MWs higher than 20 kDa; fractions collected from 20 to 25 min contained digests with MWs ranging from 4.5 kDa to 20 kDa; and fractions collected after 30 min contained digests smaller than 1 kDa (Fig. 7E). In Fig. 8A–D, the continuous lines represent the accumulation of digests generated by pepsin and chymotrypsin treatment, while the dotted lines represent the accumulation of digests generated by pepsin treatment only. The results for non-glycated BSA indicate a wide molecular weight distribution below 60 kD. Samples glycated with 1 mM GO (except for the pepsin-only treatment (most MWs > 20 kDa)) show curves similar to those of the non-glycated samples, but samples glycated with 10 mM and 50 mM GO show a substantial portion of digests with molecular weights higher than 20 kDa. Considering that the MWs of the digestive products and the particle size distributions could not be evaluated accurately via UV absorption distribution alone due to the production of cyclic compounds that can absorb ultraviolet light, UV information can be misleading to a certain extent, and a dramatic increase in the total ultraviolet absorption of the heavily glycated samples was obviously found for this reason.
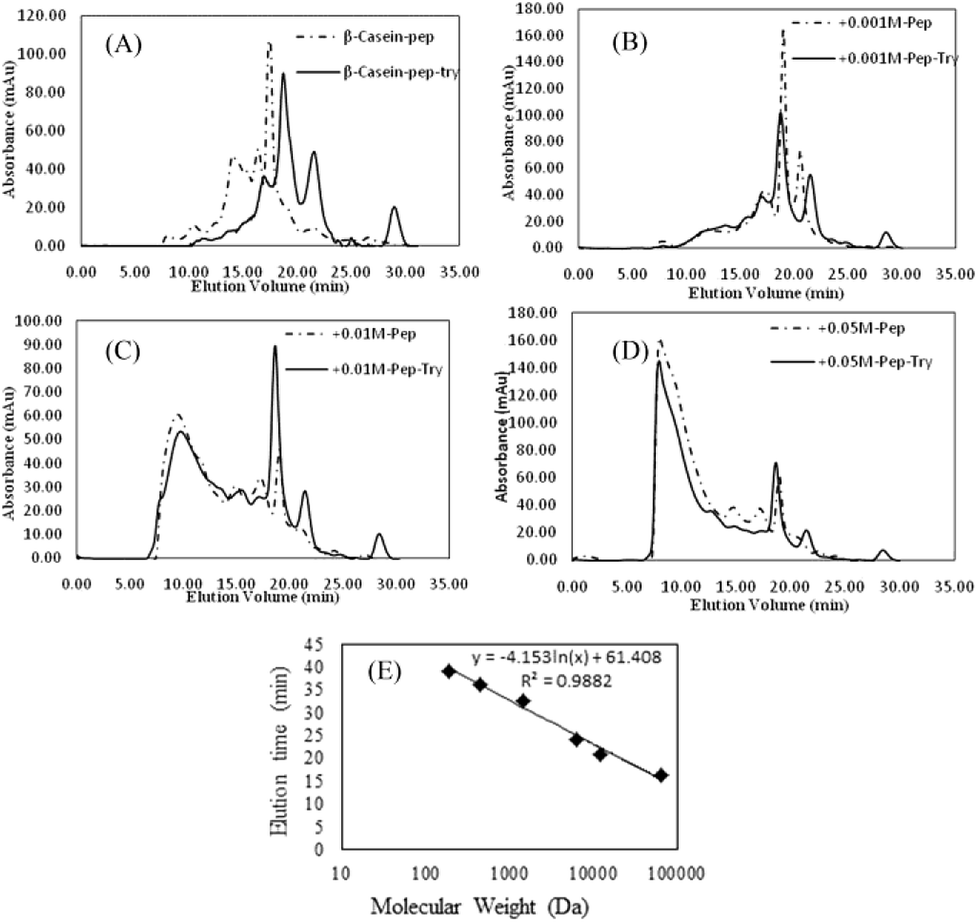 | ||
| Fig. 8 (A–D) Size-exclusion chromatograms (UV 280 nm) of peptide mixtures from non-glycated and glycated BSA after 120 min of simulated gastric digestion and 240 min of simulated intestinal digestion. (E) The calibration curve for the MW estimations of the peptides. | ||
To further analyze the MW distributions of peptide mixtures from the gastrointestinal digestion of glycated samples, CML and 7 amino acids in elute fractions collected from FPLC separation were evaluating using HPLC-MS after 24 h of HCl hydrolysis. Fig. 9 shows the CML distribution in the peptide mixtures. After the digestion of BSA glycated with 1 mM GO, 18.22 ± 3.73% CML was released into peptides bigger than 20 kDa, and this value increased to 31.29 ± 6.63% when the GO concentration increased to 10 mM (P < 0.05), which confirmed the possibility that steric hindrance caused by the formation of CML or other grafting AGEs reduced the digestibility of BSA. Then we compared the distribution of CML and Lys. Notably, a lower relative content of CML was found in the fraction with MWs < 1 kDa than that of Lys. For example, approximately 32% CML was present in the smaller digests (MWs < 1 kDa) of 10 mM GO-glycated β-LG, which is compared with approximately 41% Lys in the same fractions. This result suggested that carboxymethylation at the ε-NH2 of Lys directly reduces the digestibility of selected proteins by blocking the tryptic cleavage site (Lys) and increasing the steric hindrance. Based on this result, protein-bound AGEs seem to be less hazardous to human health, since they are not well digested compared with the original amino acids before modification.45–47
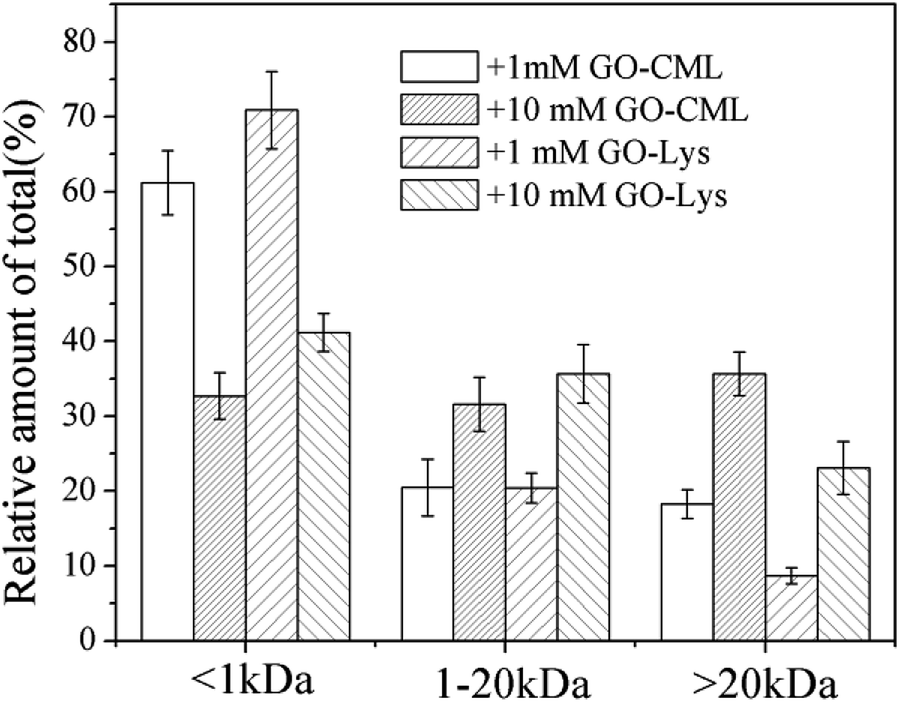 | ||
| Fig. 9 Estimates of the CML distribution in peptide mixtures from BSA glycated with various concentrations of glyoxal. | ||
After protein digestion, amino acids in smaller peptides tend to be readily absorbed by intestinal epithelial cells. The availability of an amino acid can be determined via its percentage in digests smaller than 1 kDa. Table 1 illustrates the effects of glycation on the availability of 7 kinds of amino acids. The availability of each kind of amino acid decreased significantly as the glyoxal concentration increased. Pro availability, for example, falls sharply to 11.87 ± 2.49% when 50 M glyoxal is added, which is much lower than in the non-glycated sample (69.02 ± 3.11%). Notably, the UV profiles in Fig. 3 show that digests bigger than 20 kDa account for 82.43% of UV absorption in the sample co-incubated with 50 M glyoxal. However, this percentage is much higher than the acid distribution (Phe is 33.08 ± 2.94%, Tyr is 42.06 ± 4.52%, Val is 28.13 ± 3.72% and Thr is 9.66 ± 0.63%).
| Amino acid | >20 kDa (%) | 1–20 kDa (%) | <1 kDa (%) |
|---|---|---|---|
| A | |||
| Phe | 8.36 ± 0.97 | 16.68 ± 1.28 | 74.96 ± 2.25 |
| Tyr | 15.57 ± 2.31 | 18.45 ± 1.53 | 65.98 ± 3.84 |
| Leu | 18.05 ± 2.82 | 26.92 ± 2.09 | 55.03 ± 4.91 |
| Val | 10.20 ± 1.16 | 25.60 ± 2.94 | 64.20 ± 4.10 |
| Thr | 1.94 ± 0.27 | 5.78 ± 0.62 | 92.28 ± 0.89 |
| Pro | 7.35 ± 1.17 | 23.63 ± 1.94 | 69.02 ± 3.11 |
| Asn | 18.24 ± 2.58 | 9.17 ± 1.23 | 72.59 ± 3.81 |
|
|||
| B | |||
| Phe | 30.60 ± 3.43 | 26.44 ± 0.83 | 42.96 ± 4.26 |
| Tyr | 31.68 ± 1.94 | 19.84 ± 2.37 | 48.48 ± 4.31 |
| Leu | 35.67 ± 4.34 | 30.28 ± 2.19 | 34.05 ± 6.53 |
| Val | 29.31 ± 2.10 | 36.72 ± 3.85 | 33.97 ± 1.75 |
| Thr | 7.36 ± 0.94 | 8.14 ± 1.34 | 84.50 ± 2.28 |
| Pro | 27.71 ± 3.02 | 44.63 ± 4.26 | 27.66 ± 1.24 |
| Asn | 28.53 ± 4.15 | 26.79 ± 2.35 | 44.68 ± 1.80 |
|
|||
| C | |||
| Phe | 33.08 ± 2.94 | 54.47 ± 3.75 | 12.45 ± 0.81 |
| Tyr | 42.06 ± 4.52 | 35.67 ± 2.93 | 22.27 ± 1.59 |
| Leu | 21.68 ± 3.45 | 59.68 ± 6.47 | 18.64 ± 3.02 |
| Val | 28.13 ± 3.72 | 58.07 ± 5.90 | 13.80 ± 2.18 |
| Thr | 9.66 ± 0.63 | 33.59 ± 2.41 | 56.75 ± 3.04 |
| Pro | 23.07 ± 4.34 | 65.06 ± 5.83 | 11.87 ± 2.49 |
| Asn | 24.14 ± 2.45 | 34.22 ± 3.19 | 41.64 ± 5.64 |
This phenomenon implies that a majority of the AGEs that absorb UV light are in the fractions of peptides bigger than 20 kDa, indicating that AGEs substantially hinder the cleavage of digestive protease. This result corresponds with several recent studies regarding the digestion of glycated protein.16,29,48,49
Other than blocking the Lys and arginine residues, glycation should increase the steric hindrance and other repulsion factors to hinder the digestion procedure of glycated protein.16 A related study was conducted by Homma, who investigated the metabolic fate of 15N-labeled melanoidin. He found that 26% of high-molecular weight melanoidin was excreted in the feces of mice, and no low-molecular weight melanoidin was detected in the feces.50 Crosslinking-type AGEs may play crucial roles in hindering the action of digestive proteases, considering their role in forming high molecular weight digests.20 Therefore, these AGEs in high molecular weight digests may be utilized by gut microbiota due to their low bioavailability.16 The influence of unabsorbed AGEs on the composition of gut microbiota also requires more investigation.
Conclusions
The dosages of GO used in this study are much higher than those in real food, aiming to amplify the effects of glycation derived from GO. The glyoxal-derived glycation of BSA at 98 °C breaks up the original compact structure, and new branched chains of globular aggregates are formed during this process. This structural change leads to an increased initial gastric digestion rate for the glycated aggregates. However, the overall gastric digestibility of glycated BSA drops dramatically when the glyoxal concentration is elevated above 1 mM. Steric hindrance from the generation of stem grafting-type and crosslinking-type AGEs is believed to be responsible for not only the structural changes but also the lower digestibility of the glycated samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is financially supported by the National Key R & D Program of China (no. SQ2017YFC160004/01), the National Natural Science Foundation of China (no. 31671961), the Natural Science Foundation of Guangdong Province (no. 2017A030311021) and the National Key Technology R&D Program (no. 2012BAD37B01).Notes and references
- E. D. Schleicher, E. Wagner and A. G. Nerlich, J. Clin. Invest., 1997, 99, 457–468 CrossRef CAS PubMed.
- T. Murata, R. Nagai, T. Ishibashi, H. Inomuta, K. Ikeda and S. Horiuchi, Diabetologia, 1997, 40, 764–769 CrossRef CAS PubMed.
- R. H. Nagaraj, M. Linetsky and A. W. Stitt, Amino Acids, 2012, 42, 1205–1220 CrossRef CAS PubMed.
- V. Faist and H. F. Erbersdobler, Ann. Nutr. Metab., 2001, 45, 1–12 CrossRef CAS PubMed.
- N. J. Kellow and M. T. Coughlan, Nutr. Rev., 2015, 73, 737–759 CrossRef PubMed.
- L. W. Kroh, Food Chem., 1994, 51, 373–379 CrossRef CAS.
- P. J. Thornalley, A. Langborg and H. S. Minhas, Biochem. J., 1999, 344, 109–116 CrossRef CAS PubMed.
- M. Hellwig, J. Degen and T. Henle, J. Agric. Food Chem., 2010, 58, 10752–10760 CrossRef CAS PubMed.
- a Hollnagel and L. W. Kroh, Z. Lebensm.-Unters. -Forsch. A, 1998, 207, 50–54 CrossRef CAS.
- Y. Jiang, M. Hengel, C. Pan, J. N. Seiber and T. Shibamoto, J. Agric. Food Chem., 2013, 61, 1067–1071 CrossRef CAS PubMed.
- J. Degen, M. Hellwig and T. Henle, J. Agric. Food Chem., 2012, 60, 7071–7079 CrossRef CAS PubMed.
- S. Oelschlaegel, M. Gruner, P. N. Wang, A. Boettcher, I. Koelling-Speer and K. Speer, J. Agric. Food Chem., 2012, 60, 7229–7237 CrossRef CAS PubMed.
- C. Y. Lo, W. T. Hsiao and X. Y. Chen, J. Food Sci., 2011, 76, 90–96 CrossRef PubMed.
- G. De Revel, L. Pripis-Nicolau, J. C. Barbe and A. Bertrand, J. Sci. Food Agric., 2000, 80, 102–108 CrossRef CAS.
- S. Kokkinidou and D. G. Peterson, J. Agric. Food Chem., 2014, 62, 8023–8033 CrossRef CAS PubMed.
- D. Zhao, T. T. Le, L. B. Larsen, L. Li, D. Qin, G. Su and B. Li, Food Res. Int., 2017, 102, 313–322 CrossRef CAS PubMed.
- M. O. Lederer and H. P. Bühler, Bioorg. Med. Chem., 1999, 7, 1081–1088 CrossRef CAS PubMed.
- M. A. Glomb and G. Lang, J. Agric. Food Chem., 2001, 49, 1493–1501 CrossRef CAS PubMed.
- M. W. Poulsen, R. V. Hedegaard, J. M. Andersen, B. de Courten, S. Bügel, J. Nielsen, L. H. Skibsted and L. O. Dragsted, Food Chem. Toxicol., 2013, 60, 10–37 CrossRef CAS PubMed.
- M. S. Pinto, J. Léonil, G. Henry, C. Cauty, A. Ô. F. Carvalho and S. Bouhallab, Food Res. Int., 2014, 55, 70–76 CrossRef CAS.
- Y. Wada and B. Lönnerdal, J. Agric. Food Chem., 2014, 62, 4175–4185 CrossRef CAS PubMed.
- Y. D. Livney, Curr. Opin. Colloid Interface Sci., 2010, 15, 73–83 CrossRef CAS.
- V. L. Bodiga, S. R. Eda, V. D. Veduruvalasa, L. D. Mididodla, P. K. Parise, S. Kodamanchili, S. Jallepalli, S. P. Inapurapu, M. Neerukonda and P. K. Vemuri, Int. J. Biol. Macromol., 2013, 56, 41–48 CrossRef CAS PubMed.
- B. Egelandsdal, L. P. Ren, P. Kathirvel, Y. S. Gong, M. L. Greaser and M. P. Richards, LWT--Food Sci. Technol., 2011, 44, 1005–1011 CrossRef CAS.
- H. J. Kim, S. J. Choi, W. S. Shin and T. W. Moon, J. Agric. Food Chem., 2003, 51, 1049–1056 CrossRef CAS PubMed.
- J. Liu, Z. Tu, H. Wang, L. Zhang, X. Sha and Y. Shao, Int. J. Food Eng., 2017, 13, 1–11 CAS.
- J. Uribarri, S. Woodruff, S. Goodman, W. Cai, X. Chen, R. Pyzik, A. Yong, G. E. Striker and H. Vlassara, J. Am. Diet. Assoc., 2010, 110, 911–916 CrossRef PubMed .e12.
- M. Corzo-Martínez, A. C. Soria, J. Belloque, M. Villamiel and F. J. Moreno, Int. Dairy J., 2010, 20, 742–752 CrossRef.
- D. Zhao, L. Li, T. T. Le, L. B. Larsen, G. Su, Y. Liang and B. Li, J. Agric. Food Chem., 2017, 65, 5778–5788 CrossRef CAS PubMed.
- W. S. Veraverbeke, O. R. Larroque, F. Bekes and J. A. Delcour, Cereal Chem., 2007, 77, 582–588 CrossRef.
- M. Lima, S. H. Assar and J. M. Ames, J. Agric. Food Chem., 2010, 58, 1954–1958 CrossRef CAS PubMed.
- L. Li, L. Han, Q. Fu, Y. Li, Z. Liang, J. Su and B. Li, Molecules, 2012, 17, 12758–12770 CrossRef CAS PubMed.
- P. Rondeau, G. Navarra, F. Cacciabaudo, M. Leone, E. Bourdon and V. Militello, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 789–798 CrossRef CAS PubMed.
- M. Da Silva Pinto, S. Bouhallab, A. F. De Carvalho, G. Henry, J. L. Putaux and J. Leonil, J. Agric. Food Chem., 2012, 60, 214–219 CrossRef CAS PubMed.
- G. Liu and Q. Zhong, J. Agric. Food Chem., 2012, 60, 9754–9762 CrossRef CAS PubMed.
- B. Bouma, L. M. J. Kroon-Batenburg, Y. P. Wu, B. Brünjes, G. Posthuma, O. Kranenburg, P. G. De Groot, E. E. Voest and M. F. B. G. Gebbink, J. Biol. Chem., 2003, 278, 41810–41819 CrossRef CAS PubMed.
- V. Receveur-Bréhot, J. M. Bourhis, V. N. Uversky, B. Canard and S. Longhi, Proteins: Struct., Funct., Genet., 2006, 62, 24–45 CrossRef PubMed.
- C. Moitzi, L. Donato, C. Schmitt, L. Bovetto, G. Gillies and A. Stradner, Food Hydrocolloids, 2011, 25, 1766–1774 CrossRef CAS.
- S. Chattopadhyay, D. Erdemir, J. M. B. Evans, J. Ilavsky, H. Amenitsch, C. U. Segre and A. S. Myerson, Cryst. Growth Des., 2005, 5, 523–527 CrossRef CAS.
- S. Neves and C. P. Fonseca, Electrochem. Commun., 2001, 3, 36–43 CrossRef CAS.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef CAS PubMed.
- S. R. Thorpe and J. W. Baynes, Amino Acids, 2003, 25, 275–281 CrossRef CAS PubMed.
- A. Naudí, M. Jové, D. Cacabelos, V. Ayala, R. Cabre, P. Caro, J. Gomez, M. Portero-Otín, G. Barja and R. Pamplona, Amino Acids, 2013, 44, 361–371 CrossRef PubMed.
- J. S. Fruton, Acc. Chem. Res., 1974, 7, 241–246 CrossRef CAS.
- W. A. Mckay and S. D. Memmott, Food Addit. Contam., 1991, 8(6), 781–786 CrossRef CAS PubMed.
- Y. Qiao, X. Lin, J. Odle, A. Whittaker and T. A. T. G. Van Kempen, J. Anim. Sci., 2004, 82, 1669–1677 CrossRef CAS PubMed.
- M. Hellwig, S. Geissler, R. Matthes, A. Peto, C. Silow, M. Brandsch and T. Henle, ChemBioChem, 2011, 12, 1270–1279 CrossRef CAS PubMed.
- B. Sheng, L. B. Larsen, T. T. Le and D. Zhao, Molecules, 2018, 23(4), 712 CrossRef PubMed.
- A. M. Moscovici, Y. Joubran, V. Briard-Bion, A. Mackie, D. Dupon and U. Lesmes, Food Funct., 2014, 5, 1898–1908 RSC.
- S. Homma and M. Fujimaki, Growth response of rats fed a diet containing non-dialyzable melanoidin, 1981, vol. 5 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |