
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of the solvent and the size of the nanotube in the non-covalent dispersion of carbon nanotubes with short organic oligomers – a DFT study†
Ahmad I. Alrawashdeh and
Jolanta B. Lagowski*
and
Jolanta B. Lagowski*
Department of Physics and Physical Oceanography, Memorial University of Newfoundland, St. John's, NL, Canada A1B 3X7. E-mail: jolantal@mun.ca
First published on 29th August 2018
Abstract
Among different dispersants of single-walled carbon nanotubes (SWCNTs), conjugated organic oligomers have the ability to interact strongly with SWCNTs and allow for effective dispersion in several organic solvents. Recently, we have carried out two computational investigations on the intermolecular interactions between conjugated organic oligomers and SWCNTs in order to gain insight into an important process of the non-covalent dispersion of carbon nanotubes with short oligomers. These studies highlighted the fact that two additional factors, namely, the effects of the solvent and the carbon nanotube's size on these interactions need further investigation. In this work, with the help of model compounds (which are molecular fragments of the short oligomers used in our previous investigations), we analyze the significance of these two factors. We employ three dispersion corrected density functional theory (D-DFT) approximations (B97D, wB97XD, and B3LYP-D3) to assess the effect of the DFT method, and two basis sets (6-31G(d) and 6-31++G(d,p)) to assess the importance of using a higher basis set in our computations. The main focus of this work is to assess the effect of solvation and nanotube's size on the structure, electronic properties, and binding energies of the respective pairs of model compounds and segments of carbon nanotubes. No significant differences are found between the results of (6,5) and (8,7) SWCNTs in either the geometrical parameters of interacting oligomers or the general tendency of wrapping of their long side chains (SCs) around the nanotubes. However, we find that the binding energies per atom between nanotubes and model compounds are larger for nanotubes with the smaller diameter. The results of electronic properties also show that all model compounds interact more strongly with the (6,5) SWCNT than with the (8,7) SWCNT. Polar solvents such as chloroform show lower binding energies relative to those obtained without a solvent or with non-polar solvents such as hexane. It appears that the presence of a solvent weakens the oligomer/nanotube interactions and, presumably, strengthens the oligomer/solvent and nanotube/solvent interactions to facilitate dispersion of SWCNTs.
1 Introduction
Over the last decades, a vast amount of research activities has been devoted to carbon nanotubes (CNTs) as a consequence of their superb structural, thermal, mechanical, electrical, and optoelectronic properties.1–10 Accordingly, CNTs are deemed to be novel candidates for a wide range of application in materials science,11 optics,12 electronics,13 medicine,14 and biotechnology.15 Unfortunately, the extremely poor solubility of CNTs in all types of solvents limits the applicability of as-produced CNTs in many areas.2 This is due to the large number of π–π and van der Waals interactions that cause as-produced CNTs to assemble and form bundle- or rope-like structures.5 Therefore, dispersion of CNTs is essential for their application and successful utilization of their unique properties.3,9 Covalent and non-covalent functionalization of nanotubes with dispersants are the two commonly used methods to disperse CNTs.3,16,17 The non-covalent method is preferable as it does not alter the sp2 hybridization of carbon atoms in CNTs and retains their unique properties.16,17One feasible way to non-covalently disperse CNTs is to use special polymers.17 It is also found that π-conjugated oligomers can act as efficient dispersants.18,19 Zhao and co-workers showed that phenylene ethynylene and phenylene vinylene oligomers endcapped with dithiafulvenyl (DTF) groups interact strongly with single-walled CNTs (SWCNTs) and act as good dispersants.20,21 They also showed that tetrathiafulvalene vinylogue fluorene co-oligomers interact with SWCNTs but not as strong as phenylene ethynylene and phenylene vinylene oligomers.22 In their work, the dispersion outcomes are highly solvent-dependent as the studied DTF-oligomers effectively disperse SWCNTs and form stable black suspensions in chloroform and less effectively in methylene chloride whereas in chlorobenzene, toluene, and hexane no effective dispersion is obtained.20,22 Furthermore, they found that the DTF group is a decisive factor in the dispersion of SWCNTs with studied π-conjugated oligomers by conducting a comparative study with respective oligomers endcapped with aldehyde (ALD) groups. In contrast to DTF-oligomers, no effective dispersion on SWCNTs was observed when ALD-oligomers were used.20
In our previous work on the interactions of SWCNTs with p-phenylene ethynylene oligomers (OPEs)23 and diphenylene-fluorene oligomers (DPFs),24 we showed that oligomers endcapped with DTF interact more strongly with SWCNTs than respective oligomers endcapped with ALD, therefore, the presence of the electron-donating DTF group in the oligomers is of great importance to SWCNT dispersion. We also pointed out that the oligomers backbones and side chains (SCs) play important roles in the effectiveness of OPEs and DPFs as dispersants for SWCNTs. Furthermore, our findings clearly revealed that DPFs are not as good dispersants of SWCNTs as OPEs. These computational findings agree well with experimental results.20–22
In the experimental work,20–22 the commercially available HiPCO (high-pressure carbon monoxide) SWCNTs with a diameter rage of ∼0.75–1.2 nm were used, and found to be dispersed effectively by oligo(OPE-DTF)s (but less effectively by oligo(DPF-DTF)s). The relatively large molecular sizes of both oligo(OPE-DTF)s and oligo(DPF-DTF)s facilitated the dispersion of HiPCO nanotubes with a range of chiral indices {(6,5), (7,5), (7,6), (8,3), (8,4), (8,6), (8,7), (9,4), (9,5), (10,2) and (10,3)} corresponding to diameter range of 0.75–1.05 nm. It has been found that the dispersion of HiPCO nanotubes by conjugated oligomers resulted in suspension that consisted mainly of nanotubes with (6,5), (7,5), and (7,6) chiral indices (with smaller diameters) mixed with relatively small proportion of some other nanotubes with chirality such as (8,3), (8,4), (8,6), (8,7), (9,4), (9,5), (10,2) and (10,3) (with larger diameters). Among these carbon nanotubes, the (6,5) tube is a significant component as evidenced by vis-NIR data (absorption around 1000 nm, see Fig. 7A and B in ref. 22). Our previous computations,23,24 which involved pairs consisting of an OPE or DPF oligomer and a sizable segments of (6,5) SWCNT (diameter = ∼0.75 nm), are very intensive and take a very long time to carry out even with 6-31-G(d) basis set.
In this study, our intention is to further assess the accuracy of the computations we performed in our previous work and to investigate the effect of solvation and nanotube's size on interactions between OPE and DPF oligomers and SWCNTs. To minimize our computational effort, we have carried out these additional calculations on smaller molecular subsystems (pairs) consisting of fragments of the OPE and DPF oligomers that contain SCs (see Fig. 1) and segments of (6,5) and (8,7) SWCNTs using three dispersion (D)-corrected density functional theory (D-DFT) methods (B97D,25 wB97XD,26 and B3LYP-D3 (ref. 27)) and the hybrid B3LYP method,28,29 and two basis sets (6-31G(d) and 6-31++G(d,p)). Although this work concentrates on studying the effect of solvation and nanotube's size on the interactions between the oligomers and SWCNTs, the effect of D-DFT method and basis set is also briefly investigated. It is clear from our previous works,23,24,30–32 that B97D and B3LYP-D3 methods are the most recommended for investigating properties of organic combinations at the interfacial region since they gave results that were consistent with experimental observations and/or trends. In this work, we have primarily employed the B97D method since it is less computationally intensive than the B3LYP-D3 (or wB97xD). We have also used B3LYP to determine the electronic structure energies of the molecular compounds and combinations as this method often gives a good agreement (better than D-DFT methods) with the corresponding experimental values.33 For the solvation's effect, chloroform and hexane are chosen as solvents since, in the experimental work, SWCNTs are effectively dispersed in chloroform while in hexane no effective dispersion was observed. We have chosen (representative) (6,5) and (8,7) SWCNTs for studing the effect of nanotube's size because, as mentioned before, the (6,5) tube has the smallest diameter (∼0.75 nm) among the mainly dispersed tubes {(6,5), (7,5), and (7,6)} in Zhao and co-workers work,20,22 whereas the (8,7) tube has the largest diameter (∼1.04 nm) among the other nanotubes present in the suspension.
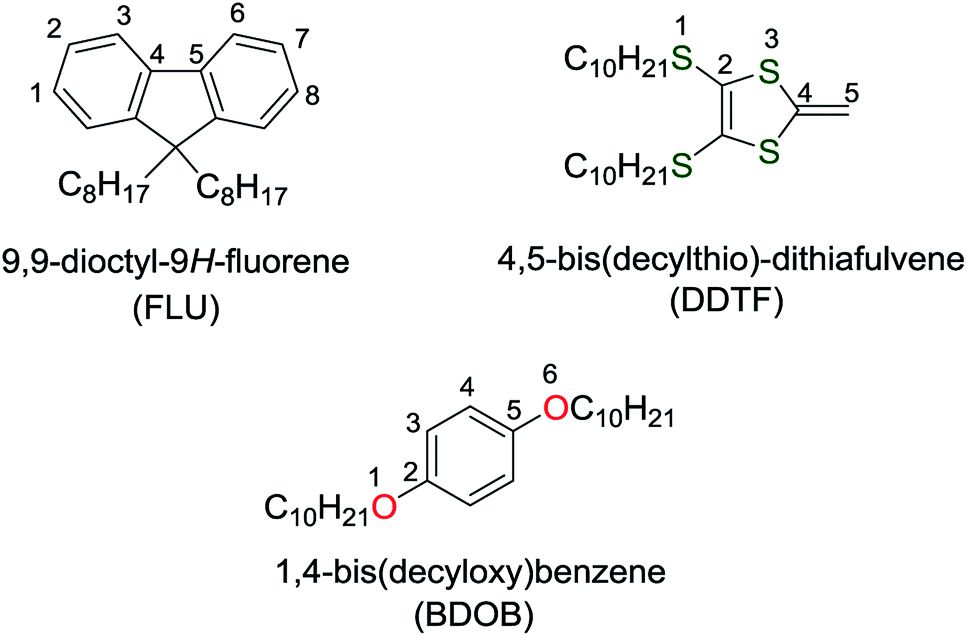 | ||
| Fig. 1 Molecular structures and the numbering of selected atoms for the investigated molecules with three side chains: C8H17, OC10H21, and SC10H21. | ||
2 Computational details
All calculations are performed with Gaussian 16, Revision A.03 software.34 Geometry optimizations for all studies systems (i.e., isolated molecules and interacting molecules with nanotubes) are carried out using three dispersion (D)-corrected DFT (D-DFT) methods: B97D, wB97XD, and B3LYP-D3 and two basis sets: 6-31G(d) and 6-31++G(d,p). The hybrid B3LYP method with the two basis sets is also used to perform geometry optimizations for the three isolated molecules and the two nanotubes. The 6-31++G(d,p) basis set is only used with B97D and B3LYP methods to assess the basis set effect. The effect of solvation is investigated by employing the polarizable continuum model (PCM)35 with chloroform and hexane as solvents.Geometries of all isolated molecular systems are fully optimized using the four DFT methods, while geometries of all interacting systems are partially optimized (by freezing the coordinates of the pre-optimized nanotube's geometries) using B97D, wB97XD, and B3LYP-D3. In addition, for comparison purposes, D-DFT binding energies are compared to those obtained using other post-HF methods such as MP2 (based on Møller–Plesset perturbation theory) by carrying out single-point (SP) computations using spin-component scaled MP2 (SCS-MP2)36 and scaled opposite-spin MP2 (SOS-MP2)37 with the 6-31G(d) basis set. The basis set superposition error (BSSE) is calculated by the counterpoise correction of Boys and Bernardi38 using B97D, wB97XD, and B3LYP-D3 with 6-31G(d) basis set.
For the structural analysis, we select a relevant subset of heavy atoms in each molecule and tabulate the bond lengths (R), bond angles (A) and dihedral angles (Dh). The selected atoms and their numbering are shown in Fig. 1. We perform the structural analysis for all studied systems. In the case of electronic analysis, we calculate electronic properties (i.e., HOMO and LUMO energies) for the isolated molecules only. Avogadro, version: 1.2.0 (ref. 39) is used to generate the initial geometries of (6,5) and (8,7) SWCNTs. GaussView 6 (ref. 40) is used to generate the initial geometries of molecules and molecule/nanotube combinations and to determine R, A, and Dh for all molecular systems.
As in our previous works,23,24 to assess the strength of molecule/nanotube interactions, the binding energies (Eb) between studied molecules and SWCNTs are computed,
| Eb = Etotal − (ESWCNT + Emolecule) | (1) |
| ΔεLA–HD = εLUMO(Acceptor) − εHOMO(Donor). | (2) |
In addition to binding energies, the intermolecular distances (Δd) between the molecular fragments and nanotubes in combinations are calculated by taking the average of the minimum distances between atoms of each molecular fragment and each nanotube. That is, for every atom in the fragment, the minimum distance is found by considering Euclidean distances between the coordinate of that atom and coordinates of all atoms of the nanotube. Then having the minimum distances for every atom in the fragment, the average is computed.
3 Results and discussion
Three molecules which are referred to as FLU, DDTF, and BDOB (shown in Fig. 1) and two segments of (6,5) and (8,7) SWCNTs are considered in this study. These molecules are fragments of the OPE and DPF oligomers used in previous computational23,24 and experimental20,22 works.With regard to the applicability of the present, short model compound study, to the previous larger model23,24 investigations, we make the following comment. In Table 3 in ref. 23, we showed that the binding (interaction) energy per benzene ring for OPEs is in the range of 0.55–0.77 eV depending on the end group of the given oligomer. Hence the binding energy scales well with the length of oligomer for the oligomers without side chains. When SCs are present, things are more complicated because the SCs contribution varies depending on their orientations relative to the nanotube. However, the analysis of the data in Table 3 (ref. 23) illustrates that the addition of two SCs adds a bit more than 1 eV to the total binding energy of the combination. These findings are consistent with our current computational results for BDOB (fragment of OPE) which consists of one ring and two SCs, its estimated binding energy is approximately 1.6 eV which is in good agreement with 1.69 eV as found in this work (see below). Based on this example, we believe that the interaction energies scale relatively predictably with the size of the model compounds. Since, in our current paper, we consistently focus on the smallest units of the oligomers in question, we believe that our results/comparisons are applicable to the full length oligomers.
The optimized structures of isolated studied molecules and the segmental nanotubes as obtained using B97D/6-31G(d) in the gas phase are shown in Fig. 2. The partially optimized geometries (front views) of interacting FLU, DDTF, and BDOB molecules with (6,5) and (8,7) SWCNTs obtained using B97D/6-31G(d) are shown in Fig. 3. For completeness, three different views (side, front, top) of these systems are given in Fig. S1–S3 in ESI.† Below we discuss effects of the DFT method, basis set, solvation, and nanotube's size on the structural parameters (R, A, and Dh), electronic properties (i.e., HOMO and LUMO eigenvalues), and binding energies of the studied systems.
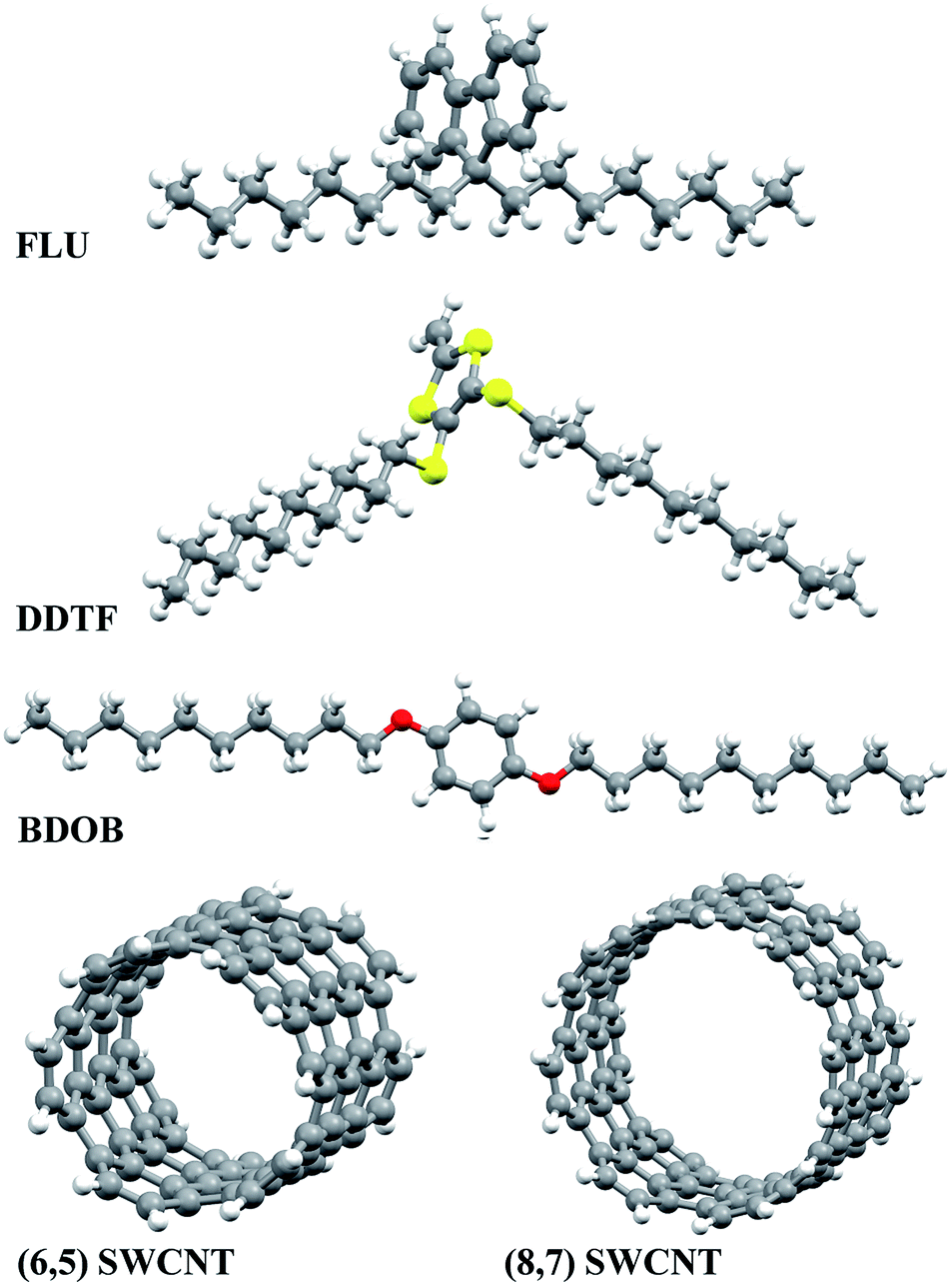 | ||
| Fig. 2 Optimized structures of isolated molecular fragments and segments of SWCNTs obtained using B97D/6-31G(d) in the gas phase (similar results are obtained with B97D/6-31++G(d,p) and other D-DFT methods with 6-31G(d) basis set). | ||
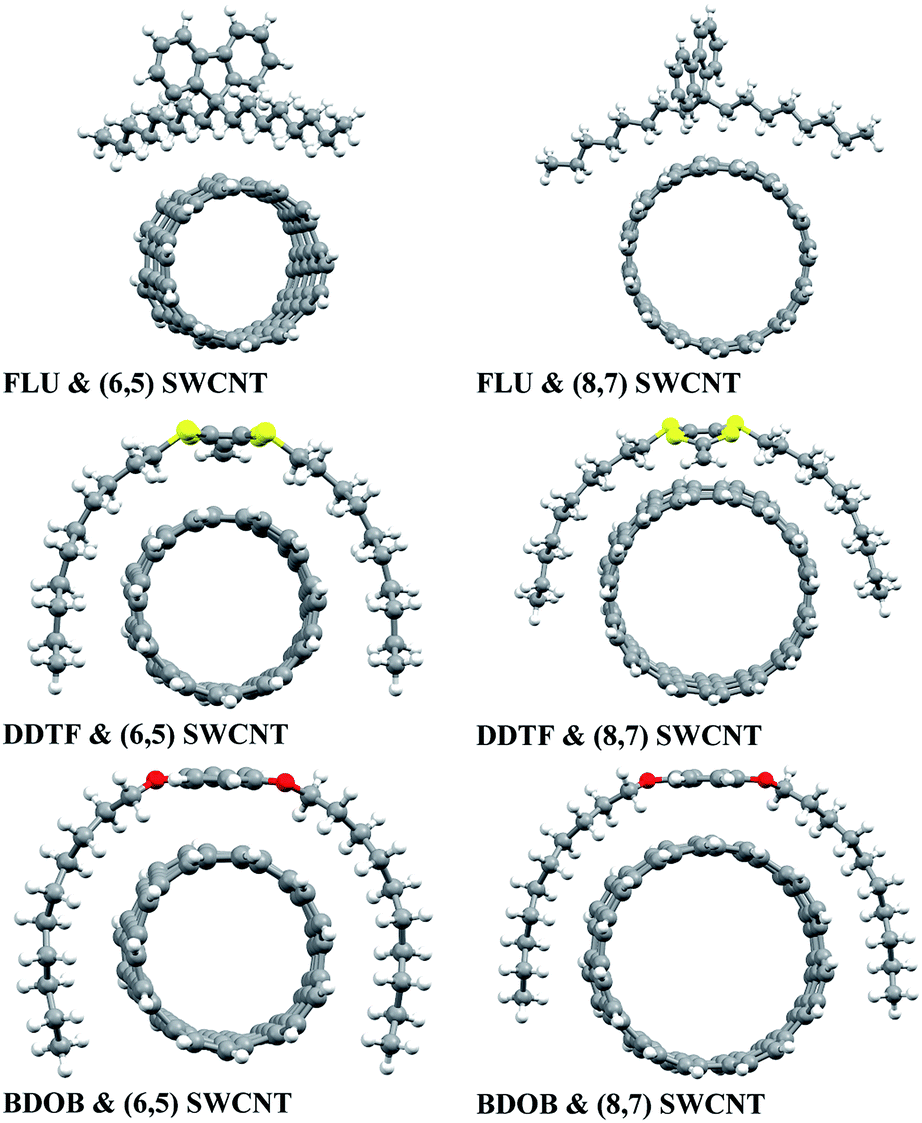 | ||
| Fig. 3 Partially optimized structures (front view) of molecules interacting with (6,5) and (8,7) SWCNTs obtained using B97D/6-31G(d) (similar results are obtained with B97D/6-31++G(d,p) and other D-DFT methods with 6-31G(d) basis set). | ||
3.1 DFT method effect
Three D-DFT methods: B97D, wB97XD, and B3LYP-D3 are employed to fully optimize geometries of the isolated molecules and to partially optimize the geometries of interacting studied systems using 6-31G(d) basis set. In all interacting systems with all D-DFT methods, SCs in each molecule wrap themselves around the nanotubes and this wrapping is stronger in both DDTF and BDOB than it in the FLU (Fig. 3 and S1–S3 in ESI†).Selected structural parameters, for the isolated molecules and for the molecules interacting with (6,5) and (8,7) SWCNTs, as obtained using B97D, wB97XD, and B3LYP-D3 with 6-31G(d) basis set are given in Tables S1–S6 and Fig. S4–S6 in ESI.† For easier comparison, their respective average values are displayed in Fig. 4. In all cases, B97D gives the longest R values followed by B3LYP-D3, while wB97XD gives the shortest R values, see Fig. 4(a) and S4(a)–S6(a).† The maximum differences between the three methods are less than 0.010 Å for the R values. Similarly, for bond and dihedral angles in all systems, the D-DFT methods give very close results which differ by less than 1.2°, see Fig. 4(b and c) and S4(b and c)–S6(b and c).† Hence, the three D-DFT methods give very similar optimized geometries for all isolated and interacting molecular fragments.
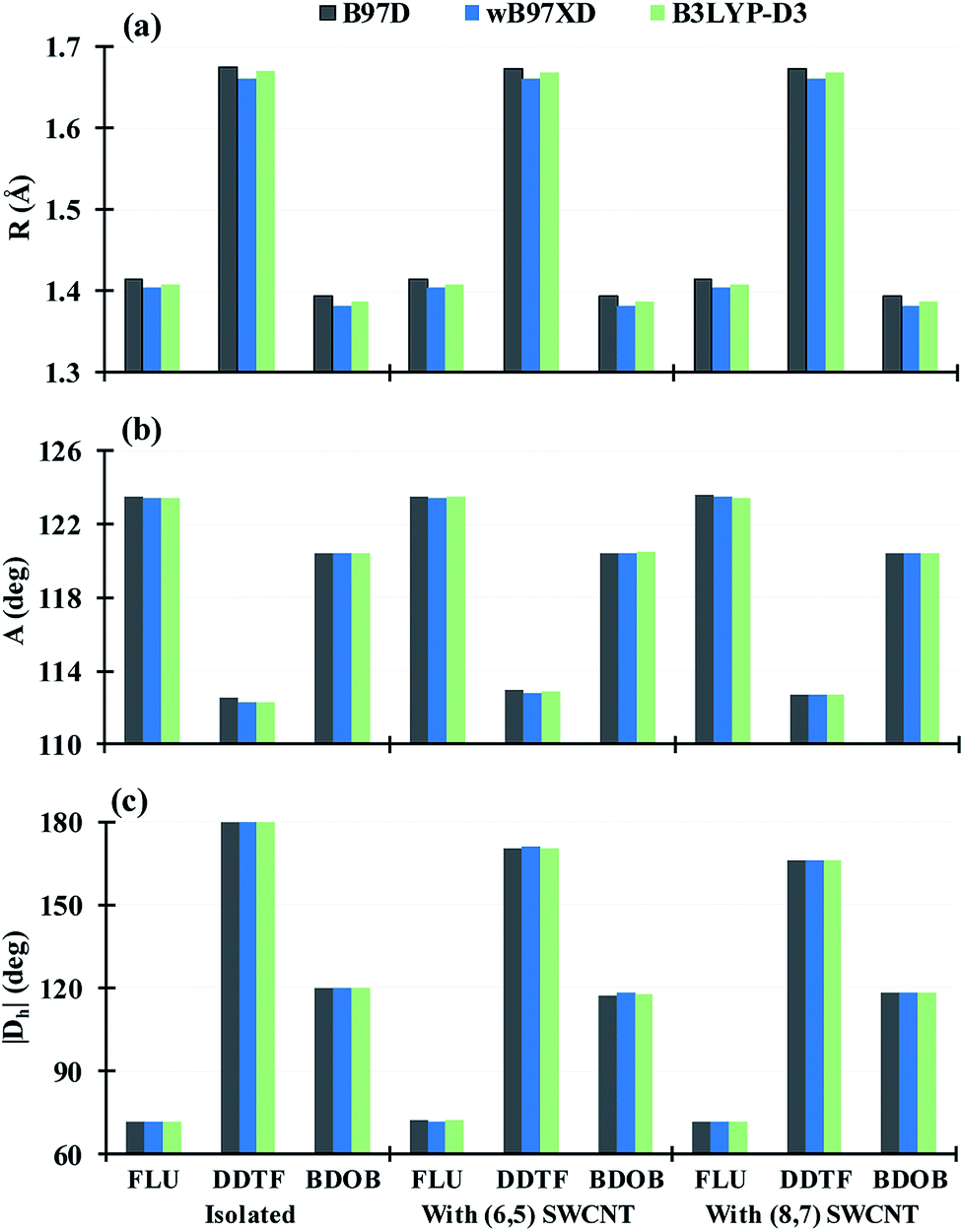 | ||
| Fig. 4 Average of (a) bond lengths (R) (in Å), (b) bond angles (A), and (c) dihedral angles (|Dh|) (in degrees) for the isolated molecules and for the molecules interacting with (6,5) and (8,7) SWCNTs obtained using B97D, wB97XD, and B3LYP-D3 with 6-31G(d) basis set. | ||
Magnitudes of total binding energies, Eb (in eV), of the molecular fragments interacting with (6,5) and (8,7) SWCNTs obtained using B97D, wB97XD, and B3LYP-D3 with 6-31G(d) basis set are shown in Fig. 5 (see also Table 1). As can be seen from this figure, all three methods give larger total binding energies for FLU, DDTF, and BDOB when interacting with the (8,7) SWCNT than with the (6,5) SWCNT. Fig. 5 also shows that the highest binding energies with (6,5) and (8,7) SWCNTs are obtained for DDTF with all three methods followed by BDOB and then FLU. Since the SCs of FLU do not strongly wrap around nanotubes, FLU has very small binding energies relative to DDTF and BDOB. For any given molecule, trends in binding energies are the same for the three methods with B97D giving the largest Eb values followed by wB97XD and B3LYP-D3.
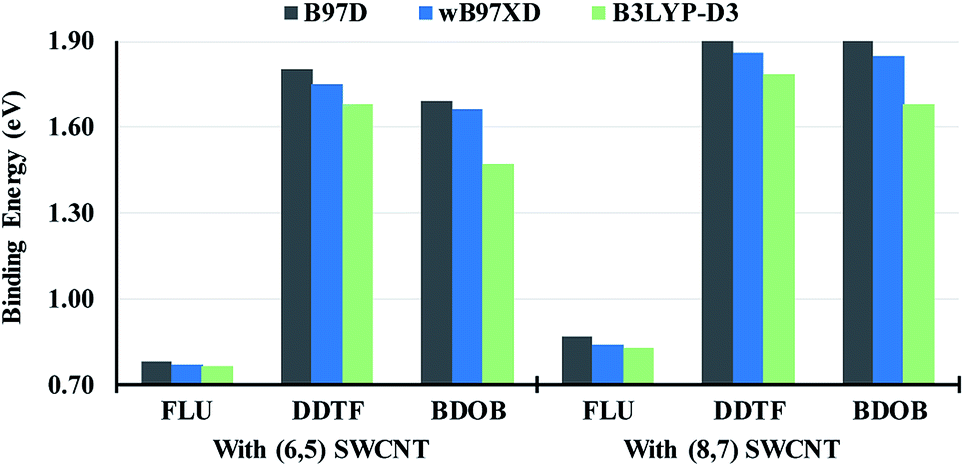 | ||
| Fig. 5 Absolute values of total binding energies, Eb (in eV), between the molecular fragments and (6,5) and (8,7) SWCNTs obtained using B97D, wB97XD, and B3LYP-D3 with 6-31G(d) basis set. | ||
| With (6,5) SWCNT | With (8,7) SWCNT | |||||
|---|---|---|---|---|---|---|
| FLU | DDTF | BDOB | FLU | DDTF | BDOB | |
| a Values in brackets are with BSSE corrections. | ||||||
| B97D | 0.78 (0.59) | 1.80 (1.63) | 1.69 (1.66) | 0.86 (0.76) | 1.90 (1.75) | 1.90 (1.75) |
| wB97XD | 0.77 (0.66) | 1.75 (1.78) | 1.66 (1.76) | 0.84 (0.81) | 1.86 (1.88) | 1.85 (1.85) |
| B3LYP-D3 | 0.76 (0.60) | 1.68 (1.49) | 1.47 (1.47) | 0.89 (0.81) | 1.78 (1.69) | 1.68 (1.58) |
| SCS-MP2 | 0.61 | 1.36 | 1.27 | 0.66 | 1.37 | 1.35 |
| SOS-MP2 | 0.62 | 1.40 | 1.31 | 0.68 | 1.41 | 1.39 |
We have computed BSSE corrections to interaction energies for the three D-DFT functionals. These BSSE corrected results as well as the results for the three D-DFT functionals without BSSE corrections are given in Table 1. With BSSE corrections, the binding energies in most cases are lowered by 0 to 0.2 eV (except in two cases involving wB97XD, the BSSE corrections slightly increase the binding energies). Most BSSE corrections are close to 0.1 eV. These results do not change our main conclusion that DDTF and BDOB interact more strongly with nanotubes than FLU and that the total binding energy is larger for (8,7) than for (6,5) SWCNT.
Also in Table 1, we have included the interaction energies obtained using SCS-MP2 and SOS-MP2 methods. For SCS-MP2 and SOS-MP2 methods, the results illustrate that the total binding energies are lowered by 0.2 to 0.4 eV indicating that both MP2 approaches include fewer intermolecular interactions than D-DFT methods. Similar to BSSE corrections, these MP2 results do not change our main conclusion that DDTF and BDOB interact more strongly with nanotubes than FLU and that the total binding energy is larger for (8,7) than for (6,5) SWCNT. However, because fewer intermolecular interactions are included in MP2, the differences between the various total binding energies are considerably smaller for the MP2 than for the D-DFT methods (for example, for DDTF the differences between the binding energies corresponding to (8,7) and (6,5) SWCNTs are 0.01 eV for the MP2 in comparison to 0.1 eV for D-DFT methods).
HOMO and LUMO eigenvalues of isolated FLU, DDTF, and BDOB obtained using B97D, wB97XD, B3LYP-D3, and B3LYP with 6-31G(d) basis set in the gas phase are depicted in Fig. 6. It is known that B3LYP produces results for HOMO and LUMO energies and their gaps in good agreement with experimental values.33 As can be seen from Fig. 6, B3LYP-D3 gives nearly the same HOMO and LUMO energies of the three isolated molecules as B3LYP since the dispersive forces do not change their geometries and electronic energies significantly when the same main functional is used in both computations. Even in the case where we consider the effect of the dispersive forces more directly by computing (SP) B3LYP//B3LYP-D3/6-31G(d) HOMO and LUMO eigenvalues of the interacting model compounds and comparing them with the B3LYP HOMO and LUMO eigenvalues of the isolated compounds, we find small changes in their respective values (see Tables S16 and S17†). Tables S16 and S17† show that DDFT (fragment that contains the DTF end group) is affected the most by the dispersive intermolecular interactions (HOMOs are changed by approximately 0.2 eV and LUMOs by approximately 0.4 eV). In contrast to B3LYP-D3, the results of B97D and wB97XD are notably different from B3LYP results. In particular, as shown in Fig. 6(a), B97D lowers LUMO energies for all molecules by ∼0.6 eV whereas wB97XD elevates these energies by ∼1.8 eV compared to B3LYP values. For HOMO energies as seen in Fig. 6(b), the opposite trend is observed for all molecules. As a result, B97D underestimates HOMO–LUMO gaps for all molecules by ∼1.5 eV whereas wB97XD overestimates these gaps by ∼3.8 eV compared to the B3LYP (or B3LYP-D3) results. These differences between HOMO and LUMO eigenvalues are primarily due to differences between the DFT functionals and are not due to the inclusion of dispersive forces (since as noted above the dispersion does not affect the geometry of the compounds significantly). These above observations regarding binding energies and electronic structures are consistent with our previous findings.23,24 Hence, as mentioned in the introduction, for the computational expediency sake, we will only use the B97D method to study structural parameters and binding energies of the model compound/nanotube combinations and B3LYP method to analyze their electronic properties for the remainder of this work.
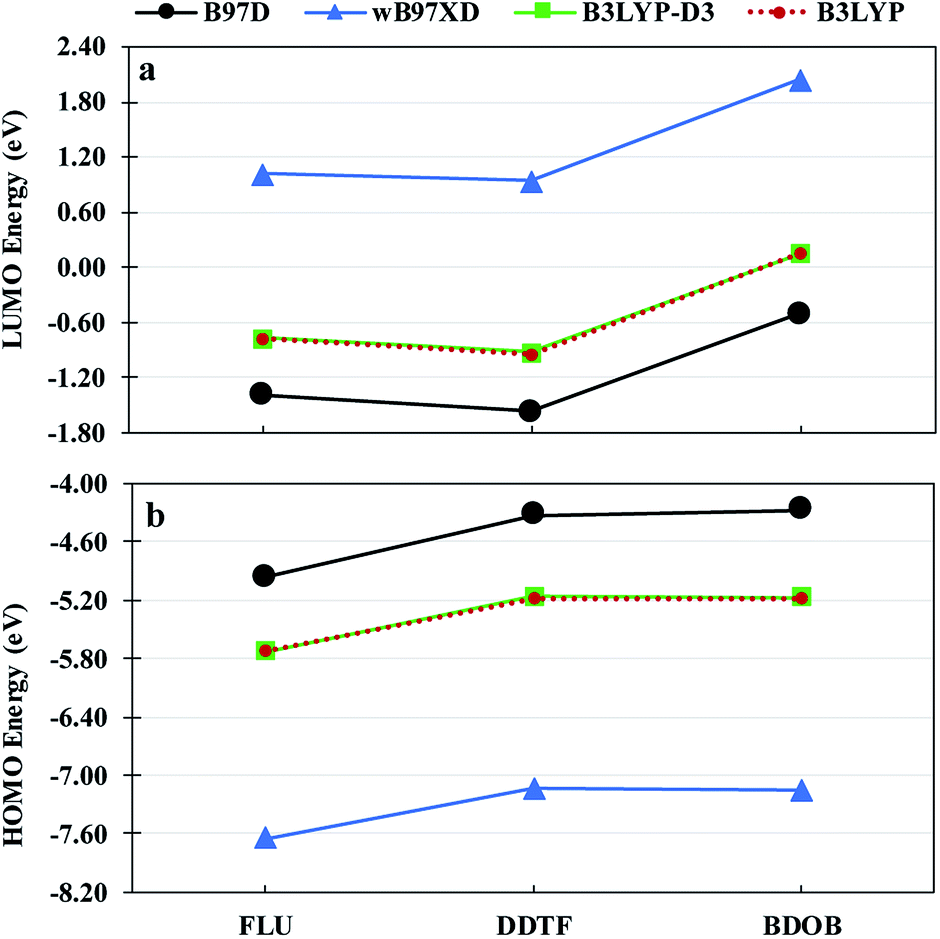 | ||
| Fig. 6 LUMO energies (a) and HOMO energies (b) for isolated FLU, DDTF, and BDOB obtained using B97D, wB97XD, B3LYP-D3, and B3LYP with 6-31G(d) basis set in the gas phase. | ||
3.2 Basis set effect
In order to investigate the effect of basis set on the studied systems, we perform calculations using B97D and B3LYP methods with both 6-31G(d) and 6-31++G(d,p) basis sets and compared their respective structural and electronic parameters and binding energies. In all cases of interacting molecule/nanotube combinations with both basis sets, the wrapping of SCs of the FLU around the two nanotubes is not as strong as the wrapping of SCs of DDTF and BDOB as shown in Fig. 3 and S1–S3 in ESI.† To assess the effect of basis set on the geometries of isolated and interacting molecular systems more accurately, their selected structural parameters (R, A, and Dh) (given in Tables S1–S3 in ESI†) are examined. As before, the labelling of atoms is shown in Fig. 1. The absolute differences between the respective structural parameters obtained using B97D/6-31G(d) and those using B97D/6-31++G(d,p) for the isolated and interacting molecules are listed in Tables S7–S9 in ESI.†It can be seen from Tables S7–S9 (and Tables S1–S3†) that both basis sets give results that are very close for all structural parameters (R, A, and Dh). The maximum absolute value differences between structural parameters obtained with B97D/6-31G(d) and those with B97D/6-31++G(d,p) are 0.005 Å, 0.5°, and 2.1° for R, A, and Dh, respectively. Based on these results, we conclude that both basis sets give very similar optimized geometries for all isolated studied molecules and SWCNTs (see Fig. 2) and for all molecule/SWCNT combinations (see Fig. 3 and S1–S3 in ESI†).
Table 2 shows absolute values of total binding energies, Eb (in eV), of the molecules with both (6,5) and (8,7) SWCNTs obtained using B97D with 6-31G(d) and 6-31++G(d,p) basis sets. For all interacting systems except for FLU/(6,5) SWCNT, the binding energies obtained with the 6-31++G(d,p) basis set are smaller than those obtained with the 6-31G(d) basis set. The differences between the respective binding energy's magnitudes are of the order of 10%. For a given nanotube, the trends in binding energies are the same for both basis sets, with DDTF interacting most strongly with nanotubes, followed by BDOB, then FLU.
| Molecule | 6-31G(d) | 6-31++G(d,p) | ||
|---|---|---|---|---|
| (6,5) NT | (8,7) NT | (6,5) NT | (8,7) NT | |
| FLU | 0.78 | 0.86 | 0.83 | 0.80 |
| DDTE | 1.80 | 1.90 | 1.58 | 1.69 |
| BDOB | 1.69 | 1.90 | 1.50 | 1.72 |
Fig. 7 shows HOMO and LUMO energies for isolated FLU, DDTF, and BDOB obtained using B3LYP with 6-31G(d) and 6-31++G(d,p) basis sets. As in the case of binding energies, the 6-31++G(d,p) basis set gives values for both HOMO and LUMO eigenvalues that are lower relative to those obtained with the 6-31G(d) basis set. The trends in the relative values of HOMO and LUMO energies are the same for both basis sets. Given the above discussion, we will use the 6-31G(d) basis set for the remainder of this work since it is computationally less costly in comparison to the 6-31++G(d,p) basis set.
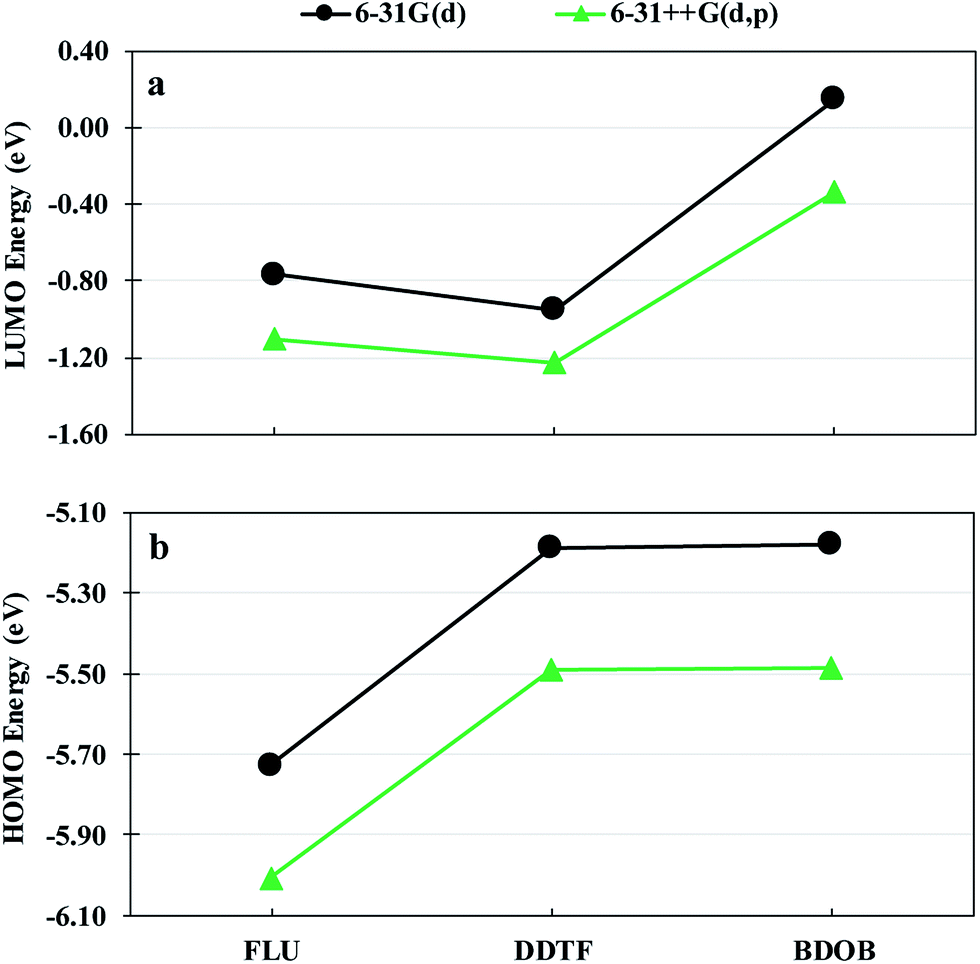 | ||
| Fig. 7 LUMO energies (a) and HOMO energies (b) for isolated FLU, DDTF, and BDOB molecules obtained using B3LYP with 6-31G(d) and 6-31++G(d,p) basis sets in the gas phase. | ||
3.3 Solvation effect
In order to investigate the effect of solvation on structural parameters, HOMO and LUMO eigenvalues, and binding energies for different systems, we optimize or partially optimize all studied systems with B97D/6-31G(d) using PCM with chloroform and hexane as solvents. We then compare the computational results obtained for the two solvents to the respective ones obtained without a solvent. Selected structural parameters for the isolated and interacting molecules with (6,5) and (8,7) SWCNTs obtained using B97D/6-31G(d) in PCM with chloroform and hexane solvents are given in Tables S10–S12 in ESI.† In Tables S13–S15 in ESI,† we list absolute differences between R, A, and Dh obtained using B97D/6-31G(d) without a solvent and those obtained using B97D/6-31G(d) in PCM/chloroform and in PCM/hexane respectively for the isolated molecules and for the molecules interacting with (6,5) and (8,7) SWCNTs. As can be seen from those tables, the effect of solvation on structural parameters is almost negligible since differences between solvated and non-solvated parameters are less than 0.004 Å, 0.4°, and 2.4° for R, A, and Dh, respectively, in all systems. We conclude that the optimized or partially optimized geometries for all system obtained with PCM in both solvents are very similar to those obtained without a solvent.In Fig. 8, we present absolute values of total binding energies, Eb (in eV), between the molecules and (6,5) and (8,7) SWCNTs obtained using B97D/6-31G(d) without a solvent and with chloroform and hexane solvents. Fig. 8 clearly shows that for a given nanotube size, the solvation, with both solvents, reduces the total binding energies (by average of 0.04 and 0.01 eV with chloroform and hexane respectively) for all systems (except for FLU/(8,7) SWCNT in hexane) compared to those obtained without a solvent. This reduction in binding energies is more pronounced in chloroform than in hexane. Hence, in general, the solvent decreases the intermolecular interactions between the molecule and a given nanotube and presumably concurrently strengthens solvent/molecule and solvent/nanotube interactions, and thus facilitating dispersion of SWCNTs with short conjugated oligomers.
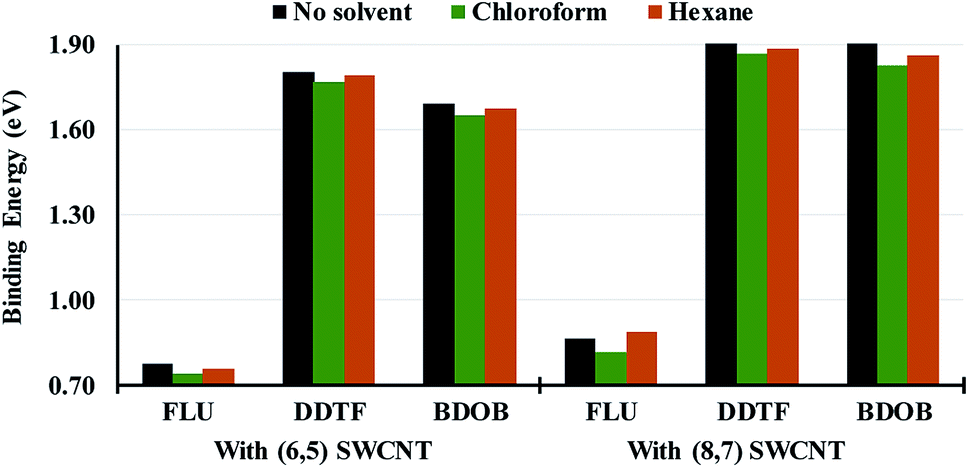 | ||
| Fig. 8 Absolute values of the total binding energies, Eb's (in eV), between the molecular fragments and (6,5) and (8,7) SWCNTs obtained using B97D/6-31G(d) without a solvent and with chloroform and hexane solvents. | ||
Fig. 9 shows LUMO and HOMO energies for isolated FLU, DDTF, and BDOB molecules obtained using B3LYP/6-31G(d) without a solvent and with chloroform and hexane solvents. As in the case of binding energies, both solvents lower LUMO and HOMO energies for all three molecules. Chloroform gives lowest eigenvalues followed by hexane and then the non-solvated phase.
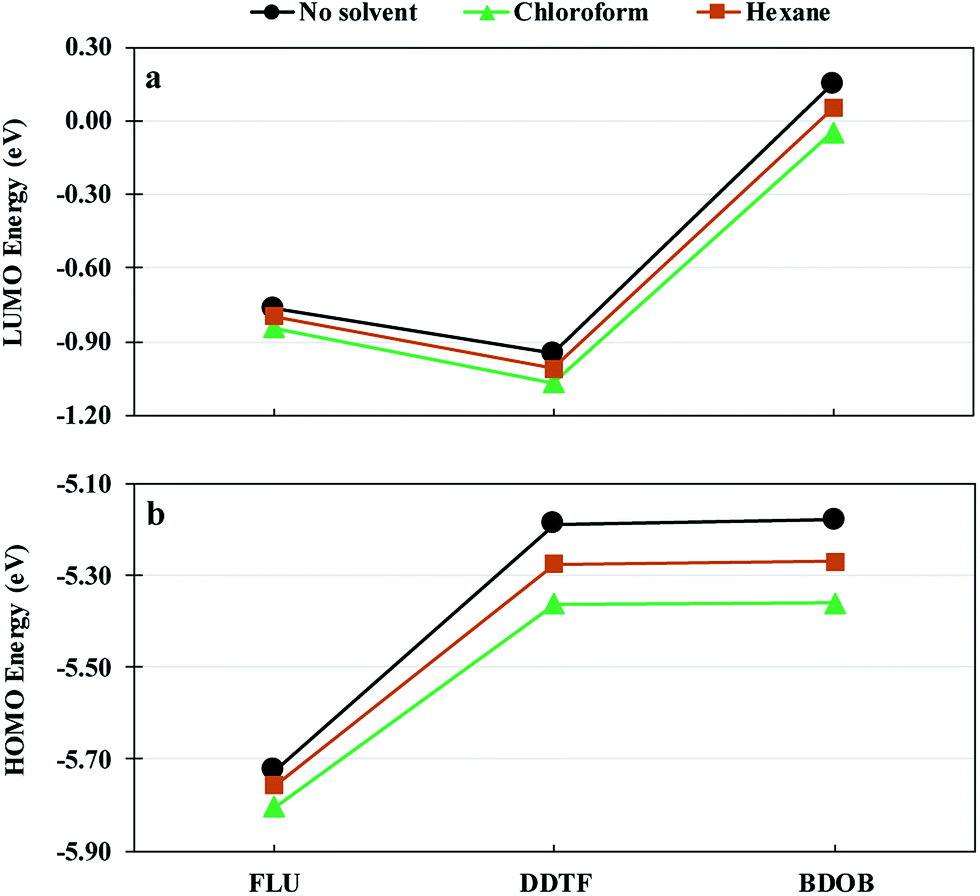 | ||
| Fig. 9 LUMO (a) and HOMO (b) energies for isolated FLU, DDTF, and BDOB obtained using B3LYP/6-31G(d) without a solvent and with chloroform and hexane solvents. | ||
Table 3 gives the ΔεLA–HD values between the εLUMO of (isolated) (6,5) and (8,7) SWCNTs and the εHOMO of isolated FLU, DDTF, and BDOB molecules obtained using B97D/6-31G(d) without a solvent and with chloroform and hexane solvents. The values of εHOMO and εLUMO that are used in calculating the ΔεLA–HD's (as given in Table 3) are listed in Table S16 in ESI.† As can be seen from Table 3, the ΔεLA–HD values for FLU with each SWCNTs without a solvent, with chloroform, and with hexane are very close to each other, whereas ΔεLA–HD values for DDTF and BDOB with both SWCNTs in chloroform are larger (indicating weaker interactions) by approximately 0.1 eV and in hexane by approximately 0.05 eV than those obtained without a solvent. These results of ΔεLA–HD's, for a given nanotube, support the above binding energy findings (see Fig. 8) in which the molecules give the lowest binding energies (again indicating weaker interactions) with SWCNT in chloroform, followed by hexane and then the non-solvated phase.
| (6,5) SWCNT | (8,7) SWCNT | |||||
|---|---|---|---|---|---|---|
| FLU | DDTF | BDOB | FLU | DDTF | BDOB | |
| No solvent | 3.17 | 2.64 | 2.63 | 3.44 | 2.90 | 2.90 |
| Chloroform | 3.18 | 2.74 | 2.74 | 3.44 | 3.00 | 3.00 |
| Hexane | 3.18 | 2.70 | 2.69 | 3.45 | 2.96 | 2.96 |
In addition, the above findings are also consistent with experimental observations.20,22 As we mentioned in the introduction, experimentally the HiPCO nanotubes were dispersed more effectively by oligo(OPE-DTF)s than by oligo(DPF-DTF)s. It is clear from Table 3 that ΔεLA–HD values for combinations of SWCNT and FLU (a fragment from oligo(DPF-DTF)s but not oligo(OPE-DTF)) do not change indicating that FLU-SWCNT intermolecular interactions are not affected by the presence of a solvent. Hence, weaker dispersion of SWCNTs was experimentally observed when oligo(DPF-DTF)s (that contained FLU) were used. In contrast in cases of DDTF and BDOB (fragments of oligo(OPE-DTF)s), the presence of solvents increases ΔεLA–HD values of both DDTF and BDOB relative to their non-solvated phase values. Therefore, the presence of a solvent weakens the oligomer/nanotube interactions (and presumably strengthens the oligomer/solvent and nanotube/solvent interactions) to facilitate dispersion of SWCNTs. This effect is more pronounced in chloroform than in hexane and this explains why the HiPCO nanotubes were dispersed effectively by oligo(OPE-DTF)s in chloroform but not in hexane.
3.4 Nanotube's size effect
To study the effect of the nanotube's size on structures and binding energies of the interacting systems, we consider two fragments of (6,5) and (8,7) chiral SWCNTs that have respective diameters of 0.75 and 1.04 nm. The (6,5) SWCNT consists of 92 carbon atoms and is terminated with 22 hydrogen atoms, whereas the (8,7) SWCNT consists of 106 carbon atoms and is terminated with 30 hydrogen atoms. The optimized structures of both nanotubes are given in Fig. 2.Each of the molecules interacts with both nanotubes in a very similar way as shown in Fig. 3 and S1–S3 in ESI.† In all cases the wrapping of SCs of the molecules around both nanotubes is observed (FLU shows the weakest wrapping). As shown in Fig. 4(a, b), Tables S1–S6 and S10–S12 in ESI,† both nanotubes give very similar results for the bond lengths (differ by <0.003 Å) and bond angles (differ by <0.7°) for all systems with all DTF methods and basis sets. For dihedral angles, with few exceptions (DDTF with wB97XD, B3LYP-D3, PCM/chloroform, and PCM/hexane, in which the largest difference is 5.3° with wB97XD) the two nanotubes also give very similar results (differ by <1.5°) for most studied systems with all DTF methods and basis sets, see Fig. 4(c), Tables S1–S6 and S10–S12 in ESI.†
As commented above, Tables 1 and 2, Fig. 5 and 8 show that the combinations with (8,7) SWCNT display slightly larger binding energies than with (6,5) SWCNT for all molecules with all D-DFT methods and basis sets (one exception is the FLU with B97D/6-31++G(d,p)). The differences between the respective binding energy's magnitudes are of the order of 10% with all D-DFT methods and basis sets. Experimentally,19,21 as we previously mentioned, the (6,5) nanotube exhibits much better dispersion by conjugated oligomers than the (8,7) nanotube. This implies that the interactions and hence binding energies of conjugated oligomers with (6,5) SWCNTs are stronger than those with (8,7) SWCNTs. In order to normalize our Eb results, we calculate binding energies per atom (Eb per atom) for all combinations. The absolute values of Eb per atom between the molecular fragments and (6,5) and (8,7) SWCNTs obtained using B97D/6-31G(d) are displayed in Table 4. As expected, based on the experimental observations, the values of Eb per atom for molecular combinations containing (6,5) SWCNT are higher than those containing (8,7) SWCNT (see Table 4). Furthermore, the energy differences (ΔεLA–HD) between the LUMO energy of the acceptor (nanotube) and the HOMO energy of the donor (oligomer) for all molecules with the (6,5) SWCNT are smaller (by 0.2–0.3 eV) than those with the (8,7) SWCNT in the non-solvated phase and in chloroform and hexane as shown in Table 3. The ΔεLA–HD results are consistent with the Eb per atom results showing that molecular fragments interact more strongly with the (6,5) SWCNTs than with the (8,7) SWCNTs, and hence the (6,5) SWCNT is dispersed more easily by conjugated oligomers than the (8,7) SWCNT.
| Molecule | With (6,5) SWCNT | With (7,6) SWCNT | With (8,7) SWCNT |
|---|---|---|---|
| a Values in brackets are for Δd. | |||
| FLU | 0.039 (4.87) | 0.038 (4.55) | 0.036 (4.86) |
| DDTE | 0.044 (3.61) | 0.040 (3.57) | 0.039 (3.57) |
| BDOB | 0.055 (3.59) | 0.053 (3.59) | 0.045 (3.58) |
To further test the above conclusions, we have carried out additional computations for Eb, Eb per atom, and Δd using a representative segment of the chiral (7,6) SWCNT. The (7,6) SWCNT segment consists of 96 carbon atoms and is terminated with 26 hydrogen atoms with an intermediate diameter of 0.87 Å. The binding energies with (7,6) SWCNT are 0.95, 1.83, 1.79 Å for FLU, DDTF, and BDOB respectively obtained using B97D/6-31G(d). These Eb values fall between those for (6,5) and (8,7) nanotubes with DDTF and BDOB but with FLU, Eb is larger than those for (6,5) and (8,7) nanotubes (see Table 2). Table 4 shows Eb per atom and Δd values obtained using B97D/6-31G(d) for the three nanotubes. As can be seen from Table 4, the Eb per atom values for (7,6) SWCNT fall between those for (6,5) and (8,7) nanotubes which is consistent with experimental observations. From the Eb values of the studied molecules with the three nanotubes (as given in Table 1 and above), it can be seen that the Eb is inversely proportional to the intermolecular distance (Δd) between the oligomer and the nanotube (see Table 4) in all cases considered.
4 Conclusions
We have investigated the interactions between conjugated organic oligomers and SWCNTs to gain insight into roles of the solvent and nanotube's size in the non-covalent dispersion of carbon nanotubes with these short oligomers. Three D-DFT methods (B97D, wB97XD, and B3LYP-D3) and two basis sets (6-31G(d) and 6-31++G(d,p)) are used to assess the effect of the DFT method and the basis set in our computations. We consider three molecules (FLU, DDTF, and BDOB) that are fragments from the OPE and DPF oligomers used in our previous computational work23,24 as well as in the experimental work20,22 and two segments of (6,5) and (8,7) SWCNTs. The computations are performed without a solvent and with chloroform and hexane as solvents.Our investigation shows that the use of higher basis set such as 6-31++G(d,p) in our D-DFT calculations does not change significantly either the geometrical parameters of oligomers or the binding energies of the combinations in comparison to the corresponding findings obtained with the 6-31G(d) basis set. The three D-DFT methods give comparable results for the geometrical parameters of oligomers and the binding energies of the combinations, whereas B3LYP-D3 gives the best results for the electronic properties of oligomers and SWCNTs. Our findings indicate that the use of larger diameter (8,7) SWCNT does not significantly change either the geometrical parameters of oligomers or the overall behaviour of SCs wrapping around nanotubes in comparison to respective values and behaviours as obtained with the smaller diameter (6,5) SWCNT. However, our results in agreement with experimental findings indicate that all molecular conjugated fragments are interacting more strongly with the (6,5) SWCNTs than with the (8,7) SWCNTs. The presence of solvent weakens the oligomer/nanotube interactions (and strengthens the oligomer/solvent and nanotube/solvent interactions) to facilitate dispersion of SWCNTs, and this effect is more pronounced in chloroform than in hexane in agreement with experimental observations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Atlantic Computational Excellence Network (ACEnet), the Western Canadian Research Grid (WestGrid), and Compute Canada for providing the computational facilities. We are grateful to the Natural Sciences and Engineering Research Council of Canada (NSERC) for financial support.Notes and references
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef.
- J. Hilding, E. Grulke, Z. G. Zhang and F. Lockwood, J. Dispersion Sci. Technol., 2003, 24, 1–41 CrossRef.
- M. Sennett, E. Welsh, J. B. Wright, W. Z. Li, J. G. Wen and Z. F. Ren, Appl. Phys. A: Mater. Sci. Process., 2003, 76, 111–113 CrossRef.
- V. N. Popov, Mater. Sci. Eng., R, 2004, 43, 61–102 CrossRef.
- X. L. Xie, Y. W. Mai and X. P. Zhou, Mater. Sci. Eng., R, 2005, 49, 89–112 CrossRef.
- D. A. Britz and A. N. Khlobystov, Chem. Soc. Rev., 2006, 35, 637–659 RSC.
- J. H. Du, J. Bai and H. M. Cheng, eXPRESS Polym. Lett., 2007, 1, 253–273 CrossRef.
- B. S. Shim, W. Chen, C. Doty, C. Xu and N. A. Kotov, Nano Lett., 2008, 8, 4151–4157 CrossRef PubMed.
- P. C. Ma, N. A. Siddiqui, G. Marom and J. K. Kim, Composites, Part A, 2010, 41, 1345–1367 CrossRef.
- S. Liang, A. V. Subrahmanyam, M. Khadem, Y. Zhao and A. Adronov, RSC Adv., 2016, 6, 25733–25740 RSC.
- C. Duan, K. Zhang, C. Zhong, F. Huang and Y. Cao, Chem. Soc. Rev., 2013, 42, 9071–9104 RSC.
- P. Avouris, M. Freitag and V. Perebeinos, Nat. Photonics, 2008, 2, 341–350 CrossRef.
- S. Park, M. Vosguerichian and Z. Bao, Nanoscale, 2013, 5, 1727–1752 RSC.
- M. Prato, K. Kostarelos and A. Bianco, Acc. Chem. Res., 2008, 41, 60–68 CrossRef PubMed.
- L. N. Cella, W. Chen, N. V. Myung and A. Mulchandani, J. Am. Chem. Soc., 2010, 132, 5024–5026 CrossRef PubMed.
- N. Karousis, N. Tagmatarchis and D. Tasis, Chem. Rev., 2010, 110, 5366–5397 CrossRef PubMed.
- T. Fujigaya and N. Nakashima, Sci. Technol. Adv. Mater., 2015, 16, 1–21 CrossRef PubMed.
- J.-Y. Hwang, A. Nish, J. Doig, S. Douven, C.-W. Chen, L.-C. Chen and R. J. Nicholas, J. Am. Chem. Soc., 2008, 130, 3543–3553 CrossRef PubMed.
- S. Liang, G. Chen, J. Peddle and Y. Zhao, Chem. Commun., 2012, 48, 3100–3102 RSC.
- K. Mulla and Y. Zhao, J. Mater. Chem. C, 2013, 1, 5116–5127 RSC.
- K. Mulla, S. Liang, H. Shaik, E. A. Younes, A. Adronov and Y. Zhao, Chem. Commun., 2015, 51, 149–152 RSC.
- M. Khadem and Y. Zhao, J. Org. Chem., 2015, 80, 7419–7429 CrossRef PubMed.
- S. Aljohani, A. I. Alrawashdeh, M. Z. H. Khan, Y. Zhao and J. B. Lagowski, J. Phys. Chem. C, 2017, 121, 4692–4702 CrossRef.
- M. Z. H. Khan, A. I. Alrawashdeh, S. Aljohani, Y. Zhao and J. B. Lagowski, Phys. Chem. Chem. Phys., 2017, 19, 28071–28082 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. J. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- S. A. Ayoub and J. B. Lagowski, Comput. Theor. Chem., 2018, 1139, 15–26 CrossRef.
- S. A. Ayoub and J. B. Lagowski, J. Phys. Chem. C, 2016, 120, 496–507 CrossRef.
- S. A. Ayoub and J. B. Lagowski, Mater. Des., 2018, 156, 558–569 CrossRef.
- S. Tortorella, M. M. Talamo, A. Cardone, M. Pastore and F. De Angelis, J. Phys.: Condens. Matter, 2016, 28, 074005 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 16, Revision A.03, 2016 Search PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- S. J. Grimme, Chem. Phys., 2003, 118, 9095–9102 Search PubMed.
- Y. Jung, R. C. Lochan, A. D. Dutoi and M. J. Head-Gordon, Chem. Phys., 2004, 121, 9793–9802 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro, Version: 1.2.0, J. Cheminf., 2012, 4, 17 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- J. S. Michael and F. R. S. Dewar, The Molecular Orbital Theory of Organic Chemistry, McGraw-Hill, Inc., New York, 1969 Search PubMed.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Selected structural parameters (R, A, and Dh) for isolated and interacting FLU, DDTF, and BDOB with (6,5) and (8,7) SWCNTs calculated using different DFT methods and basis set without a solvent and with chloroform and hexane solvents. HOMO and LUMO energies for the isolated molecules and SWCNTs. Representative optimized structure of FLU, DDTF, and BDOB interacting with (6,5) and (8,7) SWCNTs obtained using B97D/6-31G(d). See DOI: 10.1039/c8ra02460j |
| This journal is © The Royal Society of Chemistry 2018 |