
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExposure of mass-selected bimetallic Pt–Ti nanoalloys to oxygen explored using scanning transmission electron microscopy and density functional theory
Saeed Gholhaki ac,
Shih-Hsuan Hungb,
David J. H. Cantc,
Caroline E. Blackmorea,
Alex G. Shardc,
Quanmin Guoa,
Keith P. McKennab and
Richard E. Palmer*d
ac,
Shih-Hsuan Hungb,
David J. H. Cantc,
Caroline E. Blackmorea,
Alex G. Shardc,
Quanmin Guoa,
Keith P. McKennab and
Richard E. Palmer*d
aSchool of Physics and Astronomy, University of Birmingham, Birmingham, B15 2TT, UK
bDepartment of Physics, University of York, York, YO10 5DD, UK
cNational Physical Laboratory, Hampton Road, Teddington, Middlesex TW11 0LW, UK
dCollege of Engineering, Swansea University, Bay Campus, Fabian Way, Swansea, SA1 8EN, UK. E-mail: r.e.palmer@swansea.ac.uk
First published on 31st July 2018
Abstract
The response of nanoparticles to exposure to ambient conditions and especially oxidation is fundamental to the application of nanotechnology. Bimetallic platinum–titanium nanoparticles of selected mass, 30 kDa and 90 kDa, were produced using a magnetron sputtering gas condensation cluster source and deposited onto amorphous carbon TEM grids. The nanoparticles were analysed with a Cs-corrected Scanning Transmission Electron Microscope (STEM) in High Angle Annular Dark Field (HAADF) mode. It was observed that prior to full Ti oxidation, Pt atoms were dispersed within a Ti shell. However, after full oxidation by prolonged exposure to ambient conditions prior to STEM, the smaller size 30 kDa particles form a single Pt core and the larger size 90 kDa particles exhibit a multi-core structure. Electron beam annealing induced a single core morphology in the larger particles. First principles density functional theory (DFT) calculations were employed to calculate the lowest energy structure of the Pt–Ti nanoparticles with and without the presence of oxygen. It was demonstrated that, as the concentration of oxygen increases, the lowest energy structure changes from dispersed Pt to multiple Pt cores and finally a single Pt core, which is in good agreement with the experimental observations.
1 Introduction
The application of platinum (Pt) nanoparticles in catalysis is well established in technology. However, exploitation of the excellent properties of platinum is limited by the scarcity of the critical metal. This has driven both experimental and theoretical investigations of Pt-containing alloy nanoparticles,1,2 synthesised mainly by chemical but also by physical methods.3–6 Combining two or more metallic elements to form alloy nanoparticles may even improve the catalytic performance and stability of the nanoparticles.7–9 Theoretical calculations on platinum–titanium (Pt–Ti) alloy nanoparticles suggest better catalytic performance through improved reaction kinetics and minimised poisoning levels in comparison with pure platinum nanoparticles in Polymer Electrolyte Membrane (PEM) fuel cells.10,11 Experimental studies on chemically synthesised Pt rich Pt–Ti nanoparticles, such as Pt3Ti, have demonstrated increased catalytic performance in comparison with pure Pt nanoparticles.12–14 This system is also interesting for photocatalysis.15 It has been demonstrated that the addition of Pt atoms to TiO2 enhances the photoactivity of TiO2 by improving the charge separation between the photo-formed electrons and holes.16An important influence on the structure and properties of nanoparticles is oxidation. Exposure to air is inevitable in most real world applications. In particular, oxidation of alloy nanoparticles generally causes morphological and structural changes. The oxidation behaviour of Cu, Al and Pb alloy nanoparticles has been studied and the formation of hollow structures with metallic shells has been reported.17 The effect of oxidation and heat treatment on the morphology of carbon encapsulated iron carbide nanoparticles has been investigated. The electronic structure and the morphology were modified through heating at different temperatures and oxygen exposure.18 The oxidation of AgAu nanoalloys has been investigated under an electron beam and through in situ thermal heating. The morphology of the nanoparticles was modified from alloy to an Ag oxide shell and Au core structure.19 Electron beam accelerated coalescence of atoms and nanoparticles on surfaces has also been demonstrated.20,21 Previous work by Blackmore et al.22 demonstrated that the morphology of an alloy nanoparticle can be size dependent. Pt–TiO2 nanoparticles with a mass range between 30 kDa and 120 kDa were investigated and it was observed that larger nanoparticles form multiple Pt cores with a TiO2 shell, in contrast with the smaller sizes which exhibit a single Pt core. Here we investigate, both experimentally and theoretically, the morphological changes of mass-selected Pt–Ti nanoparticles induced by oxidation. The size selected Pt–Ti clusters were produced using the magnetron sputtering gas-condensation cluster beam method and investigated using aberration-corrected scanning transmission electron microscopy (STEM). Our focus was on comparing the morphology of 30 kDa and 90 kDa Pt–TiO2 nanoparticles prior to full oxidation and under the electron beam, alongside theoretical calculations on the morphological stability of Pt–Ti nanoparticles influenced by oxygen exposure.
2 Experimental methods
The nanoparticles were produced through gas condensation using a magnetron sputtering cluster source23 with a base pressure of 10−7 mbar. A target alloy of Pt–Ti was used with a Pt to Ti mass ratio of 25% to 75% (target purity 99.95%), corresponding to 13 Ti atoms for every Pt atom. The magnetron was used in DC mode. Argon gas was used to create the plasma for sputtering. In addition, helium gas was injected into the chamber to promote aggregation via three body collisions between two sputtered metal atoms and a cooling He atom. Liquid nitrogen was used to cool down the gas mixture to further enhance aggregation. The nanoparticles were mass selected using a lateral time-of-flight mass selector24 with a resolving power of M/ΔM ∼ 20. The nanoparticles were soft-landed on copper TEM grids covered with amorphous carbon films.The samples were transferred from the cluster source to the STEM using two methods. First, the transfer of samples to the STEM under an air atmosphere, which resulted in fully oxidised clusters. Second, a transfer method was designed to prevent full oxidation of the Pt–TiO2 nanoparticles. A glove box purchased from Sigma was modified so that it could be attached to the load lock through a gate valve. The glove box was filled with argon gas at atmospheric pressure to remove the oxygen. The oxygen level was constantly monitored at below the detection limit of the oxygen monitor throughout the process. The samples were loaded onto the STEM arm and transferred to the microscope under the argon gas. The samples transferred under Ar gas experienced a brief exposure to air when the STEM arm was taken out of the glove box and inserted into the STEM column. During the brief exposure it is reasonable to assume a thin layer of oxide is formed on the surface of the nanoparticle.
The TEM grids were imaged using a 200 kV JEOL 2100F STEM equipped with a spherical aberration (Cs) corrector in HAADF mode. The signal was collected using a high-angle annular dark-field detector with inner and outer collection angles of 62 and 164 mrad, respectively, at a probe convergence angle of 19 mrad. The electron beam current was 30 pA.
3 Computational methods
First principles density functional theory (DFT) calculations were carried out using the Perdew, Burke and Ernzerhof (PBE) exchange–correlation functional and a plane wave basis set, as implemented in the Vienna ab initio simulation package (VASP).25,26 The Brillouin zone (BZ) was sampled using Monkhorst–Pack (MP) grids and the plane-wave basis set truncated at an energy of 300 eV. The Γ-point was used for the BZ sampling for all nanoparticle calculations and the oxygen molecule calculation. A 7 × 7 × 7 k-point grid was used for BZ sampling for the bulk Ti and Pt primitive cell calculations. For the calculations of nanoparticles and the oxygen molecule, a cubic supercell of 30 Å length was used. The psuedopotentials included 6, 10 and 4 valence electrons for oxygen, platinum and titanium, respectively. Using these methods we predicted the lattice constants of bulk Ti (a = 2.92 and c = 4.64 Å) and bulk Pt (a = 3.97 Å) and the bond length of the oxygen molecule (1.29 Å), which were consistent with experimental results.27–29 To compare the stability of nanoparticles with different oxygen coverage, we computed the free energy,| G = Gnp(Oad) − NOμO, | (1) |
4 Experimental results
Pt–Ti nanoparticles with two different masses exposed to oxygen (air) are shown in the HAADF-STEM images in Fig. 1. It is reasonable to assume that any exposure to the air results in oxidation of the Ti into an oxide with the most likely form being TiO2. Ti has significantly lower contrast than Pt in STEM HAADF mode due to a much lower Z number.32 The smaller size, i.e. 30 kDa, Pt–Ti forms a Pt core with a TiO2 shell (Fig. 1(c) and (d)). Meanwhile the larger Pt–Ti nanoparticles, 90 kDa, form multiple Pt cores with TiO2 shells (Fig. 1(a) and (b)). The higher magnification images Fig. 1(b) and (d) show the interior structure of the nanoparticles to a better extent. The atoms of the Pt core are visible in both cases. Fig. 1(e) shows a histogram of core sizes for 30 kDa Pt–TiO2 in the single core morphology. The core size was measured to be 1.0 ± 0.2 nm where the error is the standard deviation.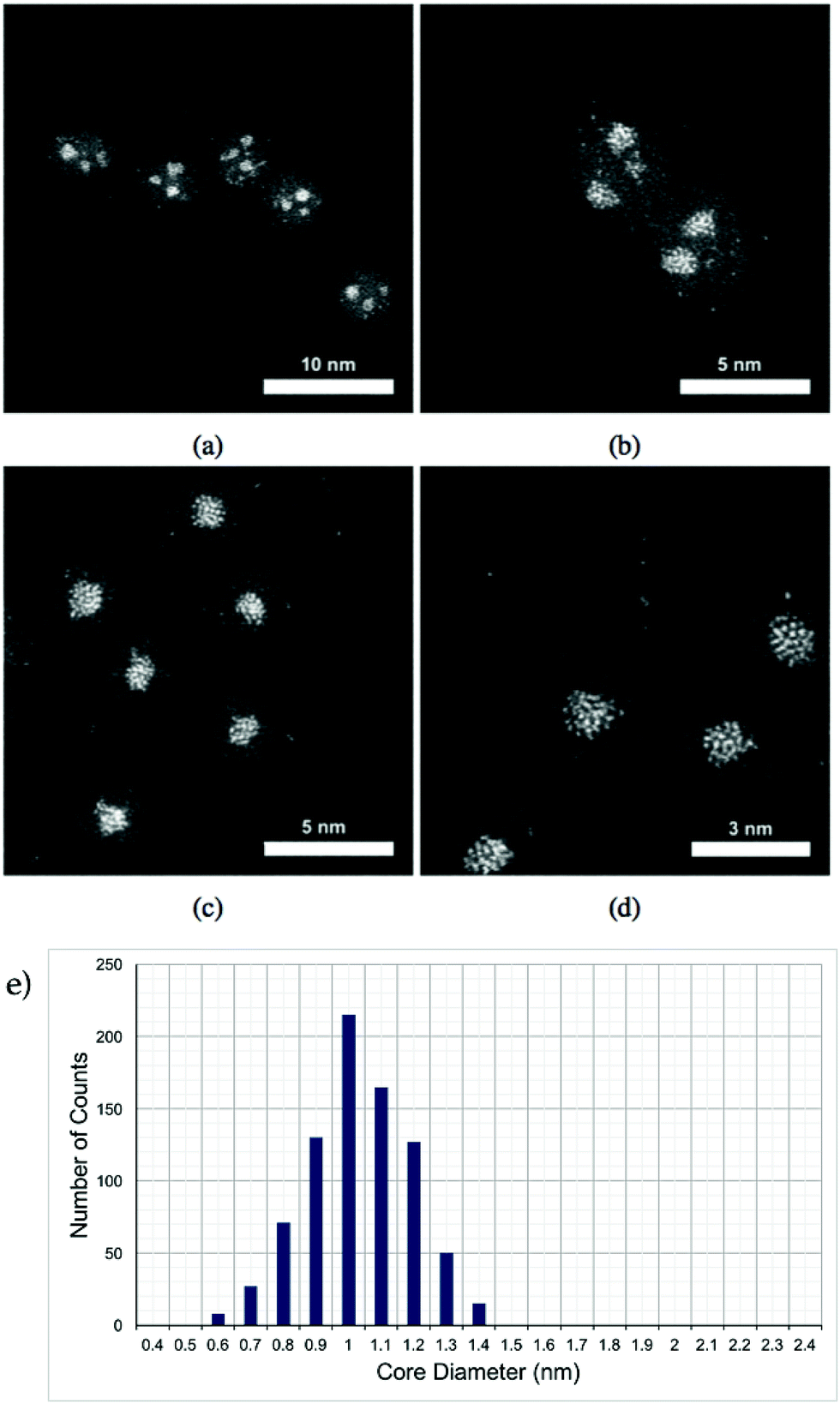 | ||
| Fig. 1 (a) and (b) are STEM HAADF images of 90 kDa Pt–TiO2 nanoparticles with two different magnifications. (c) and (d) show 30 kDa Pt–TiO2 nanoparticles. Bright dots are Pt atoms and the darker halo is a TiO2 shell. In (a) and (b), the multiple Pt core morphology of 90 kDa nanoparticles is clearly demonstrated. The higher magnification image demonstrates the interior structure of the core. (c) and (d) present the single Pt core within the smaller 30 kDa nanoparticles with the higher magnification image demonstrating the interior structure. (e) presents a histogram of the core size for 30 kDa Pt–TiO2 nanoparticles with single core morphology. | ||
When Pt–Ti nanoparticles are transferred under Ar gas to the electron microscope, they demonstrate a different morphology. HAADF-STEM images of 90 kDa Pt–Ti nanoparticles are presented in Fig. 2. They show that the initial state of 90 kDa Pt–Ti nanoparticles, i.e., without prolonged oxygen exposure, is an amorphous alloy structure of Pt and Ti. As previously mentioned, during the transfer process the nanoparticles are briefly exposed to the air. This explains the formation of a thin Ti oxide layer that can also be seen in the images around every nanoparticle. Higher magnification images presented in Fig. 2(b) and (c) show the interior structure in greater detail. The Pt atoms are randomly dispersed within the nanoparticle.
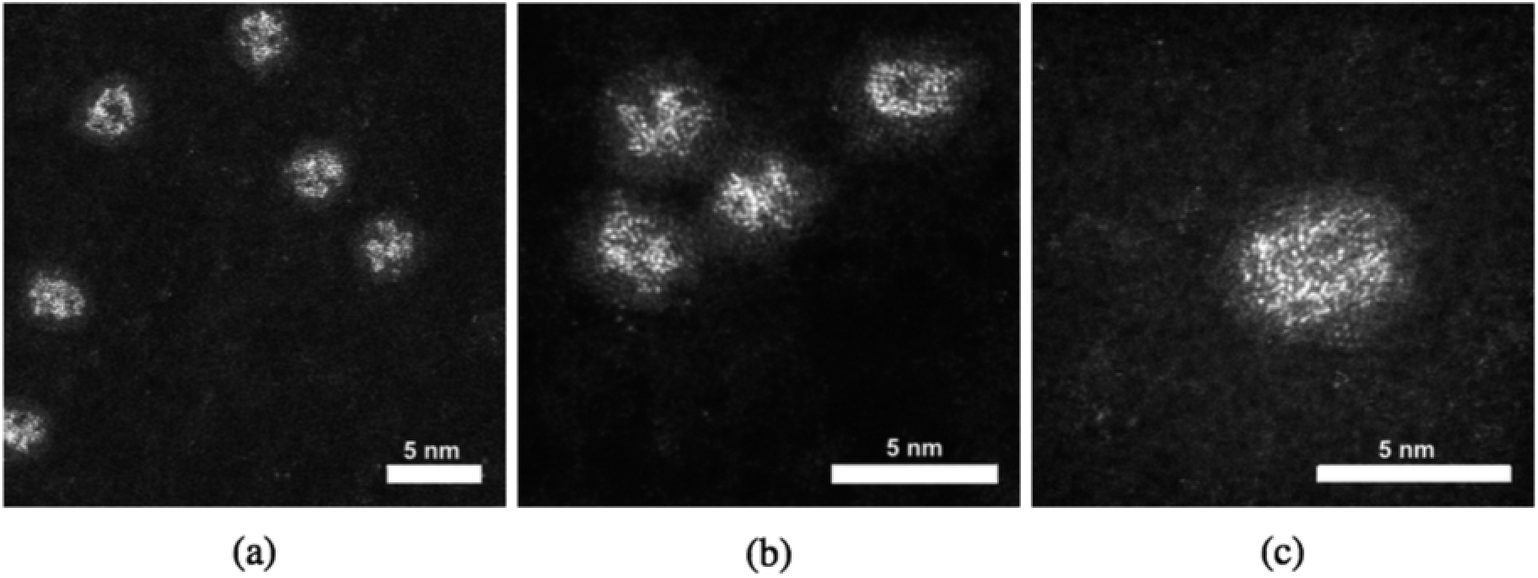 | ||
| Fig. 2 90 kDa Pt–Ti nanoparticles prior to prolonged exposure to ambient conditions. It can be seen that the Pt is dispersed throughout the nanoparticle instead of forming a Pt core morphology. (a) and (b) Present lower magnification images. Left to right shows increasing magnification. The higher magnification image, (c), shows the amorphous structure in greater detail. | ||
At the atomic level, there are two possible routes for this oxygen driven core–shell formation, observed in the experiments. Either the Ti atoms migrate to the surface where they are oxidised or oxygen penetration through the nanoparticle causes oxidation. In the latter case, one might expect to observe the Kirkandall effect (void formation) at the centre of the nanoparticles. However, due to the small size of the nanoparticles and the modest Z-contrast intensity of Ti in HAADF-STEM, it would be very difficult to confirm or exclude void formation. In any case our results show that the oxidation of the nanoparticles drives the nucleation of the Pt core. In the case of the 30 kDa clusters (Fig. 1(c) and (d)), the size of the nanoparticle is small enough that the Pt atoms are able to diffuse and form a single Pt core. In contrast, in the 90 kDa nanoparticles (Fig. 1(a) and (b)), it seems that the diffusion length of the Pt atoms inside the TiOx matrix is insufficient to form a single Pt core. Instead, several local cores of Pt atoms are nucleated within the nanoparticle.
Application of an electron beam to the nanoparticles can be used to explore the thermal effect upon the atomic structure of the Pt–Ti bimetallic nanoparticles in a vacuum environment. This irradiation was amplified by zooming the electron beam to a magnification of 50m on a single, surface oxidised 90 kDa Pt–Ti nanoparticle. Fig. 3 demonstrates the procedure. The red circled nanoparticle in Fig. 3(a) is the selected nanoparticle prior to modification. Fig. 3(b) shows the same nanoparticle circled in blue after modification, where it exhibits a large single Pt core. Fig. 3(c) demonstrates a second nanoparticle modified with the same procedure. Again, it was observed that local annealing under vacuum creates a single large Pt core in a Ti shell, an outcome that was reproducible.
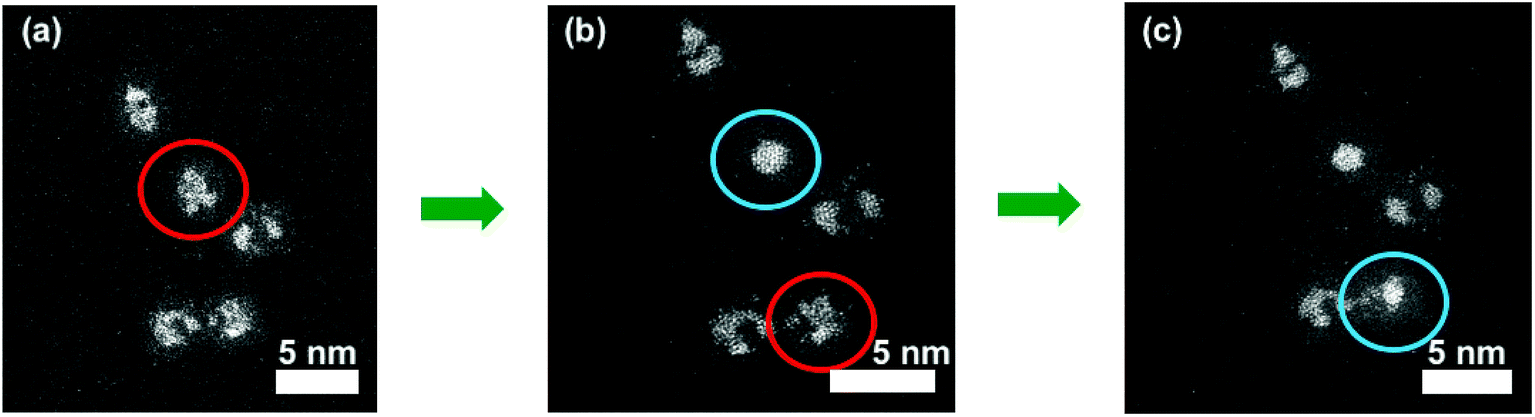 | ||
| Fig. 3 Sequential modification of two individual surface oxidised Pt–Ti nanoparticles using a focused electron beam. Initial state is an amorphous alloy Pt–Ti indicated by red circles in (a) and (b), however the interactions with the electron beam at a 30 pA current create a single Pt core indicated by a blue circle in (b) and (c). The time direction is from left to right. Red circles indicate the nanoparticle prior to modification and blue circles indicate the same nanoparticle post modification. | ||
Single core formation as a result of electron beam annealing is quite different from oxidation, since multi-core formation is observed in the latter case. It should be noted that when the electron beam was focused adjacent to the particle, single core formation was not observed, which rules out the possibility of surrounding gas excitation as a mechanism. Moreover when the beam was focused on an off-centre location of the nanoparticle, to examine if the position of the core depends on the location of the beam, the core was always formed at the centre of the nanoparticle. It seems most likely that heating by the electron beam increases the diffusion length of the Pt atoms within the nanoparticle, resulting in the formation of a single core. In order to examine if the single core formation would also be induced in a previously fully oxidised multi-core Pt–TiO2 nanoparticle, the electron beam was focused on such a particle. However single core formation was not observed. Thus electron beam reduction appears to be inactive.
5 Theoretical results
Small metallic nanoparticles exhibit a range of morphologies, including octahedral (Oh), truncated-octahedral (t-Oh), decahedral (Dh), Ino-decahedral (I-Dh), Marks-decahedral (M-Dh) and icosahedral (Ih).33,34 However, strained Ih nanoparticles are often the most stable in small size ranges (N ∼ 100, where N is the number of atoms in the nanoparticle) due to their low surface energies.35 Therefore, we consider Ih Pt–Ti nanoparticles for the investigation of the influence of oxygen adsorption on the atomic arrangement. Ih nanoparticles consist of twenty triangular close-packed (111) facets with twelve vertices on the surface. The magic numbers for the Ih morphology are 13, 55, 147, 309, etc. We considered an Ih nanoparticle with 147 atoms in this study, which offers a balance between sufficiently large nanoparticle size and computational feasibility. Since the ratio between Pt and Ti atoms in the Pt–Ti nanoparticles studied experimentally is approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13, we divided the 147 atoms into 13 Pt and 134 Ti atoms, hereafter (Pt–Ti)147. The number of the Pt atoms equals the first magic number of the Ih nanoparticles, meaning it can form a highly symmetric single core in the centre of the nanoparticle, as well as alternative configurations, such as Pt atoms distributed on or inside the nanoparticle.
:13, we divided the 147 atoms into 13 Pt and 134 Ti atoms, hereafter (Pt–Ti)147. The number of the Pt atoms equals the first magic number of the Ih nanoparticles, meaning it can form a highly symmetric single core in the centre of the nanoparticle, as well as alternative configurations, such as Pt atoms distributed on or inside the nanoparticle.
To investigate the stability of different atomic arrangements, we considered 5 different types of configurations: (I) Pt and Ti atoms randomly distributed throughout the nanoparticle (random), (II) Pt atoms at each of the vertex sites and 1 Pt atom in the central core site (vertex), (III) Pt atoms forming a single 13-atom core in the centre of the nanoparticle (single-core), (IV) Pt atoms forming two clusters inside the core of the nanoparticle (double-core) and (V) Pt atoms forming three clusters in the core of the nanoparticle (triple-core). We consider 3, 1, 1, 2 and 3 different configuration(s) for each of the above types of arrangement, respectively. Recent DFT studies show that the oxygen atoms should adsorb at the three-fold hollow sites on the surface of Ti nanoparticles (either hcp or fcc) as well as octahedral sites on the sub-surface layers.36,37 We investigated four oxygen coverages on each of the above nanoparticle arrangements and calculated the total energy to assess their stability. The four considered oxygen coverages are denoted 0 ML, 0.5 ML, 1 ML and 2 ML. 0 ML corresponds to the nanoparticle without any adsorbed oxygen. 0.5 ML has 60 oxygen atoms adsorbed on surface fcc sites (3 per facet). 1 ML has 120 oxygen atoms adsorbed on surface hcp sites (6 per facet). Additional oxygen added to the nanoparticle preferentially occupies octahedral sites in the sub-surface layer. An additional 60 oxygen atoms fully occupy the octahedral sites in the first sub-surface layer. Therefore, 2 ML corresponds to 180 adsorbed oxygen atoms in both the surface and subsurface layers.
Our results show that the vertex arrangement (Fig. 4(a)) of Pt atoms is the most stable Pt–Ti geometry in the absence of adsorbed oxygen but that the random arrangement is only slightly less stable. The energy difference between the vertex and random arrangements is less than 0.5 eV per nanoparticle in a vacuum. However, the energy difference significantly increases once oxygen atoms adsorb on the surface of the nanoparticles. The vertex arrangement of Pt atoms is the most stable for 0.5 ML oxygen coverage, as shown in Fig. 4(b). As the oxygen coverage increases further up to 1 ML, the triple-core arrangement (Fig. 4(c)) becomes the most stable. Finally, the nanoparticle with the single-core arrangement (Fig. 4(e)) becomes the most stable for the highest oxygen coverage of 2 ML. Table 1 shows the diameters of the nanoparticles (excluding oxygen atoms) and the average bond lengths for specific atom pairs. The diameter is defined as the length of the diagonal passing through the centre of the nanoparticle joining two vertices. Almost every parameter increases with increasing oxygen coverage, suggesting that oxygen adsorption induces an expansive strain in the nanoparticle. The exception is the average Pt–Ti bond length, which decreases between the 1 and 2 ML coverages. This can be understood because in this case, the Pt atoms form a single core at the centre of the nanoparticle which is more compact than other arrangements.
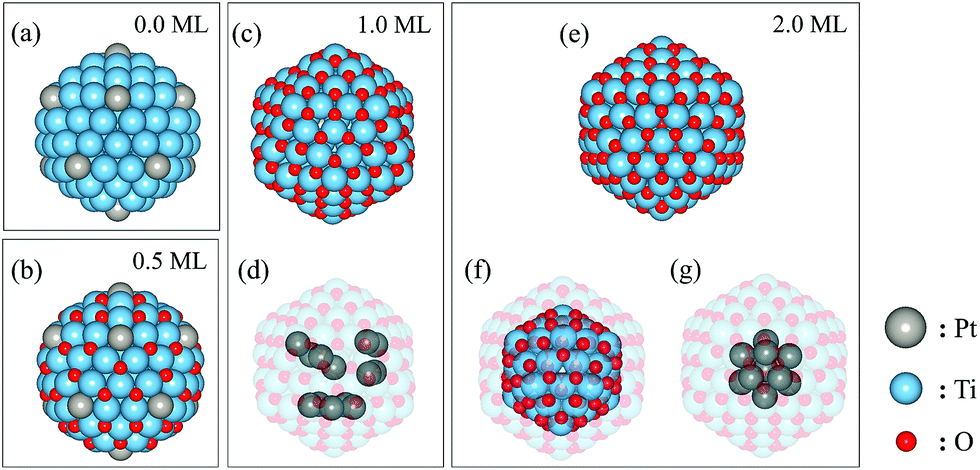 | ||
| Fig. 4 Atomic configurations of (Pt–Ti)147 clusters obtained from DFT calculations. (a) The most stable structure of bare Pt–Ti nanoparticles in the calculations. (b) The most stable configuration for 0.5 ML oxygen coverage. (c) The most stable structure with 1.0 ML oxygen coverage in the calculation and (d) the three Pt cores in the centre of the Pt–Ti nanoparticle. (e) The most stable single core arrangement for 2.0 ML oxygen coverage. (f) and (g) show the oxygen configuration in the sub-surface layer and the central Pt core, respectively. The silver, blue and red spheres represent the Pt, Ti and O atoms, respectively. | ||
| Oxygen coverage (ML) | Arrangement | Diameter of nanoparticle (Å) | Average bond length (Å) | |||
|---|---|---|---|---|---|---|
| Ti–Ti | Pt–Ti | Pt–Pt | Ti–O | |||
| 0.0 | Vertex | 15.58 | 2.85 | 2.73 | — | — |
| 0.5 | Vertex | 15.66 | 2.95 | 2.95 | — | 1.95 |
| 1.0 | Triple-core | 16.68 | 3.05 | 3.05 | 2.95 | 1.95 |
| 2.0 | Single-core | 16.75 | 3.05 | 2.75 | 3.05 | 2.15 |
In order to capture the influence of oxygen on the atomic arrangement of the Ih PtTi nanoparticle in a continuous way, we calculated the free energy as a function of the oxygen chemical potential using eqn (1). The free energies were calculated using the total energies of the most stable configurations for the four different oxygen coverages (Fig. 5). We defined the upper limit of the oxygen chemical potential relative to half the energy of the oxygen molecule (such that μO = EO2/2 = 0 eV). Below μO = −6.0 eV, the bare Pt–Ti nanoparticle with the vertex arrangement is the most stable configuration. For −6.0 eV < μO < −4.9 eV, the nanoparticle maintains the same vertex arrangement with 0.5 ML of adsorbed oxygen atoms. For μO > −4.9 eV, the 2 ML oxygen adsorption with a single core arrangement is the most stable configuration. However, the nanoparticle with 1.0 ML oxygen adsorption is not thermodynamically stable at any oxygen chemical potential in the calculation. We also show in Fig. 5 the chemical potential at which bulk rutile TiO2 is reduced to metallic hcp-Ti. We note that the surface and subsurface of the Ih nanoparticle are fully oxidized at chemical potentials lower than the bulk reduction limit, indicating that the nanoparticle is more easily oxidized than bulk TiO2 as expected. The above results show that the structural arrangement of Pt atoms within Pt–Ti alloy nanoparticles can be modified with the adsorption of oxygen, which is consistent with the experimental observation of 30 kDa Pt–TiO nanoparticles forming a Pt core with a TiO2 shell.
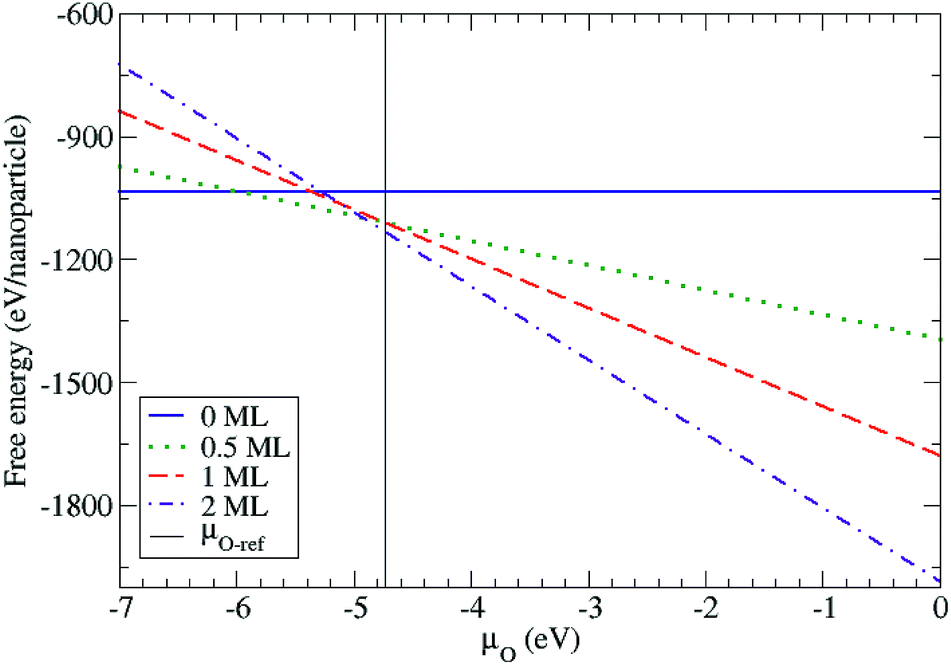 | ||
| Fig. 5 Free energy of the Pt–Ti nanoparticles for different oxygen coverages as a function of oxygen chemical potential. The oxygen chemical potential is defined relative to half the energy of the oxygen molecule (such that μO = 0 eV corresponds to oxygen rich conditions). The vertical line at μO = −4.73 eV indicates the point at which reduction of bulk rutile TiO2 to hcp-Ti occurs, i.e. (ETiO2 − EbulkTi − EO2)/2, where ETiO2, Ehcp-Ti and EO2 are the energies of bulk TiO2, hcp-Ti and O2 molecules, respectively. | ||
These results are also consistent with the fact that, in a vacuum, the predicted average surface free energy of pure Pt nanoparticles is lower than that of pure Ti nanoparticles (1.59 J m−2 versus 2.00 J m−2).38 Hence, in a vacuum, one would expect Pt to preferentially segregate to the surface. However, the oxidation of Ti is much more favourable than that of Pt (formation enthalpies of −944 kJ mol−1 and −80 kJ mol−1 respectively), explaining why Ti prefers to segregate to the surface on oxidation.39,40
6 Discussion
We have demonstrated experimentally that the process of oxidation as a result of exposure to ambient conditions has a critical impact on the morphology of Pt–Ti bimetallic nanoparticles. In order to determine the chemical state of Ti after exposure to air, data from X-ray photoelectron spectroscopy (XPS) measurements on 90 kDa Pt–Ti nanoparticles after extended air exposure i.e. fully oxidised, are presented in Fig. 6. The binding energy of the Ti 2p3/2 peak calibrated to the carbon peak at 258 eV in Fig. 6(a) is 485.55 eV. This is in accordance with the reported binding energy of TiO2.41,42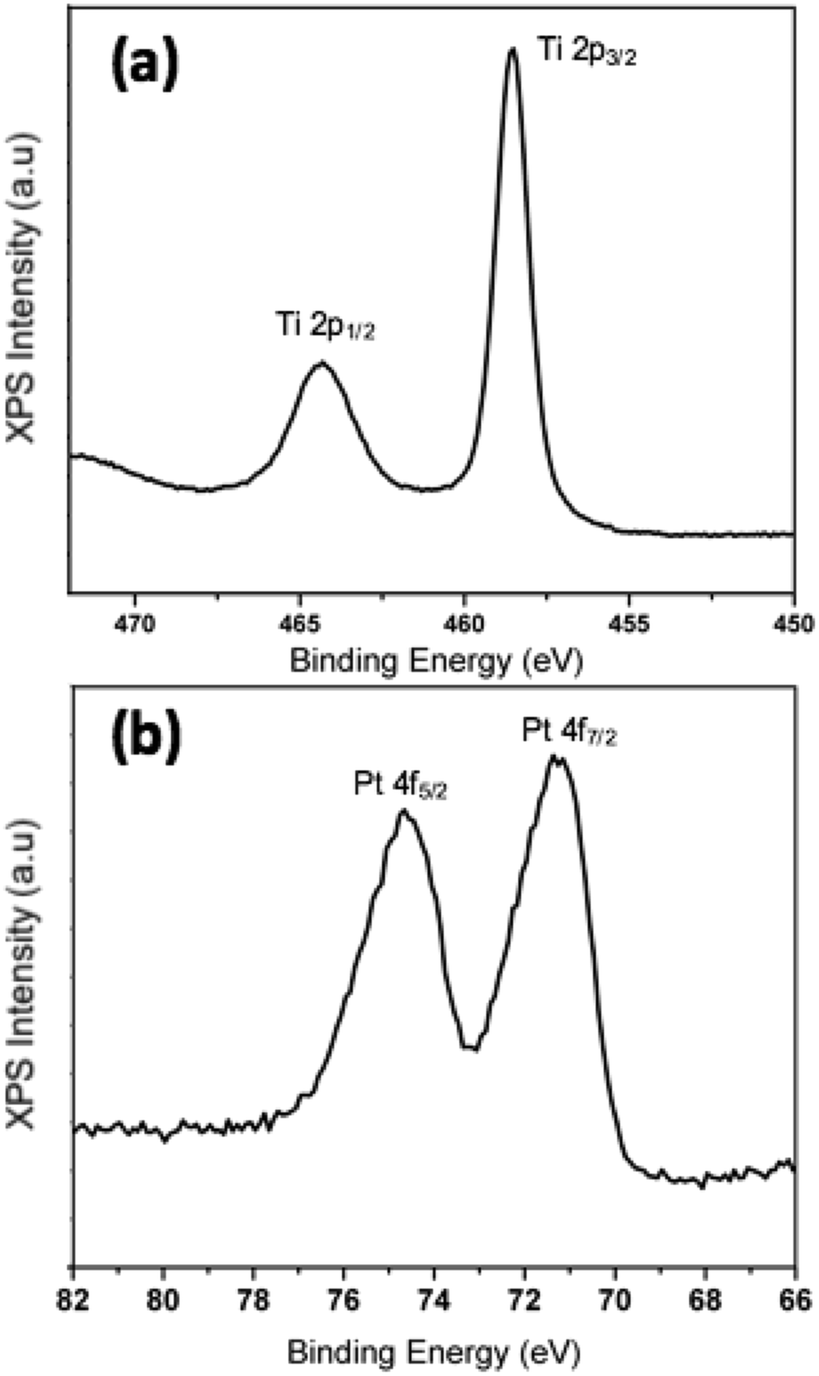 | ||
| Fig. 6 XPS spectra of the Pt core and Ti shell after extended air exposure, nanoparticle mass 90 kDa. (a) Spectrum of Ti 2p doublet demonstrating the chemical state of TiO2 after full oxidation and (b) spectrum of Pt 4f doublet. | ||
STEM observation shows that the initial morphology of Pt–Ti nanoparticles, i.e. before extended exposure to ambient conditions, is a dispersion of Pt atoms within a Ti shell. However, exposure to oxygen results in oxidation of Ti to TiO2. This process is accompanied by Pt core formation. In the case of smaller 30 kDa nanoparticles, the diffusion length of Pt atoms within the nanoparticle is comparable to the size of the particle, resulting in the coalescence of Pt atoms to form a single core. On the other hand, in larger 90 kDa nanoparticles, the diffusion length is not sufficient for complete coalescence into a single Pt core. Instead, several local cores are observed. The heating effect of the electron beam was used to increase the diffusion length of the Pt atoms in samples transferred in inert gas, which resulted in large single core formation in the 90 kDa nanoparticles.
There are several factors that may influence the accuracy of the theoretical predictions. First is the effect of the approximation for exchange and correlation (PBE). The PBE functional provides good descriptions for bulk Ti and Pt, reasonable Ti–O bond length and a formation enthalpy of bulk TiO2 comparable with experimental data.29,43,44 The well known underestimated band gap of TiO2 should not affect the formation energy and free energy in the calculations. Therefore, we believe that the PBE functional provides a reasonable approximation for this investigation. The most stable Pt–Ti nanoparticle structure is obtained by considering 10 different atomic arrangements, and four different oxygen coverages for each configuration (representing 40 distinct structures for DFT calculation). The lower symmetry random and multiple core configurations may have other possible arrangements, but it is not feasible to screen all possibilities at the DFT level due to the high computational expense. Similarly, we considered that oxygen atoms adsorb homogeneously and symmetrically on all facets of the nanoparticle whereas real configurations of oxygen adsorption may exhibit a reduced symmetry. However, we are satisfied that the subset of structures considered are sufficient to illustrate the structural trends on oxidation. First principles molecular dynamics could provide further insights into the oxygen adsorption process, but would be challenging owing to the large number of atoms in the system. The multi-core structures were not found to be stable over a significant chemical potential range for the small Ih nanoparticles we have modelled, which is consistent with the experimental observation of the different configurations between smaller and larger nanoparticles. The experimental observation of multiple cores could be a result of the increased flexibility in the size and shape with respect to larger nanoparticles and their ability to accommodate strain fields associated with multiple cores.
7 Conclusions
The morphology of mass-selected 30 kDa and 90 kDa bimetallic Pt–Ti nanoparticles was investigated using Cs corrected STEM imaging and theoretical calculations. It was observed that surface oxidised nanoparticles exhibit an alloy morphology with a thin TiO2 surface layer. However prolonged exposure to ambient conditions i.e. further oxidation of Ti, results in Pt core formation. In 30 kDa nanoparticles, a single Pt core was formed, however 90 kDa particles demonstrated a multi-core structure. This was most likely due to the fact that in larger particles the diffusion length is smaller than the nanoparticle size. Electron beam annealing of samples transferred under Ar gas resulted in a single large core structure. DFT calculations suggest that prior to any oxidation, the most energetically favourable morphology is with most of the Pt atoms on the surface. As the oxygen concentration is increased the calculated trend is towards a single core morphology, i.e. the morphology transformed from dispersed Pt to a multi-core and finally a single core structure. Note that the nanoparticles investigated experimentally have an oxide shell in all cases. The reported results highlight the importance of air exposure in the production and application of alloy nanoparticles, where detailed understanding of the morphological stability is key to harnessing the derived properties of such nanoparticles, for instance, in catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. P. M. acknowledges support from (EP/K003151/1). This work made use of the facilities of Archer, the UK’s national high-performance computing service, via our membership of the UK HPC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202/1). This work also made use of the facilities of the N8 HPC Centre of Excellence, provided and funded by the N8 consortium and EPSRC (EP/K000225/1). The Centre is coordinated by the Universities of Leeds and Manchester. All data relating to the theoretical calculations created during this research are available by request from the University of York Research database http://dx.doi.org/10.15124/a0a87163-b553-4077-9371-a97572d97d00. Work performed at the University of Birmingham was supported by the EPSRC Fellowship Grant and the European Metrology Programme for Innovation and Research (EMPIR) as part of the InNanoPart 14IND12 project. The EMPIR initiative is co-funded by the European Union Horizon 2020 research and innovation programme and by the EMPIR participating states.Notes and references
- Y. Matsuura, S. Seino, T. Okazaki, T. Akita, T. Nakagawa and A. Y. Takao, Radiat. Phys. Chem., 2016, 122, 9–14 CrossRef.
- D. Guedes-Sobrinho, R. K. Nomiyama, A. S. Chaves, M. J. Piotrowski and J. L. F. D. Silva, J. Phys. Chem. C, 2015, 119, 15669–15679 CrossRef.
- X. Teng, D. Black, N. J. Watkins, Y. Gao and H. Yanga, Nano Lett., 2003, 3, 261–264 CrossRef.
- I. Bilecka, I. Djerdj and M. Niederberger, Chem. Commun., 2008, 886–888 RSC.
- A. Caillard, S. Cuynet, T. Lecas, P. Andreazza, M. Mikikian, A. l. Thomann and P. Brault, J. Phys. D: Appl. Phys., 2015, 48(47), 475302 CrossRef.
- B. R. Cuenya, Thin Solid Films, 2010, 518(12), 3127–3150 CrossRef.
- K. Cao, X. Liu, Q. Zhu, B. Shan and R. Chen, ChemCatChem, 2016, 8, 326 CrossRef.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef PubMed.
- R. Ferrando, J. Phys.: Condens. Matter, 2015, 27(1), 013003 CrossRef PubMed.
- P. C. Jennings, B. G. Polleta and L. J. Roy, Phys. Chem. Chem. Phys., 2012, 14, 3134–3139 RSC.
- P. C. Jennings, H. A. Aleksandrov, K. M. Neyman and L. J. Roy, J. Phys. Chem. C, 2015, 119, 11031–11041 CrossRef.
- Z. Cui, H. Chen, M. Zhao, D. Marshall, Y. Yu and F. J. Disalvo, J. Am. Chem. Soc., 2014, 136(29), 10206–10209 CrossRef PubMed.
- G. Saravanan, H. Abe, Y. Xu, N. Sekido, H. Hirata, S.-i. Matsumoto, H. Yoshikawa and Y. Yamabe-mitarai, Langmuir, 2010, 26(13), 11446–11451 CrossRef PubMed.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef PubMed.
- M. Anpo, H. Yamashita and S. E. Yuichi Ichihashi, J. Electroanal. Chem., 1995, 396, 21–26 CrossRef.
- M. Anpo, Catal. Surv. Jpn., 1997, 1, 169–179 CrossRef.
- R. Nakamura, D. Tokozakura, H. Nakajima and H. Mori, J. Appl. Phys., 2007, 101, 074303 CrossRef.
- Y. Fedoseeva, L. G. Bulusheva, A. V. Okotrub, D. V. Vyalikh, J. Huo, H. Song and J. Z. X. Chen, Mater. Chem. Phys., 2012, 135, 235–240 CrossRef.
- E. A. Lewis, T. J. A. Slater, E. Prestat, A. Macedo, P. O’Brien, P. H. C. Camargo and S. J. Haigh, Nanoscale, 2014, 6, 13598 RSC.
- M. Tanaka, M. Takeguchi and K. Furuya, Micron, 2002, 33, 441–446 CrossRef PubMed.
- Y. Chen, R. E. Palmer and J. P. Wilcoxon, Langmuir, 2006, 22, 2851–2855 CrossRef PubMed.
- C. E. Blackmore, N. V. Reesb and E. P. Richard, Phys. Chem. Chem. Phys., 2015, 17, 28005–28009 RSC.
- S. Pratontepa, S. J. Carroll, C. Xirouchaki, M. Streun and R. E. Palmer, Rev. Sci. Instrum., 2005, 76, 045103 CrossRef.
- B. von Issendorff and R. E. Palmer, Rev. Sci. Instrum., 1999, 70, 4497 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B, 1996, 54, 11169 CrossRef.
- R. W. G. Wyckoff, Journal of Materials Science, 1963 Search PubMed.
- D. A. Outka, J. Stöhr, W. Jark, P. Stevens, J. Solomon and R. J. Madix, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 4119 CrossRef.
- W. P. Davey, Phys. Rev., 1925, 25, 753–761 CrossRef.
- C. Raymond, Physical chemistry for the chemical and biological sciences, University Science Books, 2000 Search PubMed.
- Y. A. Mastrikov, E. Heifets, E. A. Kotomin and J. Maier, Surf. Sci., 2009, 603, 326–335 CrossRef.
- Z. W. Wang, Z. Y. Li, S. J. Park, A. Abdela, D. Tang and R. E. Palmer, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 2–4 Search PubMed.
- L. D. Marks, Rep. Prog. Phys., 1994, 57, 603–649 CrossRef.
- J. L. Elechiguerra, J. Reyes-Gasga and M. J. Yacaman, J. Mater. Chem., 2006, 16, 3906–3919 RSC.
- F. Baletto and R. Ferrando, Rev. Mod. Phys., 2005, 77, 371 CrossRef.
- A. N. Chibisov, J. Mater. Sci., 2014, 82, 131–133 Search PubMed.
- S.-H. Hung and K. P. McKenna, J. Phys. Chem. C, 2018, 122, 3107–3114 CrossRef.
- R. Tran, Z. Xu, B. Radhakrishnan, D. Winston, W. Sun, K. A. Persson and S. P. Ong, Sci. Data, 2016, 3, 160080 CrossRef PubMed.
- J. D. Cox, D. D. Wagman and V. A. Medvedev, CODATA key values for thermodynamics, Hemisphere Pub. Corp., New York, 1989 Search PubMed.
- Y. Nagano, J. Therm. Anal. Calorim., 2002, 69, 831–839 CrossRef.
- N. Kruse and S. Chenakin, Appl. Catal., A, 2011, 391, 367–376 CrossRef.
- F. Liu, Z. Zhao, L. Qiu and L. Zhao, J. Surf. Anal., 2009, 15(3), 271–273 Search PubMed.
- G. Charlton, P. B. Howes, C. L. Nicklin, P. Steadman, J. S. G. Taylor, C. A. Muryn, S. P. Harte, J. Mercer, R. McGrath and D. Norman, et al., Phys. Rev. Lett., 1997, 78, 495 CrossRef.
- A. Janotti, J. B. Varley, P. Rinke, N. Umezawa, G. Kresse and C. G. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085212 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |