
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decarboxylative aldol reaction of α,α-difluoro-β-ketocarboxylate salt: a facile method for generation of difluoroenolate†
Atsushi Tarui,
Mayuna Oduti,
Susumu Shinya,
Kazuyuki Sato and
Masaaki Omote *
*
Faculty of Pharmaceutical Sciences, Setsunan University, 45-1 Nagaotogecho, Hirakata, Osaka 573-0101, Japan. E-mail: Omote@pharm.setsunan.ac.jp
First published on 5th June 2018
Abstract
We developed a decarboxylative aldol reaction using α,α-difluoro-β-ketocarboxylate salt, carbonyl compounds, and ZnCl2/N,N,N′,N′-tetramethylethylenediamine. The generation of difluoroenolate proceeded smoothly under mild heating to provide α,α-difluoro-β-hydroxy ketones in good to excellent yield (up to 99%). The α,α-difluoro-β-ketocarboxylate salt was bench stable and easy to handle under air, which realizes a convenient and environmentally friendly methodology for synthesis of difluoromethylene compounds.
Introduction
Organic molecules containing a difluoromethylene group are particularly useful in medicinal chemistry.1 Among these, α,α-difluoroketones show a variety of bioactivities such as cholesterol-lowering, analgesic, and GABAB agonist activities (Fig. 1).2 Thus, simple and mild strategies for accessing α,α-difluoroketone substructures should be useful for developing new therapeutic candidates.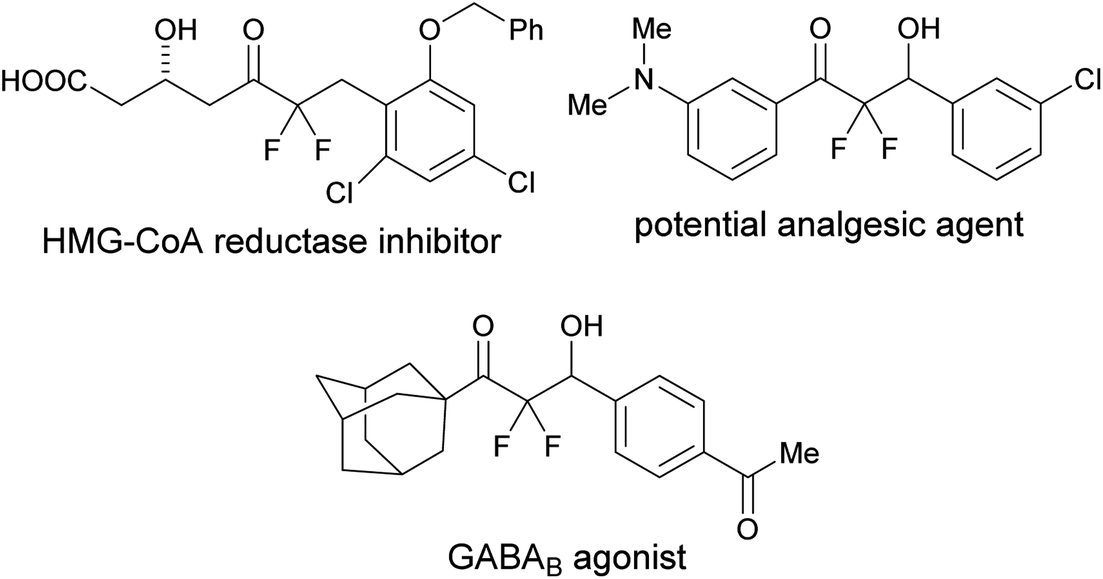 | ||
| Fig. 1 Bioactive α,α-difluoroketones. | ||
α,α-Difluoroenolate plays a key role in the construction of CF2–carbon bonds and many synthetic routes have been reported based on this nucleophilic synthon, including a metal-mediated Reformatsky reaction of halodifluoromethyl ketone (Scheme 1, eqn (a)),3 a Lewis acid-catalyzed aldol reaction of difluoroenol O-Boc esters (Scheme 1, eqn (b)),4 a copper-catalyzed reaction of α,α,α-trifluoromethylketones via β-fluoro elimination (Scheme 1, eqn (c)),5 a one-pot reaction of acylsilanes and trifluoromethyltri-methylsilane (TMSCF3) with aldehydes (Scheme 1, eqn (d)),6 an aldol reaction of halodifluoromethyl ketone via reduction of halogen process by lithium triethylborohydride (Scheme 1, eqn (e)),7 and a detrifluoroacetylative aldol reaction of trifluoromethyl α,α-difluoro-β-keto gem-diols (Scheme 1, eqn (f)).8 In the course of these studies, decarboxylation of β-keto acids is a mild and convenient method for providing the corresponding enolate and is an environmentally friendly system.9 Wennemers has reported the first decarboxylative process for the preparation of fluorinated enolate using fluoromalonic acid halfthioesters (F-MAHT).10 Recently, two groups have reported the synthesis of α,α-difluoro-β-hydroxy ketones using 2,2-difluoro-3-oxo-3-phenyl-propanoic acid (1). The first route was based on an aldol reaction of 1 with aldehydes under metal-free conditions, from the Mao group.11 The other route involved copper-catalyzed difluoroalkylation of aromatic aldehydes using 1 from the Mai group.12 However, the former reaction needed relative long reaction times and a high reaction temperature (100 °C). In the latter reaction, the scope of aldehydes was limited and only aromatic aldehydes were suitable for the transformation. In this paper, the simple potassium 2,2-difluoro-3-oxo-3-phenylpro-panoate (2a) was used as a precursor of difluoroenolate for the decarboxylative aldol reaction with carbonyl compounds under mild reaction conditions.
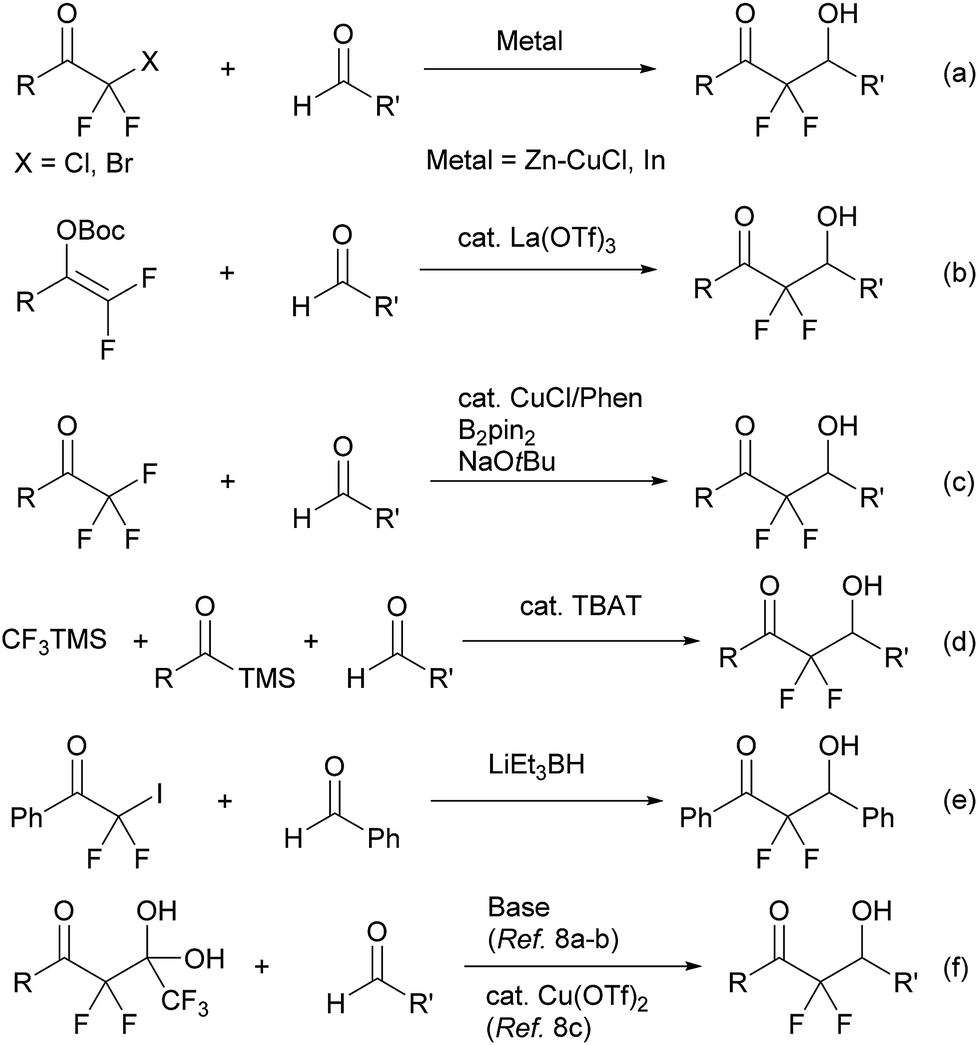 | ||
| Scheme 1 Various methods for a generation of α,α-difluoroenolate. | ||
Results and discussion
We used a potassium salt of α,α-difluoro-β-keto acid 1 (2a) as a model precursor of difluoroenolate to examine the decarboxylative aldol reaction with benzaldehyde (3a). The model substrate 2a was synthesized as following 3 steps (Scheme 2);13 Honda–Reformatsky reaction of ethyl bromodifluoroacetate with 3a gave β-hydoroxy-α,α-difluoroacetate 4a, then 4a was oxidized to β-keto-α,α-difluoroester 5a by TEMPO oxidation. The saponification of β-ketoester 5a gave rise to the potassium carboxylate 2a. The substrate 2a was isolated easily by filtration from the reaction mixture as a non-hygroscopic compound which enabled easy handling even under air atmosphere. Another substrate 1 was obtained by the acidified of 2a and was used to the desired reaction without purification. The results for the decarboxylative aldol reaction of 2a and 1 are summarized in Table 1. To examine decarboxylation under mild heating (50 °C) efficiently, ZnCl2 was used as an acceptor of the enolate (Table 1, entry 1). The reaction of 2a provided the desired product in low yield (20% yield). In the case of using carboxylic acid 1, only trace amounts of the desired product 6aa were obtained (entry 2). To improve the yield of 6aa, the reaction temperature was elevated to 80 °C and a moderate yield of product was obtained (60%, entry 3). The current screening of the reaction conditions was performed in dry THF (entries 1–3). Using wet THF in this system, we observed a considerable improvement of the yield of 6aa. Thus, when an equimolar amount of H2O for 2a was added to dry THF, the desired product was obtained in 84% yield reproducibly (entry 4). It was important to use ZnCl2 in the current system and a lower yield of 6aa was observed in the absence of ZnCl2 (entry 5). To evaluate the effects of the metal, other Lewis acids, including boron and ytterbium, were examined (entries 6–8). Zinc metal was shown to be the most effective and the bench stable reagent ZnCl2 N,N,Nʹ,Nʹ-tetramethylethylenediamine complex (ZnCl2/TMEDA) was the best metal source in this decarboxylative aldol process (Entry 8). ZnCl2/TMEDA was commercially available and was easy to handle compared with hygroscopic ZnCl2.14 Further optimization of the reaction conditions showed that the 1.2 equivalent of 2a and ZnCl2/TMEDA provided a 98% yield of 6aa (entry 9).15 Among the solvents tested, THF was the best solvent for this reaction.16 Furthermore, we examined the effects of the addition of water using dry THF solvent. Under the anhydrous conditions, a lower yield of product was observed (entry 10). When a catalytic amount of H2O (10 mol%) was added, there was no effect on the yield of product and benzaldehyde was recovered from the reaction mixture after 5 h (entry 11). Other proton sources such as ethanol and 2,2,2-trifluoroethanol were added to the reaction media. However, these additives did not markedly affect the yield of 6aa. In these cases (entries 10–13), the decarboxylation of 2a did not occur effectively and α,α-difluoro-β-keto acid (1) was detected in 19F NMR of the crude reaction mixture.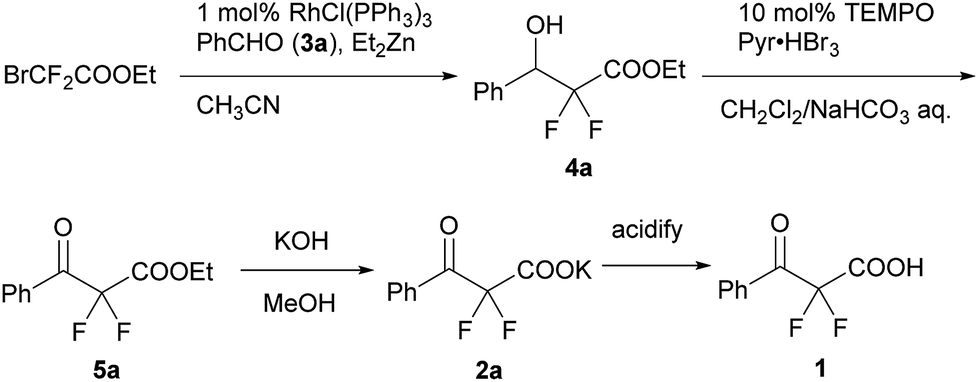 | ||
| Scheme 2 The synthesis of a potassium α,α-difluoro-β-keto carboxylate (2a) and its carboxylic acid (1). | ||
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Metal reagent | (Equiv.) | Substrates | (Equiv.) | Additive | (Equiv.) | Temp. (°C) | Time (h) | Yield of 6aa (%)a |
| a Isolated yield.b 19F NMR yields. | |||||||||
| 1 | ZnCl2 | (1.0) | 2a | (1.0) | None | 50 | 24 | 20 | |
| 2 | ZnCl2 | (1.0) | 1 | (1.0) | None | 50 | 24 | Trace | |
| 3 | ZnCl2 | (1.0) | 2a | (1.0) | None | 80 | 8 | 60 | |
| 4 | ZnCl2 | (1.0) | 2a | (1.0) | H2O | (1.0) | 80 | 8 | 84 |
| 5 | None | 2a | (1.0) | H2O | (1.0) | 80 | 5 | 46 | |
| 6 | BF3–Et2O | (1.0) | 2a | (1.0) | H2O | (1.0) | 80 | 16 | 59 |
| 7 | Yb(OTf)3 | (0.1) | 2a | (1.0) | H2O | (1.0) | 80 | 26 | 32 |
| 8 | ZnCl2–TMEDA | (1.0) | 2a | (1.0) | H2O | (1.0) | 80 | 5 | 88 |
| 9 | ZnCl2–TMEDA | (1.2) | 2a | (1.2) | H2O | (1.0) | 80 | 5 | 98 |
| 10 | ZnCl2–TMEDA | (1.2) | 2a | (1.2) | None | 80 | 5 | 42b | |
| 11 | ZnCl2–TMEDA | (1.2) | 2a | (1.2) | H2O | (0.1) | 80 | 5 | 50b |
| 12 | ZnCl2–TMEDA | (1.2) | 2a | (1.2) | EtOH | (1.0) | 80 | 7 | 59b |
| 13 | ZnCl2–TMEDA | (1.2) | 2a | (1.2) | CF2CH2OH | (1.0) | 80 | 7 | 58b |
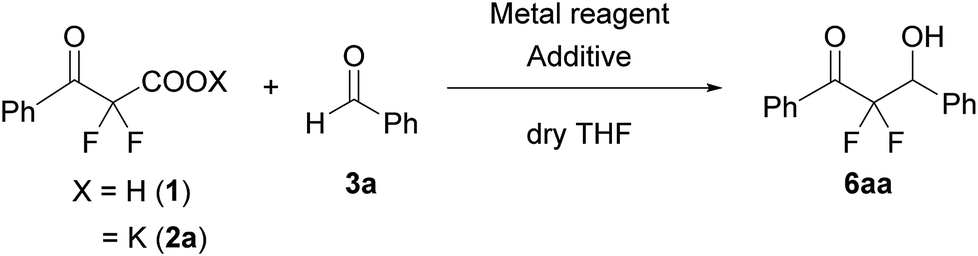
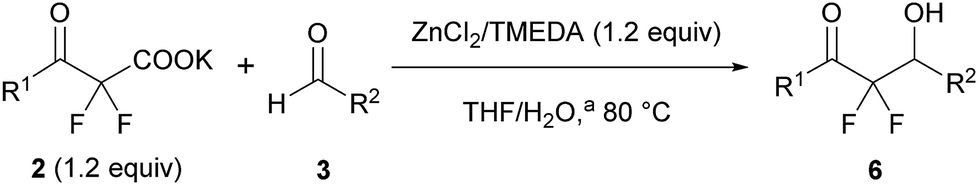
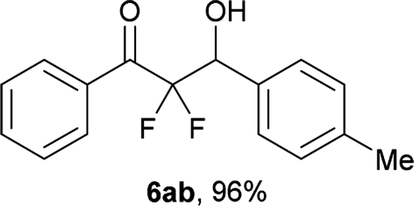
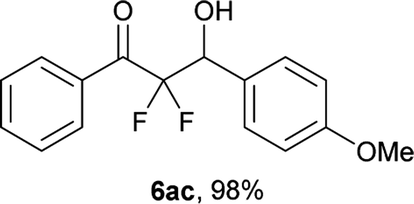
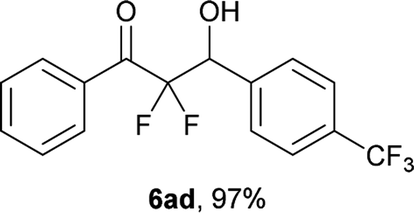
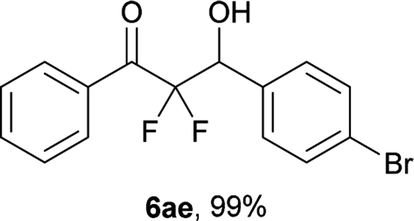
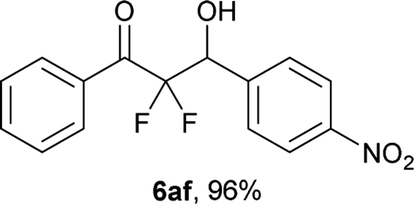
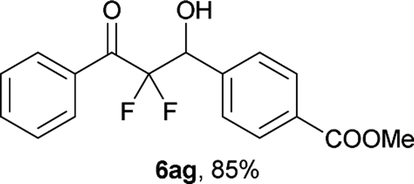
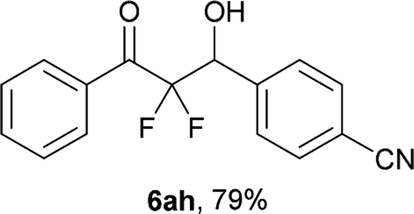
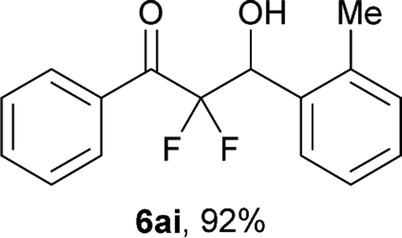
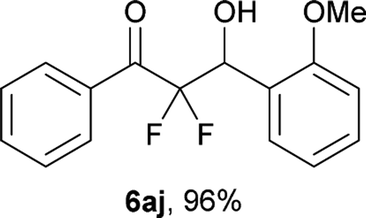
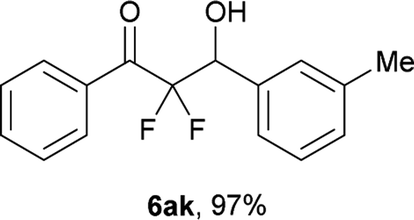
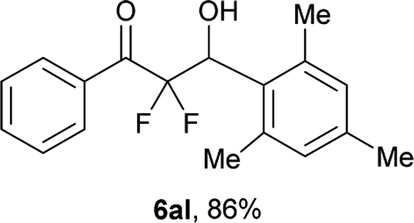
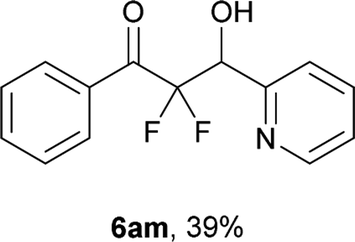
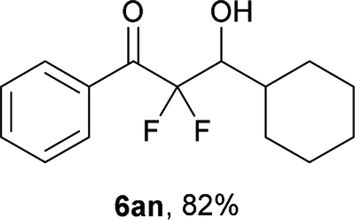
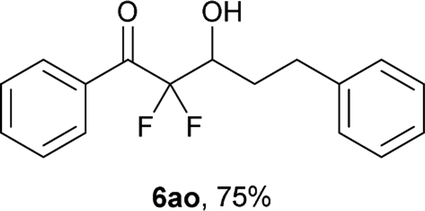
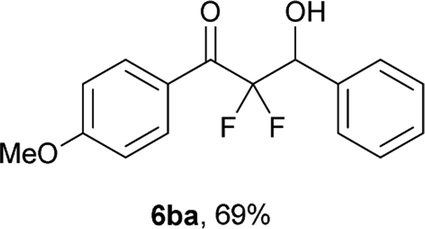
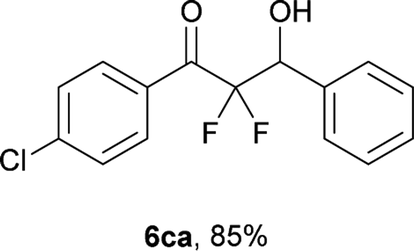
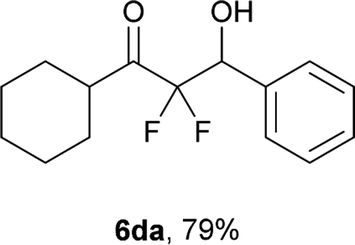
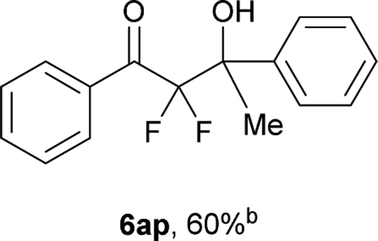
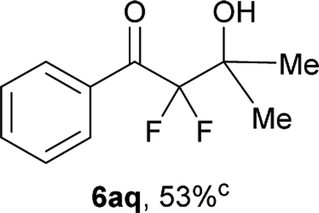
After optimization of the reaction conditions for this decarboxylative aldol reaction, various substrates were tested (Table 2). Various aldehydes 3 reacted with 2,2-difluoro-3-oxo-3-phenylpropanoate (2a) to provide the desired products in good to excellent yields. Aromatic aldehydes were especially suitable for the reaction (6ab–6al). The electroproperties and positions of the substituents on the phenyl ring of the aldehydes did not affect the yield of the reaction. Among these, the reaction of functionalized aldehydes, such as methoxycarbonyl and cyano groups, also provided the corresponding products (6ag and 6ah) in good yields. However, in the case of 2-pyridinecarboxaldehyde, only 39% of product (6am) was obtained. Furthermore, enolizable aliphatic aldehydes were also tolerated, providing the corresponding aldol products 6an and 6ao in good yields. The scope of the 2,2-difluoro-3-oxo-propanoates (2b–d) was also tested.17 Both electron-donating (CH3O) and electron-withdrawing groups (Cl) on the phenyl ring of the substrate 2 were well tolerated in the decarboxylative aldol process. Furthermore, the aliphatic substrate 2d produced the corresponding product 6da in good yield. For the reaction with ketones, when an excess amount of the ketones was used, the desired products 6ap and 6aq were obtained in moderate yields of 60% and 53%, respectively.
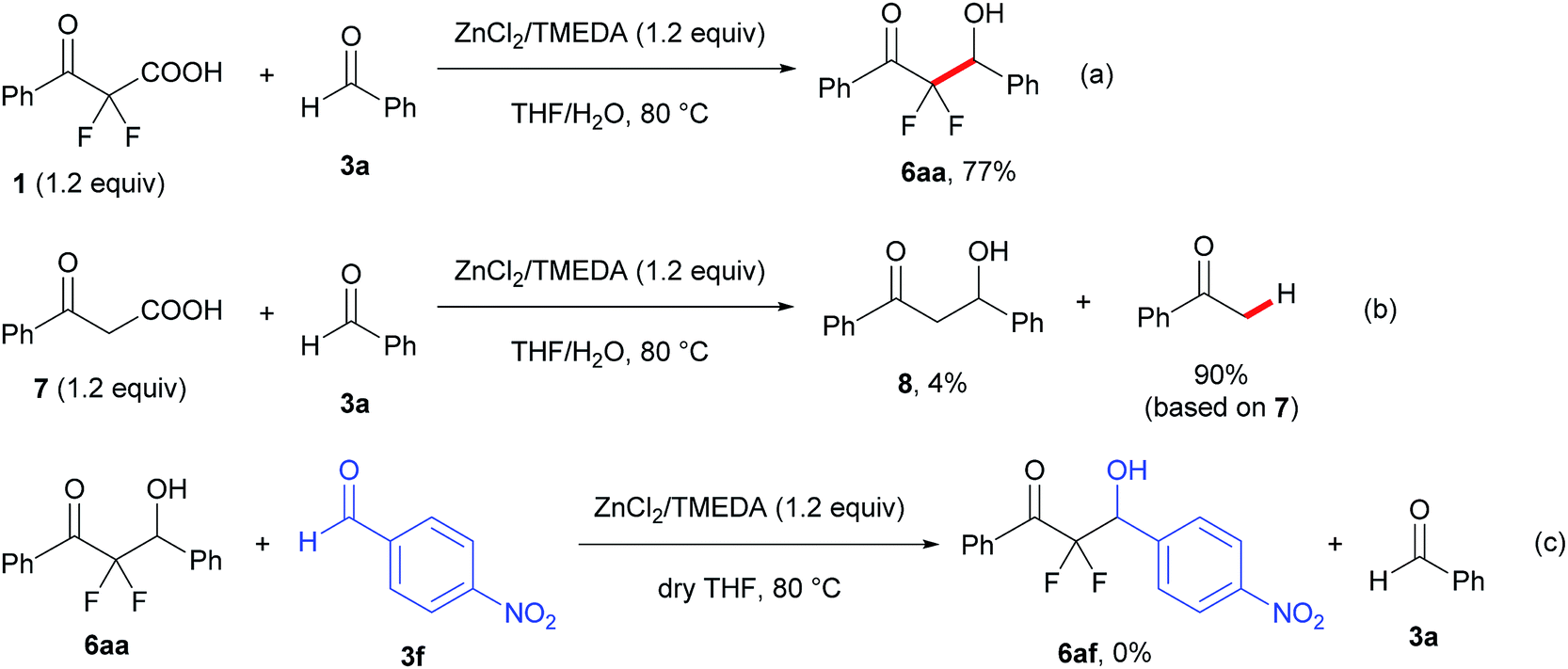
To examine the reaction mechanism, a control experiment was conducted with 2,2-difluoro-3-oxo-3-phenylpropanoic acid (1) and a non-fluorinated 3-oxo-3-phenylpropanoic acid (7) under the optimized reaction conditions (Scheme 2, eqn (a) and (b)). The reaction of compound 1 with 3a showed a decrease in the yield of product and a prolonged reaction time, which provided the aldol product 6aa in 77% yield for 19 h under the optimal conditions. However, trace amounts of the aldol product 8 were obtained from a non-fluorinated substrate 7 along with a high yield of acetophenone (90% based on 7) via a decarboxylative process. In reports on palladium-catalyzed benzylation reactions of α,α-difluoroketone enolate, Altman suggested that rehybridization of the α,α-difluorinated enolate carbanions from C (sp3) to C (sp2) actually occurs more slowly than for non-fluorinated enolates.17 This report and our results suggest that the α,α-difluorinated enolate from 2a prefers C–enolate form. As a result, the reaction of C–enolate together with the high nucleophilicity led to the aldol product more effectively than a non-fluorinated enolate generated from 7. Moreover, when a scrambling experiment of the aldol product 6aa with another aromatic aldehyde 3f was performed under the current reaction conditions (Scheme 3, eqn (c)), no retro-aldol reaction was observed and the scrambling product 6af was not formed.
 | ||
| Scheme 3 Control experiments and examination of retro-aldol reaction. | ||
On the basis of previous works on the synthesis of α,α-difluoro-β-hydroxyketones and our experiments,11,12 a tentative reaction mechanism for this decarboxylative aldol reaction of potassium α,α-difluoro-β-ketocarboxylate (2a) with benzaldehyde (3a) is proposed. First, zinc(II) is accepted by the nucleophilic enolate generated from decarboxylation of 2a. Then, the nucleophilic addition of zinc difluoroenolate to 3a occurred to lead to the formation of aldol alkoxide. Stoichiometric amount of water promotes the protonation of zinc alkoxide for the formation of the product 6aa in the equilibrium of the aldol process.
Conclusions
In conclusion, we have successfully developed a mild decarboxylative aldol reaction for potassium α,α-difluoro-β-keto carboxylate with aldehydes. The reaction is mild, with a reaction temperature below 100 °C, and a variety of α,α-difluoro-β-hydroxy ketones with biological activity could be obtained in good to excellent yields. Compared with previous methods, the substrates 2a–d used in this reaction are bench stable salts and a broad substrate scope was realized. Now we are investigating an asymmetric version of this decarboxylative aldol reaction.Experimental
General
NMR spectra were obtained from a solution in CDCl3 using 400 MHz for 1H, 100 MHz for 13C, 376 MHz for 19F. Chemical shifts of 1H and 13C NMR are reported in ppm from tetramethylsilane (TMS) as an internal standard. Chemical shifts of 19F NMR are reported in ppm from CFCl3 as an internal standard. All data are reported as follows: chemical shifts, multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, dd = double doublet, ddd = double double doublet, m = multiplet), coupling constants (Hz), and relative integration value. HRMS experiments were measured on a double-focusing mass spectrometer with an ionization mode of EI. Melting points were measured uncorrected.All experiments were carried out under argon atmosphere in flame-dried glassware using standard inert techniques for introducing reagents and solvents unless otherwise noted. Tetrahydrofuran (THF) was purchased from Kanto Chemical Co. Inc. as “Dehydrated”. All commercially available materials were used as received without further purification.
General procedure for the decarboxylative aldol reaction
To a dry and argon-flushed reaction vessel, equipped with a magnetic stirrer, were added zinc chloride N,N,N′,N′-tetramethylethylenediamine complex (151 mg, 0.6 mmol) and potassium 2,2-difluoro-3-oxopropanoate (2, 0.6 mmol). The solids were suspended in THF (4 mL), then the corresponding aldehyde (0.5 mmol) and THF solution of H2O (0.5 mL, 1 M) were added. The reaction mixture was heated at 80 °C with stirring for 5 h. The reaction was quenched with 10% aqueous HCl, and the resultant mixture was extract with AcOEt. The combined organic phases were washed with brine and dried over MgSO4. Then the extract was concentrated in vacuo, and the residue was purified by column chromatography on silica gel to give the corresponding aldol adducts 6.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4). 1H NMR (400 MHz, CDCl3) δ 8.06–8.04 (m, 2H), 7.65–7.61 (m, 1H), 7.50–7.45 (m, 4H), 7.41–7.38 (m, 3H), 5.38 (ddd, J = 18.7, 5.6, 4.6 Hz, 1H), 3.00 (d, J = 4.6 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.9 (dd, J = 31.3, 29.2 Hz), 134.7, 134.6, 132.4 (m), 130.2 (m), 129.0, 128.6, 128.3, 128.1, 115.7 (dd, J = 261.5, 256.8 Hz), 73.3 (dd, J = 29.2, 23.1 Hz); 19F NMR (376 MHz, CDCl3) δ −104.7 (dd, JFF = 293, JHF = 5.6 Hz, 1F), −116.3 (dd, JFF = 293, JHF = 18.7 Hz, 1F); HRMS (EI) m/z calcd for C15H12F2O2 [M]+ 262.0805, found 262.0799.:9). 1H NMR (400 MHz, CDCl3) δ 8.06–8.04 (m, 2H), 7.65–7.61 (m, 1H), 7.49–7.45 (m, 2H), 7.38 (d, J = 8.0 Hz, 2H), 7.20 (d, J = 8.0 Hz, 2H), 5.34 (ddd, J = 18.6, 5.6, 3.6 Hz, 1H), 2.95 (m, 1H), 2.36 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 190.9 (dd, J = 31.3, 29.5 Hz), 138.9, 134.5, 132.5, 131.7, 130.2 (m), 129.0, 128.6, 128.0, 115.8 (dd, J = 261.2, 256.1 Hz), 73.2 (dd, J = 28.2, 23.3 Hz), 21.2; 19F NMR (376 MHz, CDCl3) δ −104.8 (dd, JFF = 291, JHF = 5.6 Hz, 1F), −116.4 (dd, JFF = 291, JHF = 18.6 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O2 [M]+ 276.0962, found 276.0966.:9). 1H NMR (400 MHz, CDCl3) δ 8.06–8.04 (m, 2H), 7.65–7.61 (m, 1H), 7.49–7.45 (m, 2H), 7.42 (d, J = 8.4 Hz, 2H), 6.92 (d, J = 8.4 Hz, 2H), 5.32 (ddd, J = 18.5, 5.9, 3.8 Hz, 1H), 3.82 (s, 3H), 2.98 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 191.0 (dd, J = 30.6, 28.5 Hz), 160.1, 134.5, 132.5, 130.2 (m), 129.4, 128.6, 126.8, 115.8 (dd, J = 263.9, 256.3 Hz), 113.8, 72.9 (dd, J = 27.8, 23.1 Hz), 55.3; 19F NMR (376 MHz, CDCl3) δ −105.1 (dd, JFF = 290, JHF = 5.9 Hz, 1F), −116.4 (dd, JFF = 290, JHF = 18.5 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O3 [M]+ 292.0911, found 292.0912.:9). mp 104.0–104.5 °C (from Et2O–C6, lit. 87–89 °C); 1H NMR (400 MHz, CDCl3) δ 8.08–8.06 (m, 2H), 7.68–7.63 (m, 5H), 7.52–7.48 (m, 2H), 5.46 (ddd, J = 19.1, 5.2, 4.5 Hz, 1H), 3.23 (d, J = 4.5 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 191.5 (dd, J = 31.6, 30.0 Hz), 138.5, 134.9, 130.0 (m), 131.1 (q, J = 32.5 Hz), 130.3 (m), 128.8, 128.6, 125.1 (m), 123.9 (q, J = 271.9 Hz), 115.3 (dd, J = 273.9, 257.4 Hz), 72.6 (dd, J = 28.4, 23.0 Hz); 19F NMR (376 MHz, CDCl3) δ −62.6 (s, 3F), −103.8 (dd, JFF = 299, JHF = 5.2 Hz, 1F), −116.5 (dd, JFF = 299, JHF = 19.1 Hz, 1F); HRMS (EI) m/z calcd for C16H11F5O2 [M]+ 330.0679, found 330.0688.:4). mp 103.0–103.5 °C (from Et2O–C6, 96–97 °C); 1H NMR (400 MHz, CDCl3) δ 8.07–8.05 (m, 2H), 7.67–7.63 (m, 1H), 7.54–7.47 (m, 4H), 7.39–7.37 (m, 2H), 5.35 (ddd, J = 18.8, 5.3, 4.5 Hz, 1H), 3.08 (d, J = 4.5 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.6 (dd, J = 30.6, 29.0 Hz), 134.8, 133.6, 132.0 (m), 131.4, 130.3 (m), 129.8, 128.8, 123.2, 115.3 (dd, J = 269.1, 257.2 Hz), 72.6 (dd, J = 28.0, 22.5 Hz); 19F NMR (376 MHz, CDCl3) δ −104.2 (dd, JFF = 298, JHF = 5.3 Hz, 1F), −116.6 (dd, JFF = 298, JHF = 18.8 Hz, 1F); HRMS (EI) m/z calcd for C15H11BrF2O2 [M]+ 339.9910, found 339.9912 (16.9), 341.9895(16.8).:7). mp 114.0–115.0 °C (from Et2O–C6); 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 8.6 Hz, 2H), 8.09–8.08 (m, 2H), 7.72–7.65 (m, 3H), 7.53–7.49 (m, 2H), 5.53 (ddd, J = 19.3, 4.5, 4.1 Hz, 1H), 3.35 (d, J = 4.5 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.2 (dd, J = 31.6, 29.6 Hz), 148.3, 141.6, 135.1, 131.7 (m), 130.3 (m), 129.2, 128.8, 123.3, 115.0 (dd, J = 268.2, 257.2 Hz), 72.2 (dd, J = 28.8, 22.5 Hz); 19F NMR (376 MHz, CDCl3) δ −103.3 (dd, JFF = 301, JHF = 4.1 Hz, 1F), −116.5 (dd, JFF = 301, JHF = 19.3 Hz, 1F); HRMS (EI) m/z calcd for C15H11F2NO4 [M]+ 307.0656, found 307.0655.:4). mp 100.5–101.5 °C (from Et2O–C6); 1H NMR (400 MHz, CDCl3) δ 8.07 (m, 4H), 7.67–7.64 (m, 1H), 7.59 (d, J = 8.4, 2H), 7.51–7.47 (m, 2H), 5.46 (ddd, J = 19.0, 4.6, 4.6 Hz, 1H), 3.93 (s, 3H), 3.17 (d, J = 4.6 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.6 (dd, J = 32.0, 29.6 Hz), 166.8, 139.5, 134.8, 132.0 (m), 130.7, 130.3 (m), 129.5, 128.8, 128.2, 115.3 (dd, J = 261.8, 257.4 Hz), 72.8 (dd, J = 28.3, 23.0 Hz), 52.2; 19F NMR (376 MHz, CDCl3) δ −103.9 (dd, JFF = 297, JHF = 4.6 Hz, 1F), −116.4 (dd, JFF = 297, JHF = 19.0 Hz, 1F); HRMS (EI) m/z calcd for C17H14F2O4 [M]+ 320.0860, found 320.0864.:9). mp 94.0–95.5 °C (from CHCl3–C6, lit. 79–81 °C); 1H NMR (400 MHz, CDCl3) δ 8.08–8.07 (m, 2H), 7.71–7.63 (m, 5H), 7.52–7.48 (m, 2H), 5.46 (ddd, J = 19.1, 4.3, 4.1 Hz, 1H), 3.31 (d, J = 4.3 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.3 (dd, J = 31.4, 29.7 Hz), 139.8, 135.0, 131.9, 131.8 (m), 130.3 (m), 128.9, 128.8, 118.5, 115.1 (dd, J = 269.7, 258.1 Hz), 112.8, 72.4 (dd, J = 28.6, 23.0 Hz); 19F NMR (376 MHz, CDCl3) δ −103.5 (dd, JFF = 301, JHF = 4.1 Hz, 1F), −116.5 (dd, JFF = 301, JHF = 19.1 Hz, 1F); HRMS (EI) m/z calcd for C16H11F2NO2 [M]+ 287.0758, found 287.0760.:9). 1H NMR (400 MHz, CDCl3) δ 8.09–8.07 (m, 2H), 7.67–7.62 (m, 2H), 7.50–7.46 (m, 2H), 7.29–7.19 (m, 3H), 5.70 (ddd, J = 20.3, 4.4, 3.8 Hz, 1H), 2.91 (d, J = 4.4 Hz, 1H), 2.39 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 191.0 (dd, J = 31.9, 29.4 Hz), 136.8, 134.6, 133.3, 132.4 (m), 130.4, 130.3 (m), 128.8, 128.7, 128.1, 126.1, 116.4 (dd, J = 257.9, 255.5 Hz), 69.0 (dd, J = 29.2, 22.6 Hz), 19.6; 19F NMR (376 MHz, CDCl3) δ −104.1 (dd, JFF = 294, JHF = 3.8 Hz, 1F), −116.9 (dd, JFF = 294, JHF = 20.3 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O2 [M]+ 276.0962, found 276.0967.:4). 1H NMR (400 MHz, CDCl3) δ 8.05–8.03 (m, 2H), 7.63–7.59 (m, 1H), 7.49–7.44 (m, 3H), 7.36–7.32 (m, 1H), 7.05–7.01 (m, 1H), 6.87–6.85 (m, 1H), 5.66 (ddd, J = 17.3, 7.3, 7.1 Hz, 1H), 3.69 (s, 3H), 3.58 (d, J = 7.3 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.1 (dd, J = 30.0, 29.1 Hz), 157.4, 134.2, 132.9, 130.2, 130.1 (m), 129.6, 128.5, 122.8, 120.9, 116.8 (dd, J = 264.0, 258.2 Hz), 110.9, 70.2 (dd, J = 28.0, 24.4 Hz), 55.3; 19F NMR (376 MHz, CDCl3) δ −106.8 (dd, JFF = 275, JHF = 7.1 Hz, 1F), −114.7 (dd, JFF = 275, JHF = 17.3 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O3 [M]+ 292.0911, found 292.0912.:9). 1H NMR (400 MHz, CDCl3) δ 8.06–8.04 (m, 2H), 7.65–7.61 (m, 1H), 7.49–7.45 (m, 2H), 7.31–7.17 (m, 4H), 5.33 (ddd, J = 18.7, 5.4, 4.3 Hz, 1H), 2.96 (d, J = 4.3 Hz, 1H), 2.37 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 191.0 (dd, J = 31.0, 29.3 Hz), 138.0, 134.6, 134.5, 132.5 (m), 130.2 (m), 129.8, 128.7, 128.6, 128.2, 125.2, 115.8 (dd, J = 265.9, 256.8 Hz), 73.3 (dd, J = 28.7, 23.1 Hz), 21.4; 19F NMR (376 MHz, CDCl3) δ −104.6 (dd, JFF = 291, JHF = 5.4 Hz, 1F), −116.4 (dd, JFF = 291, JHF = 18.7 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O2 [M]+ 276.0962, found 276.0965.:9). 1H NMR (400 MHz, CDCl3) δ 8.11–8.09 (m, 2H), 7.66–7.62 (m, 1H), 7.51–7.47 (m, 2H), 6.89 (s, 2H), 5.91 (ddd, J = 26.4, 4.8, 2.7 Hz, 1H), 2.81 (d, J = 4.8 Hz, 1H), 2.45 (bs, 6H), 2.23 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 191.5 (dd, J = 30.9, 28.9 Hz), 138.2, 134.5, 132.5, 130.2 (m), 128.6, 127.6, 117.6 (dd, J = 265.6, 252.4 Hz), 70.5 (dd, J = 30.5, 22.2 Hz), 21.2, 20.8; 19F NMR (376 MHz, CDCl3) δ −102.0 (dd, JFF = 292, JHF = 2.7 Hz, 1F), −114.6 (dd, JFF = 292, JHF = 26.4 Hz, 1F); HRMS (EI) m/z calcd for C18H18F2O2 [M]+ 304.1275, found 304.1279.:7). mp 82.5–83.5 °C (from Et2O–C6); 1H NMR (400 MHz, CDCl3) δ 8.61 (m, 1H), 8.10–8.08 (m, 2H), 7.81–7.76 (m, 1H), 7.64–7.60 (m, 1H), 7.53–7.46 (m, 3H), 7.37–7.34 (m, 1H), 5.37 (dd, J = 17.9, 4.9, 1H), 1.60 (bs, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.4 (dd, J = 29.6, 26.6 Hz), 152.3, 148.1, 136.9, 134.1, 133.3, 130.2 (m), 128.5, 124.0, 123.1 (m), 116.7 (dd, J = 263.6, 257.7 Hz), 71.8 (dd, J = 29.2, 25.3 Hz); 19F NMR (376 MHz, CDCl3) δ −104.7 (dd, JFF = 272, JHF = 4.9 Hz, 1F), −116.7 (dd, JFF = 272, JHF = 17.9 Hz, 1F); HRMS (EI) m/z calcd for C14H11F2NO2 [M]+ 263.0758, found 263.0759.:9). 1H NMR (400 MHz, CDCl3) δ 8.10–8.08 (m, 2H), 7.66–7.62 (m, 1H), 7.52–7.48 (m, 2H), 4.06 (m, 1H), 2.33 (d, J = 6.9 Hz, 1H), 1.99–1.66 (m, 6H), 1.39–1.16 (m, 5H); 13C NMR (CDCl3, 100 MHz) δ 190.7 (m), 134.4, 132.5, 130.1 (m), 128.7, 117.7 (dd, J = 262.8, 257.8 Hz), 74.7 (dd, J = 26.2, 22.7 Hz), 38.1, 30.1, 27.3, 26.2, 26.1, 25.9; 19F NMR (376 MHz, CDCl3) δ −104.9 (dd, JFF = 290, JHF = 6.3 Hz, 1F), −114.4 (dd, JFF = 290, JHF = 19.8 Hz, 1F); HRMS (EI) m/z calcd for C15H18F2O2 [M]+ 268.1275, found 268.1280.:9). 1H NMR (400 MHz, CDCl3) δ 8.10–8.08 (m, 2H), 7.66–7.62 (m, 1H), 7.51–7.47 (m, 2H), 7.32–7.18 (m, 5H), 4.23–4.19 (m, 1H), 2.99 (ddd, J = 13.8, 8.8, 5.2 Hz, 1H), 2.77 (ddd, J = 13.8, 8.8, 8.5 Hz, 1H), 2.56 (d, J = 5.6 Hz, 1H), 2.13–1.96 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 190.5 (dd, J = 31.7, 30.7 Hz), 141.0, 134.7, 132.1 (m), 130.2 (m) 128.7, 128.5, 126.1, 116.4 (dd, J = 266.2, 257.2 Hz), 70.4 (dd, J = 27.3, 24.3 Hz), 31.3, 30.3; 19F NMR (376 MHz, CDCl3) δ −107.3 (dd, JFF = 298, JHF = 5.3 Hz, 1F), −116.9 (dd, JFF = 298, JHF = 17.6 Hz, 1F); HRMS (EI) m/z calcd for C17H16F2O2 [M]+ 290.1118, found 290.1121.:4). mp 84.5–85.0 °C (from Et2O–C6, lit. 68–69 °C); 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.9 Hz, 2H), 7.51–7.49 (m, 2H), 7.41–7.37 (m, 3H), 6.94 (d, J = 8.9 Hz, 2H), 5.36 (ddd, J = 19.1, 5.2, 4.3 Hz, 1H), 3.89 (s, 3H), 3.16 (d, J = 4.3 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 189.1 (dd, J = 31.3, 30.1 Hz), 164.8, 134.7, 132.9 (m), 128.9, 128.2, 128.1, 125.0, 115.7 (dd, J = 262.8, 256.8 Hz), 114.0, 73.3 (dd, J = 29.3, 23.1 Hz), 55.6; 19F NMR (376 MHz, CDCl3) δ −103.8 (dd, JFF = 295, JHF = 5.2 Hz, 1F), −115.8 (dd, JFF = 295, JHF = 19.1 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O3 [M]+ 292.0911, found 292.0912.:9). mp 115.5–116.0 °C (from CHCl3–C6, lit. 112–113 °C); 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.7 Hz, 2H), 7.49–7.38 (m, 7H), 5.36 (ddd, J = 18.5, 5.9, 4.5 Hz, 1H), 2.92 (d, J = 4.5 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 189.8 (dd, J = 31.0, 29.4 Hz), 141.3, 134.5, 131.6 (m), 130.7 (m), 129.2, 129.1, 128.4, 128.1, 115.7 (dd, J = 260.1, 256.0 Hz), 73.3 (dd, J = 27.6, 23.1 Hz); 19F NMR (376 MHz, CDCl3) δ −104.8 (dd, JFF = 291, JHF = 5.9 Hz, 1F), −116.3 (dd, JFF = 291, JHF = 18.5 Hz, 1F); HRMS (EI) m/z calcd for C15H11ClF2O2 [M]+ 296.0416, found 296.0411 (55.1), 298.0392 (18.4).:9). mp 58.0–59.0 °C (from petroleum ether, lit. 58–60 °C); NMR (400 MHz, CDCl3) δ 7.42–7.37 (m, 5H), 5.17 (ddd, J = 16.5, 7.7, 4.8 Hz, 1H), 2.77–2.72 (m, 1H), 2.69 (d, J = 4.8 Hz, 1H), 1.84–1.64 (m, 5H), 1.37–1.16 (m, 5H); 13C NMR (CDCl3, 100 MHz) δ 205.5 (dd, J = 30.3, 26.7 Hz), 134.8, 129.0, 128.4, 127.8, 115.0 (dd, J = 262.7, 256.9 Hz), 73.1 (dd, J = 27.2, 23.9 Hz), 27.8, 27.7, 25.5, 25.3, 25.2; 19F NMR (376 MHz, CDCl3) δ −112.4 (dd, JFF = 273, JHF = 7.7 Hz, 1F), −122.2 (dd, JFF = 273, JHF = 16.5 Hz, 1F); HRMS (EI) m/z calcd for C15H18F2O2 [M]+ 268.1275, found 268.1274.:9). 1H NMR (400 MHz, CDCl3) δ 7.92–7.90 (m, 2H), 7.58–7.54 (m, 3H), 7.41–7.28 (m, 5H), 3.54 (s, 1H), 1.82 (t, J = 1.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 191.6 (t, J = 31.0 Hz), 140.2, 134.3, 133.1, 130.2 (m), 128.4, 128.1, 128.0, 126.3, 116.5 (t, J = 263.0 Hz), 76.4 (t, J = 24.4 Hz), 24.0 (t, J = 2.9 Hz); 19F NMR (376 MHz, CDCl3) δ −107.4 (m, 2F); HRMS (EI) m/z calcd for C16H14F2O2 [M]+ 276.0962, found 276.0968.:3). 1H NMR (400 MHz, CDCl3) δ 8.13–8.11 (m, 2H), 7.66–7.62 (m, 1H), 7.52–7.48 (m, 2H), 2.79 (bs, 1H), 1.45 (t, J = 1.5 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ 191.2 (t, J = 31.0 Hz), 134.5, 130.0 (m), 130.4 (m), 128.6, 116.9 (t, J = 261.6 Hz), 73.3 (t, J = 24.7 Hz), 23.6 (t, J = 2.6 Hz); 19F NMR (376 MHz, CDCl3) δ −110.5 (m, 2F); HRMS (EI) m/z calcd for C11H12F2O2 [M]+ 214.0805, found 214.0799.
:4). 1H NMR (400 MHz, CDCl3) δ 8.06–8.04 (m, 2H), 7.65–7.61 (m, 1H), 7.50–7.45 (m, 4H), 7.41–7.38 (m, 3H), 5.38 (ddd, J = 18.7, 5.6, 4.6 Hz, 1H), 3.00 (d, J = 4.6 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.9 (dd, J = 31.3, 29.2 Hz), 134.7, 134.6, 132.4 (m), 130.2 (m), 129.0, 128.6, 128.3, 128.1, 115.7 (dd, J = 261.5, 256.8 Hz), 73.3 (dd, J = 29.2, 23.1 Hz); 19F NMR (376 MHz, CDCl3) δ −104.7 (dd, JFF = 293, JHF = 5.6 Hz, 1F), −116.3 (dd, JFF = 293, JHF = 18.7 Hz, 1F); HRMS (EI) m/z calcd for C15H12F2O2 [M]+ 262.0805, found 262.0799.:9). 1H NMR (400 MHz, CDCl3) δ 8.06–8.04 (m, 2H), 7.65–7.61 (m, 1H), 7.49–7.45 (m, 2H), 7.38 (d, J = 8.0 Hz, 2H), 7.20 (d, J = 8.0 Hz, 2H), 5.34 (ddd, J = 18.6, 5.6, 3.6 Hz, 1H), 2.95 (m, 1H), 2.36 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 190.9 (dd, J = 31.3, 29.5 Hz), 138.9, 134.5, 132.5, 131.7, 130.2 (m), 129.0, 128.6, 128.0, 115.8 (dd, J = 261.2, 256.1 Hz), 73.2 (dd, J = 28.2, 23.3 Hz), 21.2; 19F NMR (376 MHz, CDCl3) δ −104.8 (dd, JFF = 291, JHF = 5.6 Hz, 1F), −116.4 (dd, JFF = 291, JHF = 18.6 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O2 [M]+ 276.0962, found 276.0966.:9). 1H NMR (400 MHz, CDCl3) δ 8.06–8.04 (m, 2H), 7.65–7.61 (m, 1H), 7.49–7.45 (m, 2H), 7.42 (d, J = 8.4 Hz, 2H), 6.92 (d, J = 8.4 Hz, 2H), 5.32 (ddd, J = 18.5, 5.9, 3.8 Hz, 1H), 3.82 (s, 3H), 2.98 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 191.0 (dd, J = 30.6, 28.5 Hz), 160.1, 134.5, 132.5, 130.2 (m), 129.4, 128.6, 126.8, 115.8 (dd, J = 263.9, 256.3 Hz), 113.8, 72.9 (dd, J = 27.8, 23.1 Hz), 55.3; 19F NMR (376 MHz, CDCl3) δ −105.1 (dd, JFF = 290, JHF = 5.9 Hz, 1F), −116.4 (dd, JFF = 290, JHF = 18.5 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O3 [M]+ 292.0911, found 292.0912.:9). mp 104.0–104.5 °C (from Et2O–C6, lit. 87–89 °C); 1H NMR (400 MHz, CDCl3) δ 8.08–8.06 (m, 2H), 7.68–7.63 (m, 5H), 7.52–7.48 (m, 2H), 5.46 (ddd, J = 19.1, 5.2, 4.5 Hz, 1H), 3.23 (d, J = 4.5 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 191.5 (dd, J = 31.6, 30.0 Hz), 138.5, 134.9, 130.0 (m), 131.1 (q, J = 32.5 Hz), 130.3 (m), 128.8, 128.6, 125.1 (m), 123.9 (q, J = 271.9 Hz), 115.3 (dd, J = 273.9, 257.4 Hz), 72.6 (dd, J = 28.4, 23.0 Hz); 19F NMR (376 MHz, CDCl3) δ −62.6 (s, 3F), −103.8 (dd, JFF = 299, JHF = 5.2 Hz, 1F), −116.5 (dd, JFF = 299, JHF = 19.1 Hz, 1F); HRMS (EI) m/z calcd for C16H11F5O2 [M]+ 330.0679, found 330.0688.:4). mp 103.0–103.5 °C (from Et2O–C6, 96–97 °C); 1H NMR (400 MHz, CDCl3) δ 8.07–8.05 (m, 2H), 7.67–7.63 (m, 1H), 7.54–7.47 (m, 4H), 7.39–7.37 (m, 2H), 5.35 (ddd, J = 18.8, 5.3, 4.5 Hz, 1H), 3.08 (d, J = 4.5 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.6 (dd, J = 30.6, 29.0 Hz), 134.8, 133.6, 132.0 (m), 131.4, 130.3 (m), 129.8, 128.8, 123.2, 115.3 (dd, J = 269.1, 257.2 Hz), 72.6 (dd, J = 28.0, 22.5 Hz); 19F NMR (376 MHz, CDCl3) δ −104.2 (dd, JFF = 298, JHF = 5.3 Hz, 1F), −116.6 (dd, JFF = 298, JHF = 18.8 Hz, 1F); HRMS (EI) m/z calcd for C15H11BrF2O2 [M]+ 339.9910, found 339.9912 (16.9), 341.9895(16.8).:7). mp 114.0–115.0 °C (from Et2O–C6); 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 8.6 Hz, 2H), 8.09–8.08 (m, 2H), 7.72–7.65 (m, 3H), 7.53–7.49 (m, 2H), 5.53 (ddd, J = 19.3, 4.5, 4.1 Hz, 1H), 3.35 (d, J = 4.5 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.2 (dd, J = 31.6, 29.6 Hz), 148.3, 141.6, 135.1, 131.7 (m), 130.3 (m), 129.2, 128.8, 123.3, 115.0 (dd, J = 268.2, 257.2 Hz), 72.2 (dd, J = 28.8, 22.5 Hz); 19F NMR (376 MHz, CDCl3) δ −103.3 (dd, JFF = 301, JHF = 4.1 Hz, 1F), −116.5 (dd, JFF = 301, JHF = 19.3 Hz, 1F); HRMS (EI) m/z calcd for C15H11F2NO4 [M]+ 307.0656, found 307.0655.:4). mp 100.5–101.5 °C (from Et2O–C6); 1H NMR (400 MHz, CDCl3) δ 8.07 (m, 4H), 7.67–7.64 (m, 1H), 7.59 (d, J = 8.4, 2H), 7.51–7.47 (m, 2H), 5.46 (ddd, J = 19.0, 4.6, 4.6 Hz, 1H), 3.93 (s, 3H), 3.17 (d, J = 4.6 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.6 (dd, J = 32.0, 29.6 Hz), 166.8, 139.5, 134.8, 132.0 (m), 130.7, 130.3 (m), 129.5, 128.8, 128.2, 115.3 (dd, J = 261.8, 257.4 Hz), 72.8 (dd, J = 28.3, 23.0 Hz), 52.2; 19F NMR (376 MHz, CDCl3) δ −103.9 (dd, JFF = 297, JHF = 4.6 Hz, 1F), −116.4 (dd, JFF = 297, JHF = 19.0 Hz, 1F); HRMS (EI) m/z calcd for C17H14F2O4 [M]+ 320.0860, found 320.0864.:9). mp 94.0–95.5 °C (from CHCl3–C6, lit. 79–81 °C); 1H NMR (400 MHz, CDCl3) δ 8.08–8.07 (m, 2H), 7.71–7.63 (m, 5H), 7.52–7.48 (m, 2H), 5.46 (ddd, J = 19.1, 4.3, 4.1 Hz, 1H), 3.31 (d, J = 4.3 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.3 (dd, J = 31.4, 29.7 Hz), 139.8, 135.0, 131.9, 131.8 (m), 130.3 (m), 128.9, 128.8, 118.5, 115.1 (dd, J = 269.7, 258.1 Hz), 112.8, 72.4 (dd, J = 28.6, 23.0 Hz); 19F NMR (376 MHz, CDCl3) δ −103.5 (dd, JFF = 301, JHF = 4.1 Hz, 1F), −116.5 (dd, JFF = 301, JHF = 19.1 Hz, 1F); HRMS (EI) m/z calcd for C16H11F2NO2 [M]+ 287.0758, found 287.0760.:9). 1H NMR (400 MHz, CDCl3) δ 8.09–8.07 (m, 2H), 7.67–7.62 (m, 2H), 7.50–7.46 (m, 2H), 7.29–7.19 (m, 3H), 5.70 (ddd, J = 20.3, 4.4, 3.8 Hz, 1H), 2.91 (d, J = 4.4 Hz, 1H), 2.39 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 191.0 (dd, J = 31.9, 29.4 Hz), 136.8, 134.6, 133.3, 132.4 (m), 130.4, 130.3 (m), 128.8, 128.7, 128.1, 126.1, 116.4 (dd, J = 257.9, 255.5 Hz), 69.0 (dd, J = 29.2, 22.6 Hz), 19.6; 19F NMR (376 MHz, CDCl3) δ −104.1 (dd, JFF = 294, JHF = 3.8 Hz, 1F), −116.9 (dd, JFF = 294, JHF = 20.3 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O2 [M]+ 276.0962, found 276.0967.:4). 1H NMR (400 MHz, CDCl3) δ 8.05–8.03 (m, 2H), 7.63–7.59 (m, 1H), 7.49–7.44 (m, 3H), 7.36–7.32 (m, 1H), 7.05–7.01 (m, 1H), 6.87–6.85 (m, 1H), 5.66 (ddd, J = 17.3, 7.3, 7.1 Hz, 1H), 3.69 (s, 3H), 3.58 (d, J = 7.3 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.1 (dd, J = 30.0, 29.1 Hz), 157.4, 134.2, 132.9, 130.2, 130.1 (m), 129.6, 128.5, 122.8, 120.9, 116.8 (dd, J = 264.0, 258.2 Hz), 110.9, 70.2 (dd, J = 28.0, 24.4 Hz), 55.3; 19F NMR (376 MHz, CDCl3) δ −106.8 (dd, JFF = 275, JHF = 7.1 Hz, 1F), −114.7 (dd, JFF = 275, JHF = 17.3 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O3 [M]+ 292.0911, found 292.0912.:9). 1H NMR (400 MHz, CDCl3) δ 8.06–8.04 (m, 2H), 7.65–7.61 (m, 1H), 7.49–7.45 (m, 2H), 7.31–7.17 (m, 4H), 5.33 (ddd, J = 18.7, 5.4, 4.3 Hz, 1H), 2.96 (d, J = 4.3 Hz, 1H), 2.37 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 191.0 (dd, J = 31.0, 29.3 Hz), 138.0, 134.6, 134.5, 132.5 (m), 130.2 (m), 129.8, 128.7, 128.6, 128.2, 125.2, 115.8 (dd, J = 265.9, 256.8 Hz), 73.3 (dd, J = 28.7, 23.1 Hz), 21.4; 19F NMR (376 MHz, CDCl3) δ −104.6 (dd, JFF = 291, JHF = 5.4 Hz, 1F), −116.4 (dd, JFF = 291, JHF = 18.7 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O2 [M]+ 276.0962, found 276.0965.:9). 1H NMR (400 MHz, CDCl3) δ 8.11–8.09 (m, 2H), 7.66–7.62 (m, 1H), 7.51–7.47 (m, 2H), 6.89 (s, 2H), 5.91 (ddd, J = 26.4, 4.8, 2.7 Hz, 1H), 2.81 (d, J = 4.8 Hz, 1H), 2.45 (bs, 6H), 2.23 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 191.5 (dd, J = 30.9, 28.9 Hz), 138.2, 134.5, 132.5, 130.2 (m), 128.6, 127.6, 117.6 (dd, J = 265.6, 252.4 Hz), 70.5 (dd, J = 30.5, 22.2 Hz), 21.2, 20.8; 19F NMR (376 MHz, CDCl3) δ −102.0 (dd, JFF = 292, JHF = 2.7 Hz, 1F), −114.6 (dd, JFF = 292, JHF = 26.4 Hz, 1F); HRMS (EI) m/z calcd for C18H18F2O2 [M]+ 304.1275, found 304.1279.:7). mp 82.5–83.5 °C (from Et2O–C6); 1H NMR (400 MHz, CDCl3) δ 8.61 (m, 1H), 8.10–8.08 (m, 2H), 7.81–7.76 (m, 1H), 7.64–7.60 (m, 1H), 7.53–7.46 (m, 3H), 7.37–7.34 (m, 1H), 5.37 (dd, J = 17.9, 4.9, 1H), 1.60 (bs, 1H); 13C NMR (CDCl3, 100 MHz) δ 190.4 (dd, J = 29.6, 26.6 Hz), 152.3, 148.1, 136.9, 134.1, 133.3, 130.2 (m), 128.5, 124.0, 123.1 (m), 116.7 (dd, J = 263.6, 257.7 Hz), 71.8 (dd, J = 29.2, 25.3 Hz); 19F NMR (376 MHz, CDCl3) δ −104.7 (dd, JFF = 272, JHF = 4.9 Hz, 1F), −116.7 (dd, JFF = 272, JHF = 17.9 Hz, 1F); HRMS (EI) m/z calcd for C14H11F2NO2 [M]+ 263.0758, found 263.0759.:9). 1H NMR (400 MHz, CDCl3) δ 8.10–8.08 (m, 2H), 7.66–7.62 (m, 1H), 7.52–7.48 (m, 2H), 4.06 (m, 1H), 2.33 (d, J = 6.9 Hz, 1H), 1.99–1.66 (m, 6H), 1.39–1.16 (m, 5H); 13C NMR (CDCl3, 100 MHz) δ 190.7 (m), 134.4, 132.5, 130.1 (m), 128.7, 117.7 (dd, J = 262.8, 257.8 Hz), 74.7 (dd, J = 26.2, 22.7 Hz), 38.1, 30.1, 27.3, 26.2, 26.1, 25.9; 19F NMR (376 MHz, CDCl3) δ −104.9 (dd, JFF = 290, JHF = 6.3 Hz, 1F), −114.4 (dd, JFF = 290, JHF = 19.8 Hz, 1F); HRMS (EI) m/z calcd for C15H18F2O2 [M]+ 268.1275, found 268.1280.:9). 1H NMR (400 MHz, CDCl3) δ 8.10–8.08 (m, 2H), 7.66–7.62 (m, 1H), 7.51–7.47 (m, 2H), 7.32–7.18 (m, 5H), 4.23–4.19 (m, 1H), 2.99 (ddd, J = 13.8, 8.8, 5.2 Hz, 1H), 2.77 (ddd, J = 13.8, 8.8, 8.5 Hz, 1H), 2.56 (d, J = 5.6 Hz, 1H), 2.13–1.96 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 190.5 (dd, J = 31.7, 30.7 Hz), 141.0, 134.7, 132.1 (m), 130.2 (m) 128.7, 128.5, 126.1, 116.4 (dd, J = 266.2, 257.2 Hz), 70.4 (dd, J = 27.3, 24.3 Hz), 31.3, 30.3; 19F NMR (376 MHz, CDCl3) δ −107.3 (dd, JFF = 298, JHF = 5.3 Hz, 1F), −116.9 (dd, JFF = 298, JHF = 17.6 Hz, 1F); HRMS (EI) m/z calcd for C17H16F2O2 [M]+ 290.1118, found 290.1121.:4). mp 84.5–85.0 °C (from Et2O–C6, lit. 68–69 °C); 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.9 Hz, 2H), 7.51–7.49 (m, 2H), 7.41–7.37 (m, 3H), 6.94 (d, J = 8.9 Hz, 2H), 5.36 (ddd, J = 19.1, 5.2, 4.3 Hz, 1H), 3.89 (s, 3H), 3.16 (d, J = 4.3 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 189.1 (dd, J = 31.3, 30.1 Hz), 164.8, 134.7, 132.9 (m), 128.9, 128.2, 128.1, 125.0, 115.7 (dd, J = 262.8, 256.8 Hz), 114.0, 73.3 (dd, J = 29.3, 23.1 Hz), 55.6; 19F NMR (376 MHz, CDCl3) δ −103.8 (dd, JFF = 295, JHF = 5.2 Hz, 1F), −115.8 (dd, JFF = 295, JHF = 19.1 Hz, 1F); HRMS (EI) m/z calcd for C16H14F2O3 [M]+ 292.0911, found 292.0912.:9). mp 115.5–116.0 °C (from CHCl3–C6, lit. 112–113 °C); 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.7 Hz, 2H), 7.49–7.38 (m, 7H), 5.36 (ddd, J = 18.5, 5.9, 4.5 Hz, 1H), 2.92 (d, J = 4.5 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 189.8 (dd, J = 31.0, 29.4 Hz), 141.3, 134.5, 131.6 (m), 130.7 (m), 129.2, 129.1, 128.4, 128.1, 115.7 (dd, J = 260.1, 256.0 Hz), 73.3 (dd, J = 27.6, 23.1 Hz); 19F NMR (376 MHz, CDCl3) δ −104.8 (dd, JFF = 291, JHF = 5.9 Hz, 1F), −116.3 (dd, JFF = 291, JHF = 18.5 Hz, 1F); HRMS (EI) m/z calcd for C15H11ClF2O2 [M]+ 296.0416, found 296.0411 (55.1), 298.0392 (18.4).:9). mp 58.0–59.0 °C (from petroleum ether, lit. 58–60 °C); NMR (400 MHz, CDCl3) δ 7.42–7.37 (m, 5H), 5.17 (ddd, J = 16.5, 7.7, 4.8 Hz, 1H), 2.77–2.72 (m, 1H), 2.69 (d, J = 4.8 Hz, 1H), 1.84–1.64 (m, 5H), 1.37–1.16 (m, 5H); 13C NMR (CDCl3, 100 MHz) δ 205.5 (dd, J = 30.3, 26.7 Hz), 134.8, 129.0, 128.4, 127.8, 115.0 (dd, J = 262.7, 256.9 Hz), 73.1 (dd, J = 27.2, 23.9 Hz), 27.8, 27.7, 25.5, 25.3, 25.2; 19F NMR (376 MHz, CDCl3) δ −112.4 (dd, JFF = 273, JHF = 7.7 Hz, 1F), −122.2 (dd, JFF = 273, JHF = 16.5 Hz, 1F); HRMS (EI) m/z calcd for C15H18F2O2 [M]+ 268.1275, found 268.1274.:9). 1H NMR (400 MHz, CDCl3) δ 7.92–7.90 (m, 2H), 7.58–7.54 (m, 3H), 7.41–7.28 (m, 5H), 3.54 (s, 1H), 1.82 (t, J = 1.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 191.6 (t, J = 31.0 Hz), 140.2, 134.3, 133.1, 130.2 (m), 128.4, 128.1, 128.0, 126.3, 116.5 (t, J = 263.0 Hz), 76.4 (t, J = 24.4 Hz), 24.0 (t, J = 2.9 Hz); 19F NMR (376 MHz, CDCl3) δ −107.4 (m, 2F); HRMS (EI) m/z calcd for C16H14F2O2 [M]+ 276.0962, found 276.0968.:3). 1H NMR (400 MHz, CDCl3) δ 8.13–8.11 (m, 2H), 7.66–7.62 (m, 1H), 7.52–7.48 (m, 2H), 2.79 (bs, 1H), 1.45 (t, J = 1.5 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ 191.2 (t, J = 31.0 Hz), 134.5, 130.0 (m), 130.4 (m), 128.6, 116.9 (t, J = 261.6 Hz), 73.3 (t, J = 24.7 Hz), 23.6 (t, J = 2.6 Hz); 19F NMR (376 MHz, CDCl3) δ −110.5 (m, 2F); HRMS (EI) m/z calcd for C11H12F2O2 [M]+ 214.0805, found 214.0799.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Andrew Jackson, PhD, from Edanz Group (https://www.edanzediting.com/ac) for editing a draft of this manuscript.Notes and references
- (a) F. Xue, H. Li, S. L. Delker, J. Fang, P. Martásek, L. J. Roman, T. L. Poulos and R. B. Silvermanm, J. Am. Chem. Soc., 2010, 132, 14229 CrossRef PubMed; (b) M. O. Anderson, J. Zhang, Y. Liu, C. Yao, P.-W. Phuan and A. S. Verkman, J. Med. Chem., 2012, 55, 5942 CrossRef PubMed; (c) J. O. Link, J. G. Taylor, L. Xu, M. Mitchell, H. Guo, H. Liu, D. Kato, T. Kirschberg, J. Sun, N. Squires, J. Parrish, T. Keller, Z.-Y. Yang, C. Yang, M. Matles, Y. Wang, K. Wang, G. Cheng, Y. Tian, E. Mogalian, E. Mondou, M. Cornpropst, J. Perry and M. C. Desai, J. Med. Chem., 2014, 57, 2033 CrossRef PubMed.
- (a) G. B. Dreyer and B. W. Metcalf, Tetrahedron Lett., 1988, 29, 6885 CrossRef; (b) R. L. Hamer, B. Freed and R. C. Allen, Hoechst-Roussel Pharmaceuticals, US Pat. 5006563, 1991; (c) C. Han, A. E. Salyer, E. H. Kim, X. Jiang, R. E. Jarrard, M. S. Powers, A. M. Kirchhoff, T. K. Salvador, J. A. Chester, G. H. Hockerman and D. A. Colby, J. Med. Chem., 2013, 56, 2456 CrossRef PubMed.
- (a) M. Kuroboshi and T. Ishihara, Bull. Chem. Soc. Jpn., 1990, 63, 428 CrossRef; (b) T. Poisson, M.-C. Belhomme and X. Pannecoucke, J. Org. Chem., 2012, 77, 9277 CrossRef PubMed; (c) H. Yao, C.-R. Cao, M. Jiang and J.-T. Liu, J. Fluorine Chem., 2013, 156, 45 CrossRef.
- S. Sasaki, T. Suzuki, T. Uchiyama, S. Toyota, A. Hirano, M. Tanemura, H. Teramoto, T. Yamauchi and K. Higashiyama, J. Fluorine Chem., 2016, 192, 78 CrossRef.
- R. Doi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2016, 55, 341 CrossRef PubMed.
- (a) O. Lefebvre, T. Brigaud and C. Portella, J. Org. Chem., 2001, 66, 1941 CrossRef PubMed; (b) M. Decostanzi, J. Godemert, S. Oudeyer, V. Levacher, J.-M. Campagne and E. Leclerc, Adv. Synth. Catal., 2016, 358, 526 CrossRef.
- P. Peng, J.-j. Wu, J.-q. Liang, T.-y. Zhang, J.-w. Huang and F.-h. Wu, RSC Adv., 2017, 7, 56034 RSC.
- (a) C. Han, E. H. Kim and D. A. Colby, J. Am. Chem. Soc., 2011, 133, 5802 CrossRef PubMed; (b) P. Zhang and C. Wolf, J. Org. Chem., 2012, 77, 8840 CrossRef PubMed; (c) P. Zhang and C. Wolf, Angew. Chem., Int. Ed., 2013, 52, 7869 CrossRef PubMed.
- Selected examples for the decarboxylative reactions of β-keto acid, see: (a) N. Blaquiere, D. G. Shore, S. Rousseaux and K. Fagnou, J. Org. Chem., 2009, 74, 6190 CrossRef PubMed; (b) K. Rohr and R. Mahrwakd, Org. Lett., 2011, 13, 1878 CrossRef PubMed; (c) C.-F. Yang, C. Shen, J.-Y. Wang and S.-K. Tian, Org. Lett., 2012, 14, 3092 CrossRef PubMed; (d) H.-X. Zhang, J. Nie, H. Cai and J.-A. Ma, Org. Lett., 2014, 16, 2542 CrossRef PubMed; (e) Z. Duan, J. Han, P. Qian, Z. Zhang, Y. Wang and Y. Pan, Beilstein J. Org. Chem., 2014, 10, 969 CrossRef PubMed; (f) Z.-Y. Yang, J.-L. Zeng, N. Ren, W. Meng, J. Nie and J.-A. Ma, Org. Lett., 2016, 18, 6364 CrossRef PubMed.
- J. Saadi and H. Wennemers, Nat. Chem., 2016, 8, 276 CrossRef PubMed.
- D.-K. Huang, Z.-L. Lei, Y.-J. Zhu, Z.-J. Liu, X.-J. Hu and H.-F. Mao, Tetrahedron Lett., 2017, 58, 3394 CrossRef.
- J.-W. Yuan, S.-N. Liu and W.-P. Mai, Org. Biomol. Chem., 2017, 15, 7654 Search PubMed.
- (a) K. Sato, A. Tarui, T. Kita, Y. Ishida, H. Tamura, M. Omote, A. Ando and I. Kumadaki, Tetrahedron Lett., 2004, 45, 5735 CrossRef; (b) Z.-W. Mei, T. Omote, M. Mansour, H. Kawafuchi, Y. Takaguchi, A. Jutand, S. Tsuboi and T. Inokuchi, Tetrahedron, 2008, 64, 10761 CrossRef; (c) M.-H. Yang, D. L. Orsi and R. A. Altman, Angew. Chem., Int. Ed., 2015, 54, 2361 CrossRef PubMed.
- Bench stable ZnCl2/TMEDA complex could be treated under air..
- The effect of counter cations of 2a and the loading of ZnCl2/TMEDA were examined. See the ESI (Table S-1).†.
- See the ESI (Table S-2).†.
- Synthesis and characterizations of 2b–d were described in the ESI.†.
- J.-S. Yu, Y.-L. Liu, J. Tang, X. Wang and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 9512 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C and 19F NMR spectra for 2a–d and 6aa–6da. See DOI: 10.1039/c8ra02440e |
| This journal is © The Royal Society of Chemistry 2018 |