
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic dye-catalyzed radical ring expansion reaction†
Masato Deguchi,
Akitoshi Fujiya,
Eiji Yamaguchi,
Norihiro Tada ,
Bunji Uno and
Akichika Itoh*
,
Bunji Uno and
Akichika Itoh*
Gifu Pharmaceutical University, 1-25-4, Daigaku-nishi, Gifu 501-1196, Japan. E-mail: itoha@gifu-pu.ac.jp
First published on 27th April 2018
Abstract
Herein, we reported an attractive method for synthesizing medium-sized rings that are catalyzed by erythrosine B under fluorescent light irradiation. This synthetic approach featured mild conditions, a facile procedure, a broad substrate scope, and moderate-to-good yields.
Medium-sized rings are present in numerous important natural products and pharmaceuticals.1 Therefore, several researchers have strived to develop various methods, including olefin metathesis,2 fragmentation,3 and pericyclic reactions4 (Scheme 1(a)), for synthesizing these materials. Nevertheless, unfavorable transannular interactions and entropic factors typical of rings of this size make this task quite challenging.5 Thus, we believe that new methods need to be developed to solve these problems.
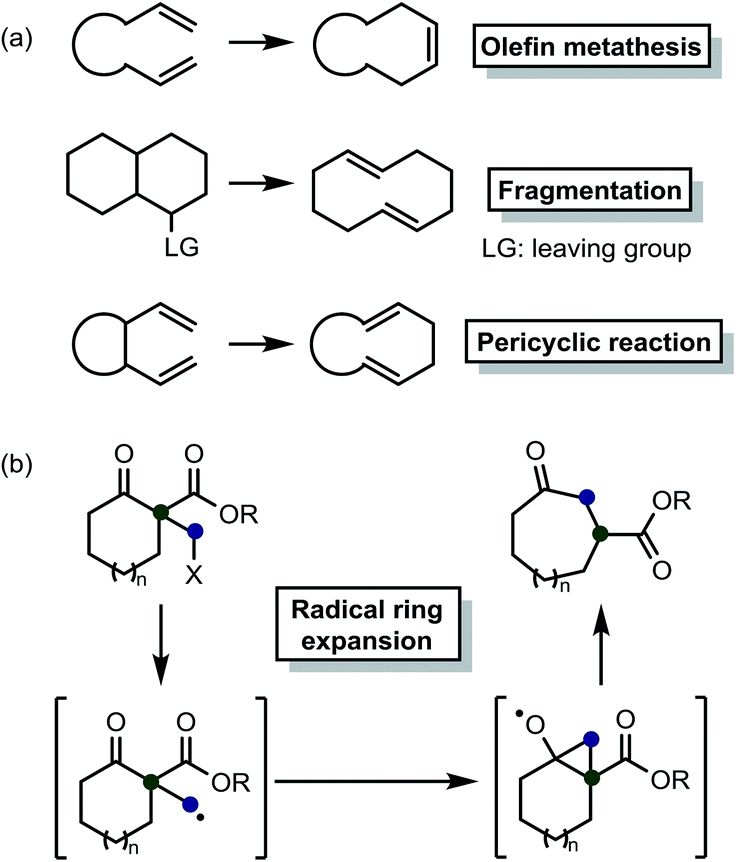 | ||
| Scheme 1 (a) Examples of conventional approaches for the synthesis of medium-sized rings. (b) Beckwith–Dowd ring expansion reaction. | ||
More than 30 years ago, the Beckwith–Dowd ring expansion reaction6 was introduced as a novel method to form medium-sized rings (Scheme 1(b)). This approach can be an attractive alternative to conventional strategies for synthesizing the abovementioned structures. Actually, the fact that no new unsaturated C–C bonds are formed as part of this method renders hydrogenation processes unnecessary, thereby minimizing the impact of transformation on the substrate. Certainly, this approach could be applicable to the synthesis of natural products.7 However, the original method requires reagents, e.g., azobisisobutyronitrile (AIBN) or tributyltin hydride (Bu3SnH), that are difficult to handle. Some related methods that employ other reagents, including Sm,8 Zn or In,9 B12–TiO2 hybrid catalyst,10 silane,11 and amines,12 have been reported. Recently, a few synthetic approaches based on photo-induced reactions have been developed.12b,12d,13 A previous study reported that even α-(ω-carboxyalkyl) β-keto esters could be employed as relevant substrates.13
In this context, we aimed to establish a more efficient and facile method than conventional ones to prepare a broad range of medium-sized rings via a ring expansion reaction. We continuously investigated various photo-initiated reactions by employing a photosensitizer and a fluorescent lamp as a light source.14 For example, the CDC cross-coupling reaction14a and 1,3-dipolar cycloaddition/aromatization reaction14d under photooxidative reaction conditions have been reported. While investigating these reactions, we found that organic dyes induced electron transfer from amine substrates to molecular oxygen.14a,14d Thus, we envisioned that C–halogen bonds can be cleaved in the presence of a sacrificial amine using photosensitized substrates as a springboard. Herein, we reported a convenient and environmentally friendly method that employs a photo-induced reaction as part of the Beckwith–Dowd ring expansion route to synthesize medium-sized rings.
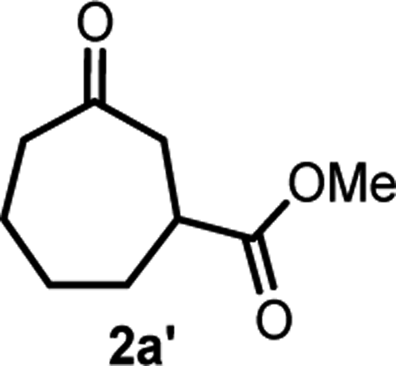
We initiated our study with the optimization of the reaction conditions (Table 1). Methyl 1-(iodomethyl)-2-oxocyclohexane-1-carboxylate (1a) was chosen as a substrate. A mixture of 1a and different types of amines and photocatalysts was irradiated with four fluorescent lamps under a nitrogen atmosphere for 20 h in DMSO. After extensive investigations, we found that a combination of erythrosine B (EB) and iPr2NEt was the most efficient combination for this transformation, affording the desired product 2a in 83% isolated yield (entry 1).15 This result can be explained by two facts: (a) the maximum absorption of EB (∼520 nm)16 coincides with the wavelength of the fluorescent lamp and (b) iPr2NEt is an effective reductive quencher (Eox(iPr2NEt˙+/iPr2NEt) = +0.68 V vs. SCE; e.g., Eox(NEt3˙+/NEt3) = +0.99 V vs. SCE).17a,17b In fact, when we employed other combinations of photocatalysts, amines, and solvents, 2a was obtained in a relatively lower yield. Furthermore, in our experiments, we recovered 1a and/or its undesired hydrogenation product 1a′ from the reaction mixture in substantial amounts (entries 2–9).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Photocatalyst | Solvent | Amine | 2aa (%) | 1a′a (%) | 1aa (%) |
| a Yields are determined by 1H NMR spectroscopy using 1,1,2,2-tetrachloroethane as an internal standard. The number in parentheses denotes isolated yield. | ||||||
| 1 | Erythrosine B (EB) | DMSO | iPr2NEt | 78(83) | 5 | 10 |
| 2 | Eosin Y | DMSO | iPr2NEt | 50 | 18 | 0 |
| 3 | AQN-2-Cl | DMSO | iPr2NEt | 12 | 10 | 63 |
| 4 | EB | DMSO | Et3N | 51 | 26 | 22 |
| 5 | EB | DMSO | 1-Methyl imidazole | 11 | 15 | 74 |
| 6 | EB | DMSO | iPr2NH | 36 | 21 | 40 |
| 7 | EB | MeCN | iPr2NEt | 70 | 0 | 26 |
| 8 | EB | DMF | iPr2NEt | 18 | 15 | 18 |
| 9 | EB | CHCl3 | iPr2NEt | 0 | 79 | 14 |
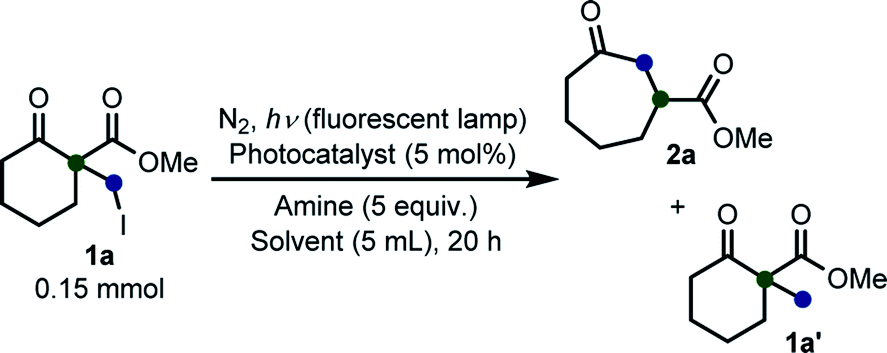
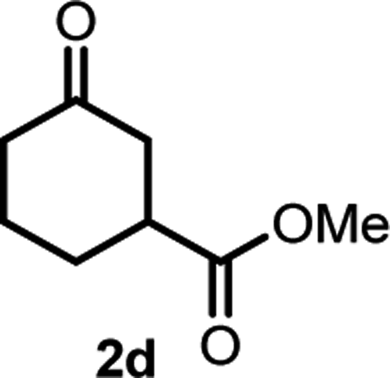
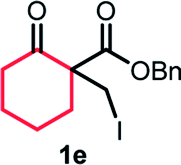
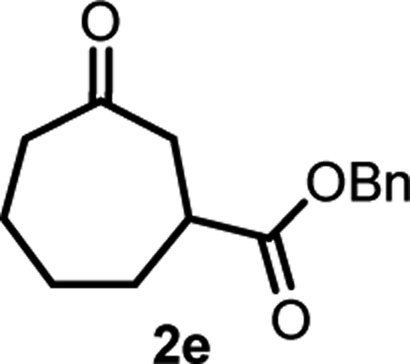
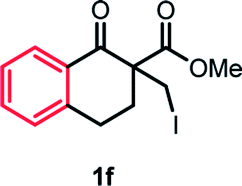
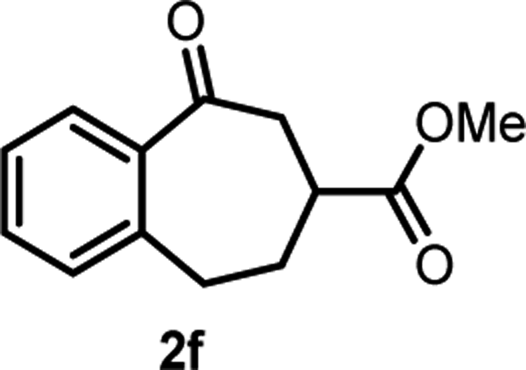
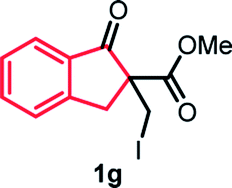
With the optimized reaction conditions in hand, we next explored the substrate scope of this transformation (Table 2). We found that compounds comprising 5–8-membered rings were suitable substrates for the reaction and gave the desired products in good yields (2a–2e). When we employed 1,2,3,4-tetrahydro naphthalene-type substrate 1f, the desired reaction proceeded smoothly. On the other hand, indane-based substrate 1g gave a mixture of the desired product and the aromatized product (3g). Due to the difficulty of achieving complete separation of the desired product and 3g, we further investigated the reaction conditions in order to obtain a single product. Although the changes in the reaction time and in the composition of the overhead atmosphere did not substantially affect the course of the reaction, we found that the addition of 3 equiv. of LiOH led to the formation of the aromatized product 3g exclusively in moderate yield. Furthermore, we tested whether compounds that contain the C–Br bond could also be used as substrates for this reaction. When we applied the standard reaction conditions to substrate 1h, the yield of the desired product 2a′ was around 10%. However, adding a catalytic amount of Ag2CO3 and extending the reaction time increased the yield. Additionally, it is worth noting that this reaction could be carried out in an air atmosphere with almost no decrease in product yield. This result indicates that the presence of oxygen or water in the atmosphere above the reaction mixture has a negligible impact on in situ radical generation and subsequent steps to furnish the ring expanded product.

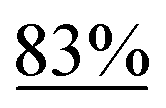
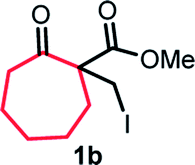
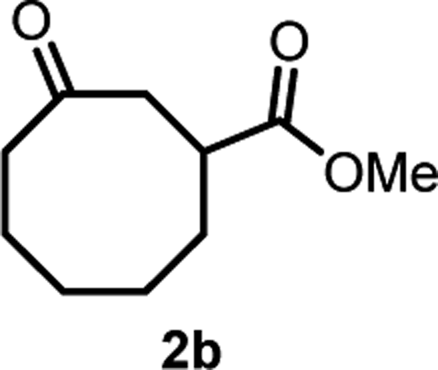
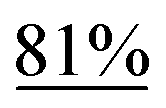
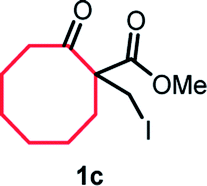
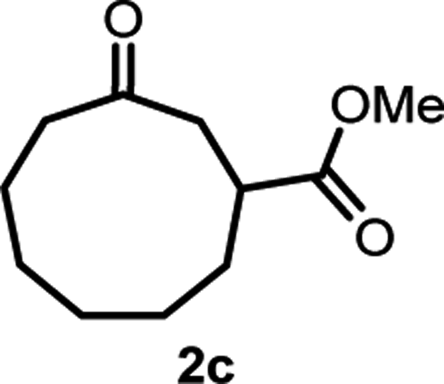
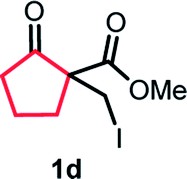
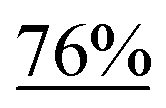
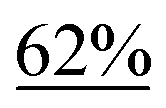
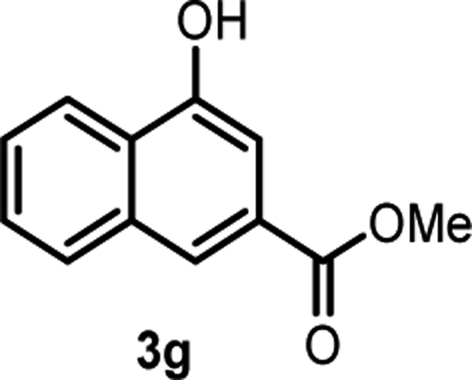

Scheme 2 summarizes the results of additional experiments conducted to investigate the substrate scope. We tested a substrate with an iodopropyl side chain, 1i, and found that the addition of a silver salt and extension of the reaction time led to the formation of a tertiary alcohol with a fused ring, 3i, instead of the corresponding cyclononanone (eqn (1)). Additionally, we found that this reaction is applicable to substrates with ketones in open-chain moieties, such as 1j and 1k. Particularly, although 2j was volatile, and we needed to treat it carefully during isolation, these substrates gave the rearranged product in high yield (eqn (2) and (3)).
 | ||
| Scheme 2 Applying the reaction conditions on substrates with three-carbon lateral chain and acyclic β-keto esters. | ||
Next, we performed some experiments to elucidate the reaction mechanism (Scheme 3). We observed that no ring expansion product 2a was obtained when we omitted EB or iPr2NEt or photo-irradiation from the fluorescent lamps. Additionally, if we added 1 equiv. of TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl free radical), the desired product was obtained in low yield and the presence of a TEMPO-adduct was detected.18 This result confirms the idea that this reaction proceeds via a radical pathway.
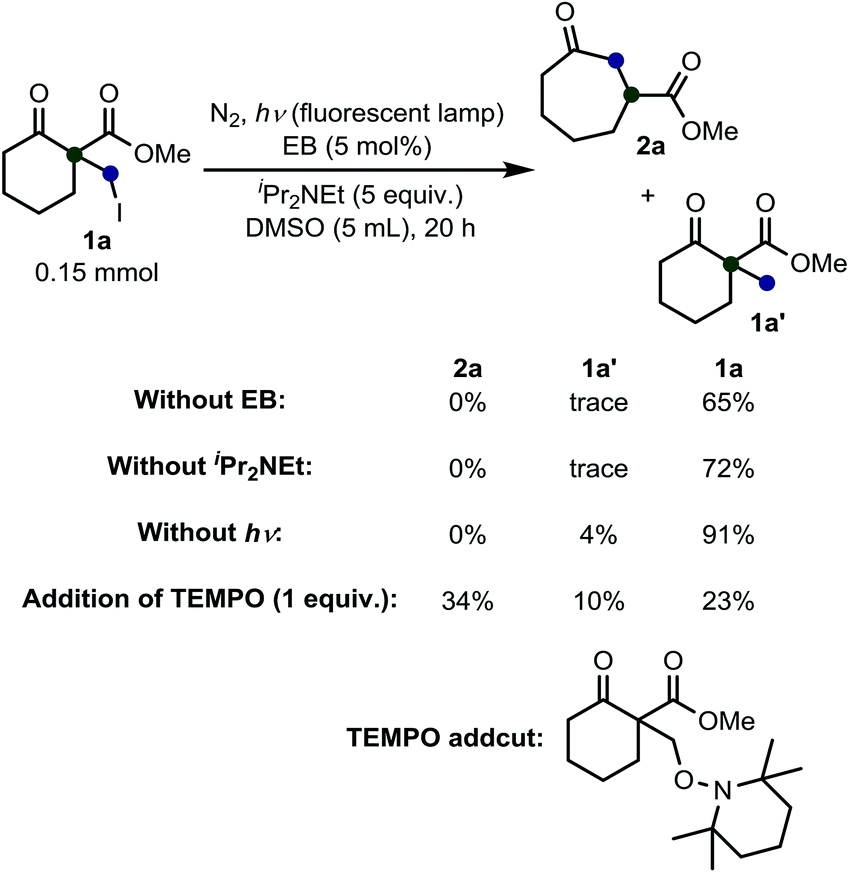 | ||
| Scheme 3 Results of the control experiments. | ||
Based on these results, previous reports on the Beckwith–Dowd ring expansion reaction,6 and recent reports of radical reactions set off by C–halogen bond cleavage,17b,19 we inferred a plausible mechanistic pathway for the reaction, which is depicted in Scheme 4. The first step in the C–I bond cleavage reaction of 1a is photo-induced single electron transfer (SET) from the excited state of EB. In the experimental conditions employed, EB is considered to be a monoanionic (EB−) or dianionic (EB2−) species because its commercially available disodium salt (EBNa2) is added to the reaction mixture containing DIPEA without acidic treatment. To gain further insight into this SET step, we calculated the Gibbs energies involved in the reaction between 1a and EB in a solution environment (solvent: DMSO; ε = 46.826) simulated by the IEF-PCM model using the M06-2X functional.20 The standard Gibbs energy change (ΔG°) calculated for the cleavage reaction of 1a with EB2− (1a + EB2− → 4a + I− + EB˙−) and with EB− (1a + EB− → 4a + I− + EB˙) in the absence of light irradiation from the fluorescent lamp is 1.72 and 1.27 eV, respectively, as shown in Scheme 5. This indicates that these reactions are thermodynamically uphill electron-transfer processes. On the other hand, it is well known that the ΔG° values for excited EB2−(EB2−*)/EB˙− and excited EB−(EB−*/EB˙) redox pairs are given by the amended Rehm–Weller equation with the excited-state energies (Ehν) as ΔG°(EB2−*/EB˙−) = ΔG°(EB2−/EB˙−) − Ehν and ΔG°(EB−*/EB˙) = ΔG°(EB−/EB˙) − Ehν, respectively.21,22 The reported excited-state energy of EB is 2.34 eV;16a thus, the ΔG° values for the two mentioned C–I bond cleavage reactions (1a + EB2−* → 4a + I− + EB˙− and 1a + EB−* → 4a + I− + EB˙) involving photo-induced SET are −0.62 and −1.07 eV, respectively, which indicate that the Ehν value is sufficiently high for both bond cleavage reactions to be thermodynamically feasible. These results indicate that the cleavage reaction of 1 is governed by an exergonic C–I bond cleavage mechanism involving SET from EB2−* or EB−*. The primary radical 4 thus generated attacks the ketone, and then a rapid cyclopropane ring opening occurs. Meanwhile, the oxidized EB is reduced by the sacrificial amine, and the catalytic cycle of EB is completed. No particular H donor was necessary for this reaction to proceed; therefore, we assumed that an “extra” hydrogen atom of the product is derived from iPr2NEt. It believed that abstraction of a hydrogen atom from the trialkylammounium radical cation could occur,23 which strongly supports the above mechanism.
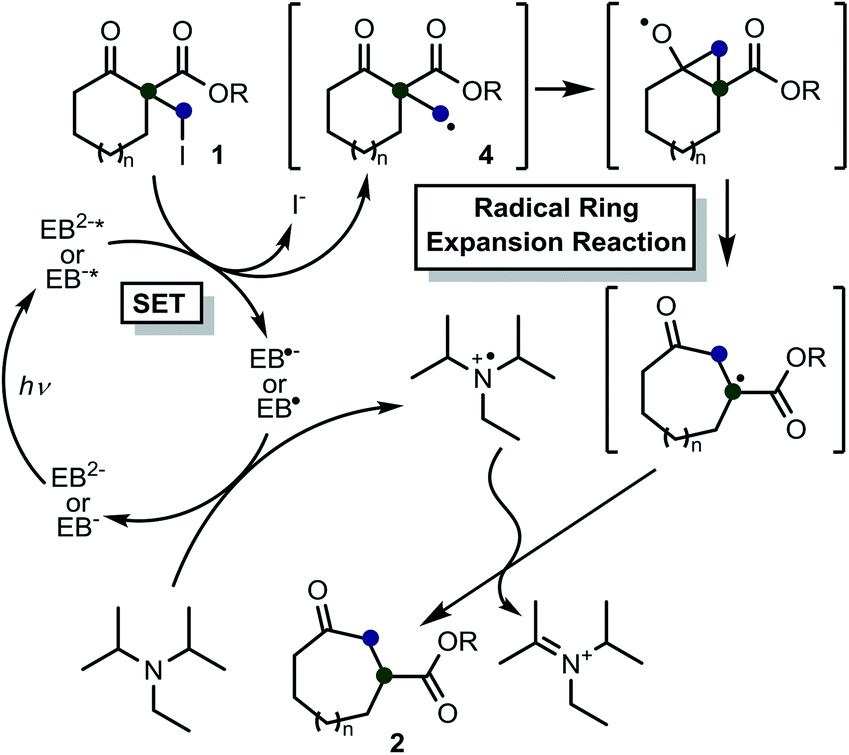 | ||
| Scheme 4 Plausible reaction mechanism. | ||
 | ||
| Scheme 5 Standard Gibbs energies calculated for the species involved in the C–I bond cleavage reaction with EB2− (1) and EB− (2) using the M06-2X/PCM method with 6-31G (d) basis sets for hydrogen, carbon, and oxygen and the MIDI! basis set for iodine. Free energy corrections are made at standard conditions of 1 atm and 298.15 K. | ||
Conclusions
In conclusion, we have developed an environmentally friendly ring expansion reaction to synthesize medium-sized rings. This facile method involves the use of a fluorescent lamp as a source of irradiated light and an easy-to-handle photocatalyst. Attractively, this reaction proceeds under very mild conditions and is applicable to a broad spectrum of substrates. We believe that our method using a photo-triggered reaction course affords access to various pharmaceuticals and natural products. Studies to determine the mechanistic details of the reaction and expand its applicability to other useful substrates are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Enago (www.enago.jp) for the English language review.Notes and references
- (a) M. Hesse, Ring Enlargements in Organic Chemistry, VCH, Weinheim, 1991 Search PubMed; (b) M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef CAS PubMed; (c) M. Norte, F. Cataldo, A. Sánchez and A. G. González, Tetrahedron Lett., 1993, 34, 5143–5146 CrossRef CAS; (d) D.-F. Chen, S.-X. Zhang, L. Xie, J.-X. Xie, K. Chen, Y. Kashiwada, B.-N. Zhou, P. Wang, L. M. Cosentino and K.-H. Lee, Bioorg. Med. Chem., 1997, 5, 1715–1723 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. Cossy, S. Arseniyadis and C. Meyer, Metathesis in Natural Product Synthesis, Wiley-VCH, Weinheim, 2010 Search PubMed; (b) M. E. Maier, Angew. Chem., Int. Ed., 2000, 39, 2073–2077 CrossRef CAS; (c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490–4527 CrossRef CAS PubMed; (d) R. R. Schrock, Chem. Rev., 2009, 109, 3211–3226 CrossRef CAS PubMed; (e) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed.
- For recent examples of fragmentation reaction for construction of medium-sized rings, see: (a) J. Tummatorn and G. B. Dudley, Org. Lett., 2011, 13, 1572–1575 CrossRef CAS PubMed; (b) C. Kitsiou, J. J. Hindes, P. I'Anson, P. Jackson, T. C. Wilson, E. K. Daly, H. R. Felstead, P. Hearnshaw and W. P. Unsworth, Angew. Chem., Int. Ed., 2015, 54, 15794–15798 CrossRef CAS PubMed; (c) J. E. Hall, J. V. Matlock, J. W. Ward and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 11153–11157 CrossRef CAS PubMed; (d) J. Koo, J. Kim and S. B. Park, Org. Lett., 2017, 19, 344–347 CrossRef CAS PubMed.
- For recent examples of pericyclic reaction for construction of medium-sized rings, see: (a) Y.-S. Lee, J.-W. Jung, S.-H. Kim, J.-K. Jung, S.-M. Peak, N.-J. Kim, D.-J. Chang, J. Lee and Y.-G. Suh, Org. Lett., 2010, 12, 2040–2043 CrossRef CAS PubMed; (b) Y. Zou, L. Zhou, Z. Li and Q. Wang, Angew. Chem., Int. Ed., 2012, 51, 5647–5651 CrossRef CAS PubMed; (c) L. Zhou, Z. Li, Y. Zou, Q. Wang, I. A. Sanhueza, F. Schoenebeck and A. Goeke, J. Am. Chem. Soc., 2012, 134, 20009–20012 CrossRef CAS PubMed; (d) J. D. Osler, W. P. Unsworth and R. J. K. Taylor, Org. Biomol. Chem., 2013, 11, 7587–7594 RSC; (e) J. C. Orejarena Pacheco and T. Opatz, J. Org. Chem., 2014, 79, 5182–5192 CrossRef CAS PubMed; (f) B. Zhou, L. Li, X.-Q. Zhu, J.-Z. Yan, Y.-L. Guo and L.-W. Ye, Angew. Chem., Int. Ed., 2017, 56, 4015–4019 CrossRef CAS PubMed.
- (a) G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95–102 CrossRef CAS; (b) M. A. Casadei, C. Galli and L. Mandolini, J. Am. Chem. Soc., 1984, 106, 1051–1056 CrossRef CAS; (c) G. Molander, Acc. Chem. Res., 1998, 31, 603–609 CrossRef CAS; (d) D. J. Faulkner, Nat. Prod. Rep., 1999, 16, 155–198 RSC; (e) B. M. Fraga, Nat. Prod. Rep., 2003, 20, 392–413 RSC; (f) J. Chang, J. Reiner and J. Xie, Chem. Rev., 2005, 105, 4581–4609 CrossRef CAS PubMed; (g) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (h) J. R. Donald and W. P. Unsworth, Chem.–Eur. J., 2017, 23, 8780–8799 CrossRef CAS PubMed.
- (a) A. L. J. Beckwith, R. Kazlauskas and M. R. Syner-Lyons, J. Org. Chem., 1983, 48, 4718–4722 CrossRef CAS; (b) A. L. J. Beckwith, D. M. O'Shea, S. Gerba and S. W. Westwood, J. Chem. Soc., Chem. Commun., 1987, 666–667 RSC; (c) P. Dowd and S. C. Choi, J. Am. Chem. Soc., 1987, 109, 3493–3494 CrossRef CAS; (d) P. Dowd and S. C. Choi, J. Am. Chem. Soc., 1987, 109, 6548–6549 CrossRef CAS; (e) A. L. J. Beckwith, D. M. O'Shea and S. W. Westwood, J. Am. Chem. Soc., 1988, 110, 2565–2575 CrossRef CAS; (f) P. Dowd and S. C. Choi, Tetrahedron Lett., 1989, 30, 6129–6132 CrossRef CAS; (g) P. Dowd and S. C. Choi, Tetrahedron, 1989, 45, 77–90 CrossRef CAS; (h) P. Dowd and S. C. Choi, Tetrahedron Lett., 1991, 32, 565–568 CrossRef CAS; (i) W. Zhang and P. Dowd, Tetrahedron Lett., 1992, 33, 3285–3288 CrossRef CAS; (j) H.-S. Oh, H. I. Lee and J. K. Cha, Org. Lett., 2002, 4, 3707–3709 CrossRef CAS PubMed; (k) J. Hierold and D. W. Lupton, Org. Lett., 2012, 14, 3412–3415 CrossRef CAS PubMedfor reviews, see: (l) P. Dowd and W. Zhang, Chem. Rev., 1993, 93, 2091–2115 CrossRef CAS; (m) L. Yet, Tetrahedron, 1999, 55, 9349–9403 CrossRef CAS.
- (a) E. Piers, M. Gilbert and K. L. Cook, Org. Lett., 2000, 2, 1407–1410 CrossRef CAS PubMed; (b) H. Watanabe, M. Takano, A. Umino, T. Ito, H. Ishikawa and M. Nakada, Org. Lett., 2007, 9, 359–362 CrossRef CAS PubMed; (c) Y. Liu and Y.-Y. Yeung, Org. Lett., 2017, 19, 1422–1425 CrossRef CAS PubMed.
- (a) E. Hasegawa, T. Kitazume, K. Suzuki and E. Tosaka, Tetrahedron Lett., 1998, 39, 4059–4062 CrossRef CAS; (b) S. H. Chung, M. S. Cho, Y. Choi, D. W. Kwon and Y. H. Kim, Synlett, 2001, 1266–1268 CrossRef CAS; (c) H. Tsuchida, M. Tamura and E. Hasegawa, J. Org. Chem., 2009, 74, 2467–2475 CrossRef CAS PubMed.
- M. Sugi, D. Sakuma and H. Togo, J. Org. Chem., 2003, 68, 7629–7633 CrossRef CAS PubMed.
- (a) H. Shimakoshi, M. Abiru, S. Izumi and Y. Hisaeda, Chem. Commun., 2009, 6427–6429 RSC; (b) S. Izumi, H. Shimakoshi, M. Abe and Y. Hisaeda, Dalton Trans., 2010, 39, 3302–3307 RSC.
- (a) M. Sugi and H. Togo, Tetrahedron, 2002, 58, 3171–3175 CrossRef CAS; (b) E. Hasegawa, Y. Ogawa, K. Kakinuma, H. Tsuchida, E. Tosaka, S. Takizawa, H. Muraoka and T. Saikawa, Tetrahedron, 2008, 64, 7724–7728 CrossRef CAS.
- (a) E. Hasegawa, Y. Tamura and E. Tosaka, Chem. Commun., 1997, 1895–1896 RSC; (b) E. Hasegawa, A. Yoneoka, K. Suzuki, T. Kato, T. Kitazume and K. Yanagi, Tetrahedron, 1999, 55, 12957–12968 CrossRef CAS; (c) E. Hasegawa, S. Takizawa, K. Iwaya, M. Kurokawa, N. Chiba and K. Yamamichi, Chem. Commun., 2002, 1966–1967 RSC; (d) E. Hasegawa, T. Ohta, S. Tsuji, K. Mori, K. Uchida, T. Miura, T. Ikoma, E. Tayama, H. Iwamoto, S. Takizawa and S. Murata, Tetrahedron, 2015, 71, 5494–5505 CrossRef CAS.
- K. Nishikawa, T. Ando, K. Maeda, T. Morita and Y. Yoshimi, Org. Lett., 2013, 15, 636–638 CrossRef CAS PubMed.
- (a) T. Yamaguchi, T. Nobuta, N. Tada, T. Miura, T. Nakayama, B. Uno and A. Itoh, Synlett, 2014, 25, 1453–1457 CrossRef; (b) A. Fujiya, T. Nobuta, E. Yamaguchi, N. Tada, T. Miura and A. Itoh, RSC Adv., 2015, 5, 39539–39543 RSC; (c) A. Okada, H. Yuasa, A. Fujiya, N. Tada, T. Miura and A. Itoh, Synlett, 2015, 26, 1705–1709 CrossRef CAS; (d) A. Fujiya, M. Tanaka, E. Yamaguchi, N. Tada and A. Itoh, J. Org. Chem., 2016, 81, 7262–7270 CrossRef CAS PubMed; (e) T. Yamaguchi, E. Yamaguchi and A. Itoh, Org. Lett., 2017, 19, 1282–1285 CrossRef CAS PubMed.
- Time course of the substrate 1a and the product 2a was shown in the ESI.†.
- (a) M. A. Jhonsi, A. Kathiravan and R. Renganathan, J. Mol. Struct., 2009, 921, 279–284 CrossRef; (b) G. Sharifzade, A. Asghari and M. Rajabi, RSC Adv., 2017, 7, 5362–5371 RSC; (c) Y. Okuno and S. Cavagnero, J. Magn. Reson., 2018, 286, 172–187 CrossRef CAS PubMed.
- (a) U. Pischel, X. Zhang, B. Hellrung, E. Haselbach, P.-A. Muller and W. M. Nau, J. Am. Chem. Soc., 2000, 122, 2027–2034 CrossRef CAS; (b) N. Esumi, K. Suzuki, Y. Nishimoto and M. Yasuda, Org. Lett., 2016, 18, 5704–5707 CrossRef CAS PubMed.
- See the ESI† for the detail..
- (a) E. Yoshioka, S. Kohtani, T. Jichu, T. Fukazawa, T. Nagai, Y. Takemoto and H. Miyabe, Synlett, 2015, 26, 265–270 CAS; (b) E. Yoshioka, S. Kohtani, T. Jichu, T. Fukazawa, T. Nagai, A. Kawashima, Y. Takemoto and H. Miyabe, J. Org. Chem., 2016, 81, 7217–7229 CrossRef CAS PubMed.
- C.-C. Chen, M.-Y. Wu, H.-Y. Chen and M.-J. Wu, J. Org. Chem., 2017, 82, 6071–6081 CrossRef CAS PubMed . The detailed method and results of the calculation are shown in the ESI.†.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- (a) S. Farid, J. P. Dinnocenzo, P. B. Merkel, R. H. Young and D. Shukla, J. Am. Chem. Soc., 2011, 133, 4791–4801 CrossRef CAS PubMed; (b) S. Farid, J. P. Dinnocenzo, P. B. Merkel, R. H. Young, D. Shukla and G. Guirado, J. Am. Chem. Soc., 2011, 133, 11580–11587 CrossRef CAS PubMed.
- (a) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed; (b) A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed; (c) Y. Q. Zou, J. R. Chen, X. P. Liu, L. Q. Lu, R. L. Davis, K. A. Jørgensen and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784–788 CrossRef CAS PubMed; (d) M. O. Ratnikov and M. P. Doyle, J. Am. Chem. Soc., 2013, 135, 1549–1557 CrossRef CAS PubMed; (e) S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286–13289 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, product characterization, time course of 1a and 2a, emission spectrum of the fluorescent lamp, and detailed information of the DFT calculation. See DOI: 10.1039/c8ra02383b |
| This journal is © The Royal Society of Chemistry 2018 |