
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmorphous red phosphorus incorporated with pyrolyzed bacterial cellulose as a free-standing anode for high-performance lithium ion batteries†
Hongyu Yanga,
Yu Liacd,
Peng Longa,
Junkai Hana,
Chen Caoa,
Fengnan Yaoa and
Wei Feng *abcd
*abcd
aSchool of Materials Science and Engineering, Tianjin University, Tianjin 300072, P. R. China. E-mail: weifeng@tju.edu.cn; Fax: +86-22-27404724; Tel: +86-22-87402059
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, P. R. China
cKey Laboratory of Advanced Ceramics and Machining Technology, Ministry of Education, Tianjin 300072, P. R. China
dTianjin Key Laboratory of Composite and Functional Materials, Tianjin 300072, P. R. China
First published on 11th May 2018
Abstract
Amorphous red phosphorus/pyrolyzed bacterial cellulose (P-PBC) free-standing films are prepared by thermal carbonization and a subsequent vaporization-condensation process. The distinctive bundle-like structure of the flexible pyrolyzed bacterial cellulose (PBC) matrix not only provides sufficient volume to accommodate amorphous red-phosphorus (P) but also restricts the pulverization of red-P during the alternate lithiation/delithiation process. When the mass ratio of raw materials, red-P to PBC, is 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the free-standing P-PBC film anode exhibits high reversible capacity based on the mass of the P-PBC film (1039.7 mA h g−1 after 100 cycle at 0.1C, 1C = 2600 mA g−1) and good cycling stability at high current density (capacity retention of 82.84% after 1000 cycles at 2C), indicating its superior electrochemical performances.
:1, the free-standing P-PBC film anode exhibits high reversible capacity based on the mass of the P-PBC film (1039.7 mA h g−1 after 100 cycle at 0.1C, 1C = 2600 mA g−1) and good cycling stability at high current density (capacity retention of 82.84% after 1000 cycles at 2C), indicating its superior electrochemical performances.
1. Introduction
Since rechargeable Li-ion batteries (LIBs) were commercialized in the 1990s, they have become privileged energy storage devices because of their superior energy density accompanied with satisfactory power density and operation life.1–4 However, the energy density of commercial LIBs has fallen behind the requirements for future applications in large scale energy storage systems,5 so the exploration of electrode materials beyond conventional compounds based on the intercalation reaction is imperative. Several candidate materials based on different reaction mechanisms such as alloying and conversion reactions6–12 have been investigated as alternative anode materials for Li-ion batteries. Among these materials, phosphorus (P), a light, low cost and environmentally friendly matter shows an ultrahigh theoretical capacity of 2596 mA h g−1, which makes it a promising anode candidate for LIBs.13 However, the poor electrical conductivity (10−14 S cm−1) and large volume expansion during lithiation processes limit its practical application. Therefore, various carbonaceous materials have been introduced as the frameworks to accommodate P and improve its electrical conductivity, including porous carbon,13–17 activated carbon,18,19 graphite,20–23 carbon black,24,25 graphene nanosheets,26–32 carbon nanotubes,33–35 and three-dimensional (3D) carbon architectures.36,37At the same time, free-standing electrodes, which do not require any metal collector, conductive additive or binder, have also received research interest, because of the capability to improve the energy density of LIBs further as well as the potential application to flexible and wearable devices.38 To date, carbonaceous materials constructed by entangled one-dimensional and stacked two-dimensional nanostructures, such as bucky-paper or graphene paper, have been most widely applied as the flexible matrixes to accommodate electrochemically active materials.36,37,39–44 Free-standing electrodes based on P were mainly prepared through the vaporization-condensation method by taking advantage of the sublimation nature of P. For example, Du et al. prepared a flexible hybrid film of red-P and graphene oxide and this film delivered a reversible capacity of 910 mA h g−1 after 200 cycles at the current density of 260 mA g−1.36 Li et al. also synthesized a crystalline red-P/porous carbon nanofibers free-standing electrode and it showed a reversible capacity of ∼850 mA h g−1 after 100 cycles at the current density of 260 mA g−1.37 Free-standing films can be also obtained through the method of vacuum filtration by using the processability of nanostructured carbons. Flexible black-P nanoparticle-graphene hybrid paper and black-P/graphene hybrid flexible film have been obtained through the vacuum filtration of their homogenously dispersed solutions, which both exhibited impressive electrochemical performances due to the good compatibility of components.29,32
Bacterial cellulose (BC), a typical biomass material, is composed of interconnected nanofiber bundles and presents a unique 3D network structure and narrow distribution of fiber diameters.45 Furthermore, the pyrolyzed bacterial cellulose (PBC) not only retains the distinctive bundle-like structure and highly porous 3D networks but also exhibits good electrical conductivity.46,47 As a result, PBC has been demonstrated as an ideal carbonaceous matrix for free-standing electrodes.48,49 Herein, we prepared a series of red-P/PBC (P-PBC) hybrid films with different mass loading of red-P by a vaporization-condensation process, during which red-P turns into white-P gaseous and penetrates into the intervals of PBC nanofiber bundles then turns back into the red-P to form a uniform and compact nanostructure. The as prepared free-standing P-PBC films, tested in half-cells, delivered the maximum initially reversible capacity of 1271.9 mA h g−1 based on the mass of P-PBC film at 0.1C (1C = 2600 mA g−1), and exhibited both excellent cycling stability (capacity retention of 82.55% after 100 cycles at 0.1C) and satisfactory rate capability (reversible capability of 539.7 mA h g−1 at 2C).
2. Experimental
2.1. The preparation of PBC film
BC hydrogels (Hainan Yeguo Foods Co., Ltd., China) were washed by deionized water (DIW) thoroughly and then cut into 3 × 4 cm2 rectangle slices with a sharp blade. The sliced pieces of BC hydrogels were frozen in liquid nitrogen and then transferred into a bulk tray dryer (Labconco Corporation, USA) for the subsequent freeze-drying process. The freeze-dried BC aerogel was pyrolyzed under the Ar atmosphere in a tubular furnace to get PBC film. Briefly, the temperature was slowly increased to 350 °C at a rate of 1 °C min−1 and held at this temperature for 1 h to stabilize the BC structure. And then, the temperature was continuously increased to 1000 °C at a rate of 3 °C min−1 and held at this temperature for 1 h to complete the carbonization process. After the carbonization process, the thickness of PBC film is 0.06 mm.2.2. The preparation of P-PBC hybrid films
Some amount of red-P powder (purity ≥ 99%, Sinopharm Chemical Reagent Co., Ltd.) and the obtained PBC film were put into a stainless steel vessel at a certain mass ratio and sealed in an argon-filled glove box. The stainless steel vessel was heated at 450 °C (the sublimation temperature of red P) for 24 h. Then the temperature was slowly decreased to 260 °C and held for another 24 h for the transformation of white-P to red-P. After the vessel cooled to the room temperature, the vessel was opened in the argon-filled glove box and the obtained P-PBC films were washed with CS2 (purity ≥ 99%, Sinopharm Chemical Reagent Co., Ltd.) to remove the residual white-P that haven't turned back into red-P during the vaporization-condensation process. Subsequently, these films were dried under vacuum at 60 °C overnight. The schematic illustration of P-PBC preparation procedure is shown in Fig. 1. A series of free-standing P-PBC films were obtained, which were detonated as P-PBC-80, P-PBC-70, P-PBC-60 and P-PBC-50 by the initial mass ratio of raw materials, mred-P:mPBC, respectively. The thickness of P-PBC-50, P-PBC-60, P-PBC-70 and P-PBC-80 are 0.06, 0.08, 0.10 and 0.10 mm, respectively. The final mass loading of red-P in the P-PBC film was determined by the thermogravimetry analysis (TGA) under N2 atmosphere in the temperature range of 40–800 °C at a heating rate of 10 °C min−1 (Fig. S1, ESI†) and the mass loading of red-P in P-PBC-80, P-PBC-70, P-PBC-60 and P-PBC-50 are 81.42, 68.56, 46.50 and 14.01 wt%, respectively.
 | ||
| Fig. 1 Schematic illustration of the preparation process for the P-PBC electrode. | ||
2.3. Characterizations
The morphology was studied by the scanning electron microscopy (SEM, Hitachi S-4800) and the transmission electron microscopy (TEM, JEM-2100F, Japan) techniques. X-ray diffraction (XRD) measurements were performed on a Philips diffractometer, which was composed of a quartz monochromator, a Cu Kα radiation source at a scan rate of 5° min−1 and a goniometric plate. Fourier transform infrared (FT-IR) spectroscopy data were obtained in the range from 1500 to 800 cm−1 using a Bruker Vector 22 spectrometer. Raman spectrum was recorded on a Raman spectrometer (DXR Microscope, Thermo Electron) using laser excitation at 532 nm. X-ray photoelectron spectroscopy (XPS) spectra were recorded on PERKINELMZR PHI 3056 using a polychromatic Al Kα X-ray source, wherein the binding energy was calibrated taking C 1s at 285.0 eV.2.4. Electrochemical characterization
The pristine red-P (80%) was mixed with acetylene black (10%) and ploy(vinylidene) binder (10%) to make a homogeneous slurry. Then the slurry was casted onto copper foil and dried under vacuum at 60 °C overnight. The mass loading of active material was about 3 mg cm−2. The pristine red-P electrode and freestanding P-PBC (PBC) films were cut into disks with the diameter of 12 mm and then transferred into a glove box filled with argon (Mikrouna Co., Advanced 2440/750) for the cell assembly. The coin cell (size 2032) was assembled with a P-PBC film as the working electrode directly and a metallic Li disc as the counter electrode, a Celgard 3500 film as the separator and 1 M LiPF6 in ethylene carbonate–dimethyl carbonate (EC–DMC, 1:1 vol.) as the electrolyte. The coin cells were charged and discharged in the potential range from 0.01 to 2.5 V (vs. Li/Li+) under constant current densities (Land CT2001A, Wu Han JinNuo Electronics Co., China) at a constant temperature of 27 °C. Cyclic voltammetry (CV) were tested on an electrochemical working station (CHI 660D) at a scan rate of 0.2 mV s−1 in the same potential range according to the charge–discharge tests. Electrochemical impedance spectroscopy (EIS) measurements were carried out on Advanced Electrochemical System Parstat 2263 in the frequencies ranging from 10−2 to 106 Hz with a perturbation voltage of 5 mV.
3. Results and discussion
The crystal structures of red-P, PBC and P-PBC are characterized by XRD and the corresponding diffraction patterns are shown in Fig. 2a. The two major broad diffraction peaks located around 24° and 56° in the XRD pattern of PBC are assigned to the (002) and (004) planes, respectively. The enlarged interlayer distance of PBC (d002 = 0.350 nm), by comparing with theoretical graphite planes (d002 = 0.3345 nm), is generated by the formation of turbostratic carbon structures at the pyrolized temperature of 1000 °C probably.50 The XRD pattern of commercial red-P exhibits one sharp peak at 16.5° and two broad humps at 35° and 55°, which are consistent with the monoclinic P, and the first sharp diffraction peak demonstrates the present medium-range ordered structure.51 The XRD patterns of prepared P-PBC seem like the simple superposition of components' XRD patterns, indicating the absence of crystalline impurity during the preparation process. However, the lower intensities of diffraction peaks at 16.5° and 35° in P-PBC than that of commercial red-P reveal the destruction of the medium-range ordered structure of red-P.33 | ||
| Fig. 2 (a) XRD patterns (b) FT-IR spectra and (c) Raman spectra of red-P, PBC and P-PBC. | ||
The FT-IR spectra of red-P, PBC and P-PBC are shown in Fig. 2b. The peaks at 1465, 1050 and 900 cm−1 in the FT-IR spectrum of PBC are assigned to the C–H vibration, C–C stretching and C–O vibration, respectively.52 The FT-IR spectrum of red-P exhibits two typical peaks at 1160 and 1000 cm−1, corresponding to P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and P–O bonds, respectively.19,53 These signals are in agreement with previous reports and confirm the oxidation of red-P in air.54 In the FT-IR spectra of prepared P-PBC, the appearance of a new peak at 1427 cm−1 indicates the formation of chemical bond between P and C atoms, and the intensity of this peak is enhanced with the increase of mred-P:mPBC.32 The molecular structures of red-P, PBC and P-PBC are further characterized by Raman spectroscopy, as shown in Fig. 2c. The Raman spectrum of PBC exhibited the characteristic G and D bands at 1578.4 and 1332.5 cm−1, respectively. The Raman spectrum of red-P shows several well defined peaks in the region of 300–500 cm−1, like previous report.13 However, the extremely weakened intensities of these peaks in the Raman spectra of P-PBC indicate the destroyed medium-range ordered structure of red-P and the physical adsorption of amorphous red-P in PBC matrix.55 In addition, both G and D bands in the Raman spectra of P-PBC gradually shift to low wavenumber with the increase of mred-P:mPBC, by comparing with those peaks in the pristine PBC. This phenomenon is assigned to the electron transfer from C to P, leading to the weakened C–C bond intensity and enlarged C–C bond length simultaneously.15
O and P–O bonds, respectively.19,53 These signals are in agreement with previous reports and confirm the oxidation of red-P in air.54 In the FT-IR spectra of prepared P-PBC, the appearance of a new peak at 1427 cm−1 indicates the formation of chemical bond between P and C atoms, and the intensity of this peak is enhanced with the increase of mred-P:mPBC.32 The molecular structures of red-P, PBC and P-PBC are further characterized by Raman spectroscopy, as shown in Fig. 2c. The Raman spectrum of PBC exhibited the characteristic G and D bands at 1578.4 and 1332.5 cm−1, respectively. The Raman spectrum of red-P shows several well defined peaks in the region of 300–500 cm−1, like previous report.13 However, the extremely weakened intensities of these peaks in the Raman spectra of P-PBC indicate the destroyed medium-range ordered structure of red-P and the physical adsorption of amorphous red-P in PBC matrix.55 In addition, both G and D bands in the Raman spectra of P-PBC gradually shift to low wavenumber with the increase of mred-P:mPBC, by comparing with those peaks in the pristine PBC. This phenomenon is assigned to the electron transfer from C to P, leading to the weakened C–C bond intensity and enlarged C–C bond length simultaneously.15
The morphology of BC, PBC and P-PBC were investigated by SEM and TEM, respectively. The SEM image of BC aerogel (Fig. S2a, ESI†) shows numerous interconnected microfibers with many junctions. Those microfibers are composed of dense nanofiber bundles, as shown in the dashed framed area in the magnified SEM image (Fig. S2b, ESI†). After the pyrolysis treatment, the interconnected structure in PBC film was preserved (Fig. S2c, ESI†) and the dashed framed area in the magnified SEM image of PBC aerogel (Fig. S2d, ESI†) still displays the bundle-like structure, though the average diameter of these nanofiber bundles were thinner, which was demonstrated by the size survey of BC and PBC bundles (Fig. S3, ESI†). The decrease of diameter can be explained by the evaporation of volatile species such as CO, CO2, methanol, and acetic acid during the pyrolysis of BC aerogels.56 The SEM images of prepared P-PBC are illustrated in Fig. 3a–d. The morphology of interconnected nanofibers is well preserved, but the diameter of these nanofibers increases with the raised mred-P:mPBC based on the size survey of P-PBC nanofibers (Fig. S3, ESI†). It is notable that the bundle-like structure can't be observed in the cases of P-PBC-70 and P-PBC-80, and even some individual red-P particles appear in P-PBC-80 (as arrowed in Fig. 3d). The appearance of individual red-P particles indicates the excessive mass loading of red-P for PBC matrix when the value of mred-P:mPBC reaches to 80. The subsequent TEM images of prepared P-PBC (Fig. 3e–h) further reveal that the intervals of nanofiber bundles were gradually filled in by red-P with the ever-increasing mred-P:mPBC, by comparing with the TEM image of pristine PBC (Fig. S4, ESI†). During which the gaseous white-P molecules penetrate into the intervals of PBC nanofiber bundles and occupy their volume partially or even entirely and then turned back into the red-P without the destruction of the original 1D morphology of PBC matrix.45 Similarly, some individual red-P particles are also apparent in the TEM image of P-PBC-80 (as arrowed in Fig. 3h), which is in consistent with its SEM image.
 | ||
| Fig. 3 (a–d) SEM images and (e–h) TEM images of P-PBC-50, P-PBC-60, P-PBC-70 and P-PBC-80, respectively; (i) elemental mapping images of P, C, and O components; (j) HRTEM image (inset: corresponding SAED pattern with a scale bar of 5 1/nm) of P-PBC-70 composite. | ||
Because P-PBC-70 is the one that has the maximum red-P content without the appearance of red-P particles, it was selected as the typical one for the further characterization of red-P distribution in PBC nanofiber bundles. The elemental mapping of P-PBC-70 (Fig. 3i) shows a uniform distribution of P, C and O elements over the entirety of the PBC matrix and P element occupied a relative lager proportion than other two elements, which is in consistent with the previous TGA results. The high resolution TEM (HRTEM) image shows an amorphous structure and the dispersed diffraction rings can be observed in the corresponding selected area electron diffraction (SAED) pattern (inset of Fig. 3j) that indicates the amorphous structure of P-PBC-70,24 which is consistent well with its XRD pattern.
For the sake of investigating the bonding state between PBC and red-P, P-PBC-70 was selected as the typical one for the further XPS characterization. The C1s peak of P-PBC-70 (Fig. 4a) can be split into two peaks, which are attributed to the C–C bonds at 284.8 eV and C–O or C–P bonds at 286.4 eV, respectively. The corresponding O 1s peak (Fig. 4b) can be divided into three peaks at 533.5, 532.5 and 531.5 eV, which are assigned to PO, C–O & P–O and CO bonds, respectively. The P 2p peak of P-PBC-70 (Fig. 4c) can be fitted to the 2p1/2 and 2p3/2 doublet. The peak at 130.7 eV is assigned to the P–P bond. The peak at 134.0 eV corresponds to the C–P bond, which confirms the formation of tight contact between red-P and PBC substructure during the vaporization-condensation process. Besides, the peaks at 135.4 and 134.5 eV represent the existence of P–O bond, which are also in accordance with the O 1s spectrum.21,23,32
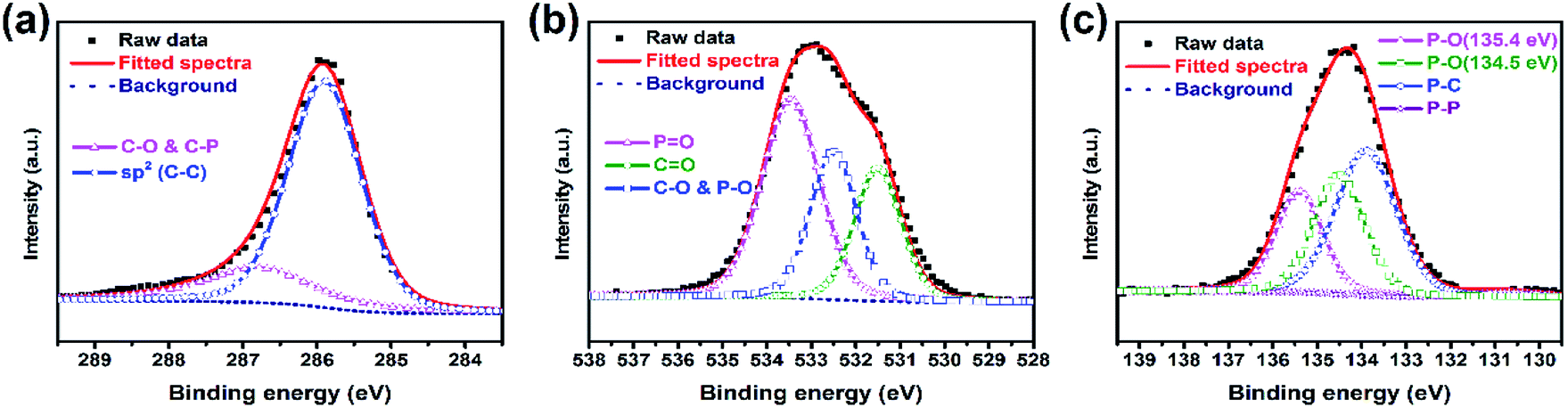 | ||
| Fig. 4 (a–c) High-resolution C 1s, O 1s and P 2p XPS spectra of P-PBC-70. | ||
Cyclic Voltammetry (CV) was tested at a scan rate of 0.2 mV s−1 in the potential window ranged from 0.01 to 2.5 V (vs. Li/Li+) to investigate lithiation/delithiation behavior. In the first cathodic sweep of PBC (Fig. S5, ESI†), three cathodic peaks appeared at 0.61, 0.98 and 1.3 V (vs. Li/Li+), which are associated with the formation of solid electrolyte interface (SEI).35 A sharp peak appeared at 0.0 V (vs. Li/Li+) is attribute to the process of lithium intercalation. For the pristine red-P (Fig. S6, ESI†), a large bulge appeared around 0.58 V (vs. Li/Li+) in the first cathodic sweep, which could be assigned to the formation LiP and Li2P. As the sweep continued, a sharp peak appeared at 0.0 V (vs. Li/Li+), which was caused by the formation of Li3P. In the following cycles, the anodic peak at 1.25 V (vs. Li/Li+) is attribute to the reduction of Li+ to Li atom.14,15 The CV curves of typical P-PBC-70 film in the first three cycles are shown in Fig. 5a. In the first cathodic process, the broad reduction peak from 0.6 V to 1.1 V (vs. Li/Li+) is related to the formation of solid electrolyte interface (SEI) film, which is consistent with the CV curves of pristine PBC film. In the following cycles, there cathodic peaks at 1.3, 0.5 and 0.35 V (vs. Li/Li+) are attributed to the stepwise formation of LiP, Li2P and Li3P, respectively.15 An anodic peak at 1.76 V (vs. Li/Li+) is owning to the oxidation reaction from P3− to amorphous red-P.14,15 It's obviously that the potential of cathodic and anode peaks of P-PBC-70 are higher than pristine red-P. The CV curves in the first three cycles of P-PBC-50, P-PBC-60 and P-PBC-80 films (Fig. S7a–c, ESI†) are all similar to P-PBC-70 film. However, the areas surrounded by the CV curve of P-PBC-50 and P-PBC-80 in the first sweep process are both larger than that in following sweep processes, which are caused by the irreversibility of the PBC and individual red-P particles, respectively.
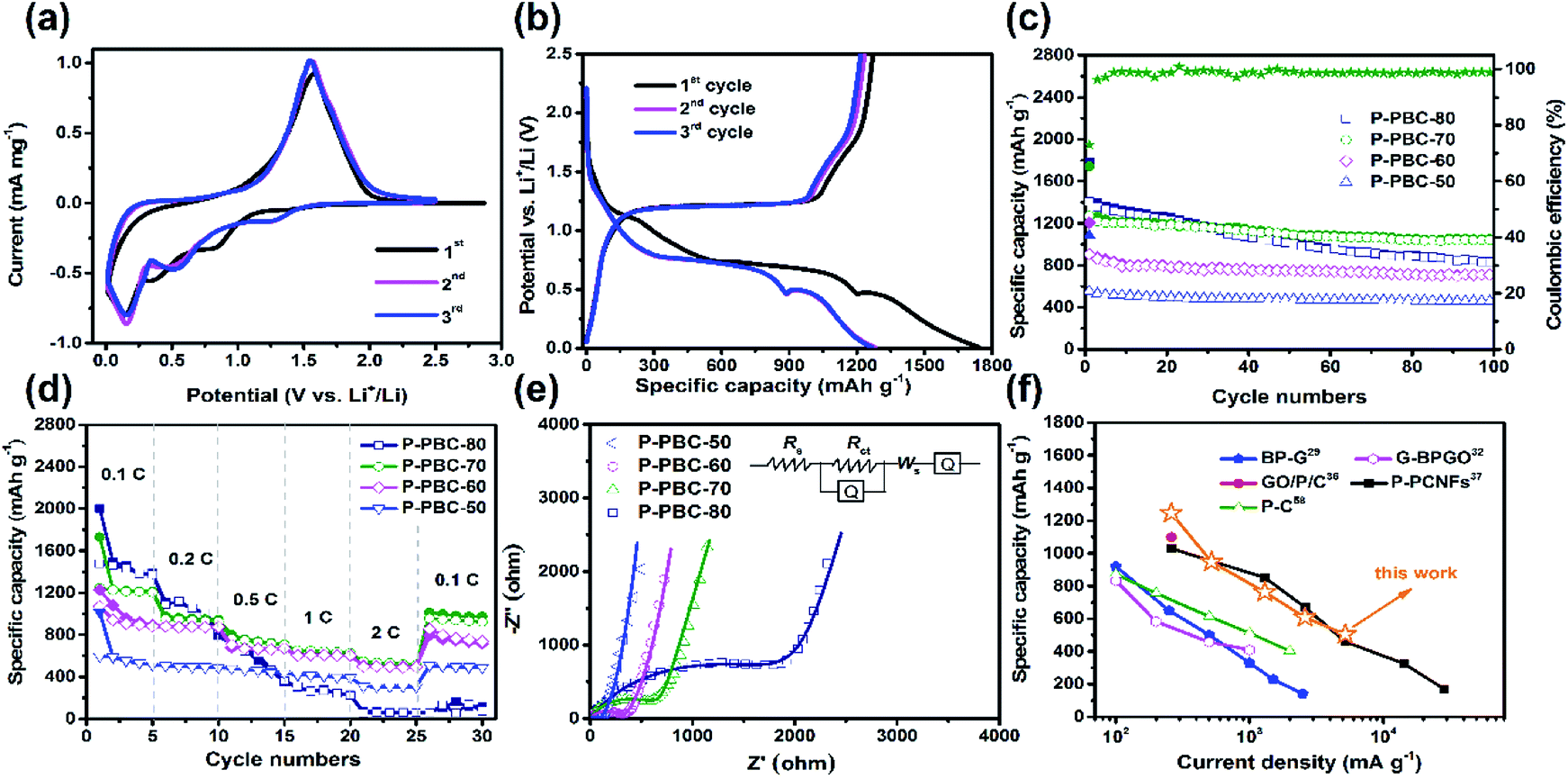 | ||
| Fig. 5 (a) CV curves of P-PBC-70 film at the scan rate 0.2 mV s−1 in the first three cycles; (b) charge–discharge profiles of P-PBC-70 film at 0.1C in the first three cycles; (c) cycling stability test for P-PBC films at 0.1C for 100 cycles (the coulombic efficiency corresponds to the sample of P-PBC-70); (d) rate capabilities of P-PBC films from 0.1 to 2C; (e) Nyquist plots of P-PBC films at the charge state after 100 cycles at 0.1C, and the inset shows the equivalent circuit; (f) the comparison of the rate capability of P-PBC-70 film with other P-based free-standing anodes for LIBs. | ||
The corresponding galvanostatic charge–discharge profiles of P-PBC-70 film in the first three cycles at the rate of 0.1C in the potential window from 0.01 to 2.5 V (vs. Li/Li+) are shown in Fig. 5b. In the first discharge process, the first slope with a plateau-like step between 1.1 and 0.8 V (vs. Li/Li+) corresponds to the formation of SEI film.37,57 The other three discharge plateaus around 1.4, 0.75 and 0.55 V (vs. Li/Li+) are ascribed to the formation of LiP, Li2P and Li3P, respectively, which are higher than the discharge plateaus of pristine red-P around 0.5 and 0.25 V (vs. Li/Li+) (Fig. S8, ESI†). After the completion of lithiation process, the potential decreases slowly until the total Li+ storage capacity of 1742.0 mA h g−1 is obtained based on the total mass of P-PBC-70 film, and the capacity equals to 2541.0 mA h g−1 based on the mass of loaded red-P, which is close to the theoretical value. In the subsequent charging process, the potential gradually increases, and a relatively flat slope appears around 1.2 V (vs. Li/Li+), demonstrating the corresponding reaction from P3− to amorphous red-P. The first charge capacity reaches 1271.9 mA h g−1, with an initial coulombic efficiency of 70.03%. The initial capacity loss is caused by the incomplete re-conversion reaction and the irreversible Li+ consumed by the formation of SEI film. P-PBC-80 and P-PBC-60 films exhibit the similar charge–discharge profiles to P-PBC-70 in the first three cycles (Fig. S9a and b, ESI†), indicating the same electrochemical reaction mechanism, and the initial discharge capacities are proportional to the value of mred-P:mPBC. However, the first discharge profile of P-PBC-50 film (Fig. S9c, ESI†) exhibits the slope-like behavior during the whole process, without any apparent plateau, which is ascribed to its low red-P content that makes the discharge profile of red-P covered by that of pristine PBC film (Fig. S10, ESI†).
The cycling stability of P-PBC films were measured at the rate of 0.1C between 0.01 and 2.5 V (vs. Li/Li+) for 100 cycles and the specific capacities as the function of cycle number are shown in Fig. 5c. When the mred-P:mPBC is not higher than 70, the prepared P-PBC films exhibit stable performances after the initial several cycles, accompanied with the coulombic efficiencies nearly 100%. The discharge capacities of P-PBC-50, P-PBC-60 and P-PBC-70 films are 459.3, 709.4 and 1039.7 mA h g−1 after 100 cycles, corresponding with the capacity retention of 82.87, 89.04 and 82.55% based on their initial reversible capabilities, respectively. However, the specific capacity of P-PBC-80 film fades consecutively during the cycling test, which decreases to 831.1 mA h g−1 after 100 cycles with the capacity retention of 59.62%, which is caused by the irreversibility of bulk red-P (Fig. S11, ESI†). Therefore, the appropriate mred-P:mPBC film is crucial to take advantage of both the high capacity of red-P and grate cycling stability of PBC film (Fig. S12, ESI†).
Electrochemical performances of electrodes at high rates are crucial for high-power LIB, the rate capabilities of prepared P-PBC films were thus evaluated at different rates for five cycles ranging from 0.1 to 2C. As shown in Fig. 5d, the specific capacities of P-PBC films drop in response to the stepwise increased charge–discharge rates. The corresponding charge–discharge profiles of P-PBC films at different rates are presented in Fig. S13 (ESI†). The discharge capacities of P-PBC-50, P-PBC-60, P-PBC-70 and P-PBC-80 films are 309.6, 504.3, 539.7 and 69.7 mA h g−1 at 2C, respectively. After the deep charge–discharge at 2C, the reversible capacities of P-PBC-50, P-PBC-60, P-PBC-70 return to 505.3, 795.6 and 1013 mA h g−1 at 0.1C, respectively. The recovered specific capacities to the original values further approve the satisfactory cycling stabilities of prepared P-PBC films at high rates. However, the reversible capacities of P-PBC-80 still remain at a low value of 64.4 mA h g−1 when the charge–discharge rate turn back to 0.1C. The superior rate capabilities of P-PBC-50, P-PBC-60 and P-PBC-70 to that of P-PBC-80 further demonstrate the dominant influence of the mred-P:mPBC on the electrochemical performances of P-PBC films, and relatively low mass loading of red-P in P-PBC film is beneficial to utilize the excellent rate capability of PBC film, which exhibits excellent capacity retention even at high rate (Fig. S14, ESI†). Because P-PBC-70 shows the highest specific capacity at 2C, it was selected as the typical one to measure the cycling stability at high rate, and its capacity retention achieves to 62.28% after 1000 cycles at 2C (Fig. S15, ESI†) without any macroscopic structural destruction reveal by the digital photo (Fig. S16, ESI†).
EIS was carried out to investigate the electrochemical reaction kinetics of prepared P-PBC films, and their Nyquist plots in the charge state after 100 cycles at 0.1C are shown in Fig. 5e. The large semicircle at high frequency region corresponds to the charge transfer resistance (Rct), and a straight line at low frequency region is assigned to the ionic diffusion impedance through the active bulk material. The simulation parameters based on the equivalent circuit (inset of Fig. 5e) are listed in Table S1 (ESI†), and the Rct values of P-PBC films enhance along with the increasing mred-P:mPBC. However, the abruptly enlarged Rct value of P-PBC-80 film is caused by the unembedded red-P particles outside PBC matrix. The Li+ diffusion coefficients (DLi+) of P-PBC-50, P-PBC-60, P-PBC-70 and P-PBC-80 are calculated as 2.83 × 10−14, 4.86 × 10−15, 6.32 × 10−16 and 3.30 × 10−17 cm2 s−1, respectively, according to the Randles plots (Fig. S17, ESI†). It is obviously that the DLi+ of P-PBC film is reduced with the increase of red-P content in the hybrid films. The significant lower DLi+ of P-PBC-80 film is caused by the individual red-P particles which block the Li+ diffusion into the 3D PBC networks probably.
By comparing with previous studies about free-standing film anodes based on P for LIBs (Table 1),29,32,36,37,58 P-PBC-70 film exhibits a superior cycling stability, associated with the comparable rate capability (Fig. 5f). The resultant electrochemical performances of prepared P-PBC films are benefit from the relatively high mass loading of red-P as well as the 3D interconnected structure of PBC matrix which is composed of unique nanofiber bundles (Fig. 6a). At the optimal ratio of mred-P:mPBC, amorphous red-P can be accommodate in the intervals of PBC nanofiber bundles, which is beneficial for the improvement of cycling stability, because the red-P was effectively constrained in PBC nanofiber bundles during the repeated lithiation/delithiation process. At the same time, the 3D interconnected structure of electrical conductive PBC matrix accelerates the electrons transfer as well as Li+ diffusion, improving the rate capability consequently. However, when the quantity of red-P exceeds the maximum permitted mass loading of PBC nanofiber bundles, the existence of red-P particles outside the 3D interconnected PBC matrix leads to the drastic deterioration of cycling stability and rate capability.
| Electrode components reference | Electrolyte | Preparation method | Potential window (V) | Initial discharge/charge capacity (mA h g−1); (coulombic efficiency (%)) | Retained capacity (mA h g−1) (cycle number/current density (mA g−1)) |
|---|---|---|---|---|---|
| BP nanosheets/few-layer graphene powder (8:2)29 |
1 M LiPF6 EC/DMC/EMC (1:1:1, v/v/v) |
Scalable mineralizer-assisted gas-phase transformation method/mechanical exfoliation | 0.001–3 | ∼950.0/∼830.0; (87.36%) | 402.0(500/500) |
| Black phosphorus/graphene (43.31 wt%)32 | Unknown | Vacuum filtration method | 0.01–3.0 | 1245.0/∼913.0; (∼70.35%) | 731.0(200/100) |
| Phosphorus/hollow carbon cloth/graphene oxide (GO) (71 wt%)36 | 1 M LiPF6 EC/DMC (1:1, v/v) |
Vaporization-condensation (Ar, 500 °C, 30 min, 300 °C, 10 h) | 0.02–2.5 | Unknown/∼1100.0; (unknown) | 910.0(200/260) |
| Crystalline red phosphorus/porous carbon nanofibers composite (34.44 wt%)37 | 1 M LiPF6 EC/DEC (1:1, v/v) |
Vaporization-adsorption-transformation(Ar, 450 °C, 2 h, 260 °C, 18 h) | 0.001–2.5 | 1402.0/1088.0; (∼78%) | 850.0(100/260) |
| Red phosphorus/cross-link-structural carbon (21 wt%)58 | 1 M LiPF6 EC/DEC (1:1, v/v) |
Vapor phase polymerization with pyrolysis process | 0.01–3.0 | 1511.0/921.7; (∼61.0%) | 903.2(640/100) |
| Red phosphorus/pyrolyzed bacterial cellulose (68.56 wt%) this work | 1 M LiPF6 EC/DMC (1:1, v/v) with 2 vol% FEC |
Vaporization-condensation method | 0.01–2.5 | 1742.0/1271.9; (70.03%) | 1039.7(100/260) |
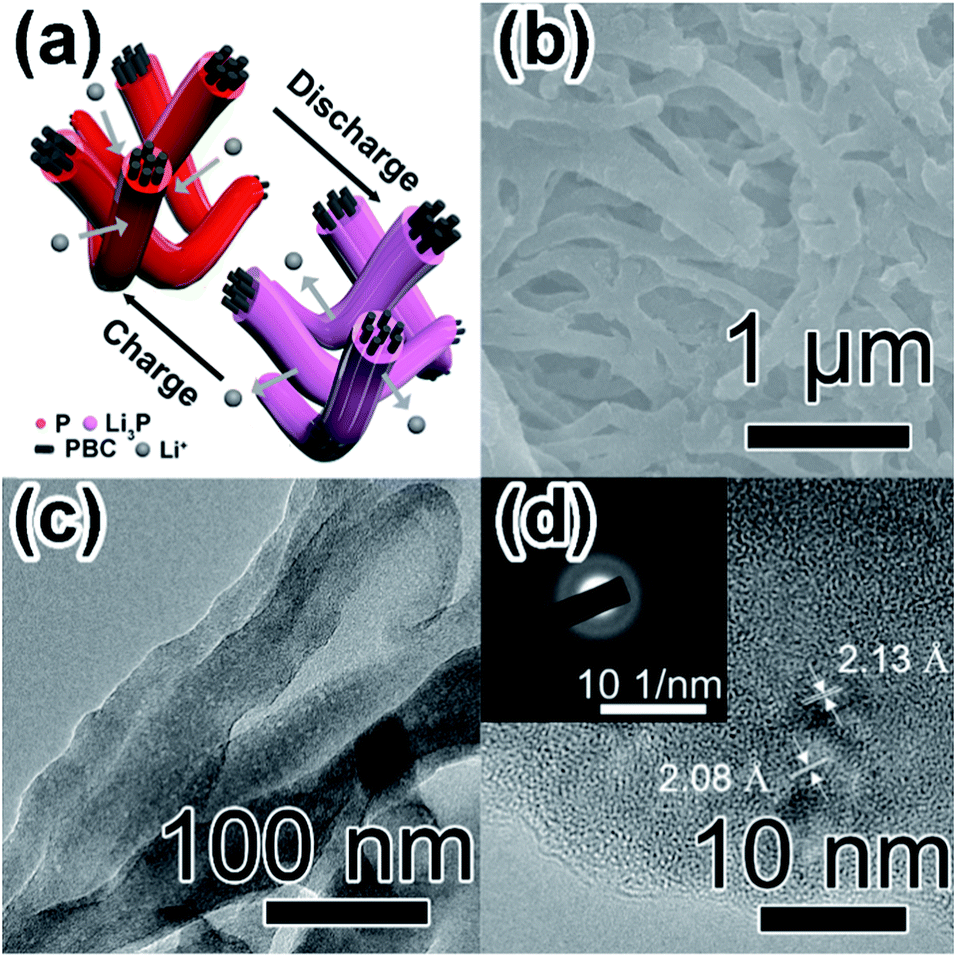 | ||
| Fig. 6 (a) The schematic illustration of morphological and volumetric changes during cycles for nanostructure in P-PBC film; (b) SEM image (c) high-magnification TEM image and (d) HRTEM image (inset: corresponding SAED pattern) of P-PBC-70 film after 1000 cycles at 2C. | ||
The SEM image of P-PBC-70 film at the charged state after 1000 cycles at 2C (Fig. 6b) exhibits the well preserved 3D networks of PBC matrix coated by red-P uniformly and tightly. However, the average diameter (Fig. S18, ESI†) of P-PBC-70 nanofiber bundles is enlarged after the cycling test because of the volume expansion caused by the formation of a part of irreversible Li3P. The corresponding TEM image (Fig. 6c) reveals the maintained bundle-like structure with embedded red-P. The majority of amorphous red-P revealed by the corresponding HRTEM image (Fig. 6d) demonstrates the desirable reversibility of P-PBC-70, but there are some crystal nanodomains with interplanar spacings of the main lattice fringes as 2.13 and 2.08 Å, consistent with the (110) and (103) planes of Li3P, respectively. The diffraction rings in the relevant SAED pattern (inset of Fig. 6d) can be also indexed to the (110) and (103) crystal planes of Li3P. These Li3P nanodomains are formed during the lithiation process and unable to revert to amorphous red-P in the subsequent charge process, which is responsible for the loss of the specific capacity during the first cycle and the slight capacity fading in the subsequent cycles.14
4. Conclusions
In conclusion, a series of free-standing P-PBC films were prepared through the vaporization-condensation method and served as the anodes for LIB directly. The electrochemical performances of P-PBC films could be adjusted by the mred-P:mPBC effectively. P-PBC-70 film delivered a maximum reversible capacity of 1039.7 mA h g−1 based on the mass of free-standing film after 100 cycles at 0.1C and the specific capacity of 418.2 mA h g−1 at 2C. The special 3D network structure of PBC frameworks constructed by interconnected nanofiber bundles is crucial to improve the electrochemical performances of red-P based film. The adequate intervals in the bundle-like structure provide enough volume to accommodate amorphous red-P so as to guarantee the large specific capacity. Meanwhile, the PBC framework can not only enable the fast access of Li+ but also grant the electrode with good electrical conductivity and high mechanical flexibility. This kind of free-standing P-PBC film has great potential in the field of flexible LIB and large scale energy storage devices.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by National Key R&D Program of China (No. 2016YFA0202302), National Natural Science Funds for Distinguished Young Scholars (No. 51425306), the State Key Program of National Natural Science Foundation of China (No. 51633007), and National Natural Science Foundation of China (No. 51573125 and 51773147).References
- Y. Nishi, J. Power Sources, 2001, 100, 101 CrossRef CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- H. Li, Z. X. Wang, L. Q. Chen and X. J. Huang, Adv. Mater., 2009, 21, 4593 CrossRef CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- C. X. Zu and H. Li, Energy Environ. Sci., 2011, 4, 2614 CAS.
- F. C. Lyu, Z. F. Sun, B. Nan, S. C. Yu, L. J. Cao, M. Y. Yang, M. C. Li, W. X. Wang, S. F. Wu and S. S. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 10699 CAS.
- X. Zhao, H. E. Wang, J. Cao, W. Cai and J. H. Sui, ChemComm, 2017, 53, 10723 RSC.
- H. E. Wang, X. Zhao, K. L. Yin, Y. Li, L. H. Chen, X. Y. Yang, W. J. Zhang, B. L. Su and G. Z. Cao, ACS Appl. Mater. Interfaces, 2017, 50, 43665 Search PubMed.
- H. E. Wang, X. Zhao, X. C. Li, Z. Y. Wang, C. F. Liu, Z. G. Lu, W. J. Zhang and G. Z. Cao, J. Mater. Chem. A, 2017, 5, 25056 CAS.
- Y. Cai, H. E. Wang, X. Zhao, F. Huang, C. Wang, Z. Deng, Y. Li, G. Z. Cao and B. L. Su, ACS Appl. Mater. Interfaces, 2017, 9, 10652 CAS.
- R. Malini, U. Uma, T. Sheela, M. Ganesan and N. G. Renganathan, Ionics, 2009, 15, 301 CrossRef CAS.
- S. Xin, Z. W. Chang, X. B. Zhang and Y. G. Guo, Natl. Sci. Rev., 2016, 4, 54 Search PubMed.
- L. Wang, X. M. He, J. J. Li, W. T. Sun, J. Gao, J. W. Guo and C. Y. Jiang, Angew. Chem., Int. Ed., 2012, 51, 9034 CrossRef CAS PubMed.
- C. Marino, L. Boulet, P. Gaveau, B. Fraisse and L. Monconduit, J. Mater. Chem., 2012, 22, 22713 RSC.
- C. Marino, A. Debenedetti, B. Fraisse, F. Favier and L. Monconduit, Electrochem. Commun., 2011, 13, 346 CrossRef CAS.
- W. H. Li, Z. Z. Yang, M. S. Li, Y. Jiang, X. Wei, X. W. Zhong, L. Gu and Y. Yu, Nano Lett., 2016, 16, 1546 CrossRef CAS PubMed.
- J. Y. Li, L. Wang, Y. M. Ren, Y. Zhang, Y. F. Wang, A. G. Hu and X. M. He, Ionics, 2016, 22, 167 CrossRef CAS.
- Y. L. Wang, L. Y. Tian, Z. H. Yao, F. Li, S. Li and S. H. Ye, Electrochim. Acta, 2015, 163, 71 CrossRef CAS.
- J. Y. Li, L. Wang, X. M. He and J. L. Wang, ACS Sustainable Chem. Eng., 2016, 4, 4217 CrossRef CAS.
- A. J. Bai, L. Wang, Y. Li, X. M. He, J. X. Wang and J. L. Wang, J. Power Sources, 2015, 289, 100 CrossRef CAS.
- J. Sun, G. Y. Zheng, H. W. Lee, N. Liu, H. T. Wang, H. B. Yao, W. S. Yang and Y. Cui, Nano Lett., 2014, 14, 4573 CrossRef CAS PubMed.
- T. Ramireddy, T. Xing, M. M. Rahman, Y. Chen, Q. Dutercq, D. Gunzelmann and A. M. Glushenkov, J. Mater. Chem. A, 2015, 3, 5572 CAS.
- J. T. Xu, I. Y. Jeon, J. M. Ma, Y. H. Dou, S. J. Kim, J. M. Seo, H. K. Liu, S. X. Dou, J. B. Baek and L. M. Dai, Nano Res., 2017, 4, 1268 CrossRef.
- C. M. Park and H. J. Sohn, Adv. Mater., 2007, 19, 2465 CrossRef CAS.
- J. F. Qian, D. Qiao, X. P. Ai, Y. L. Cao and H. X. Yang, Chem. Commun., 2012, 48, 8931 RSC.
- L. Y. Wang, H. L. Guo, W. Wang, K. Y. Teng, Z. W. Xu, C. Chen, C. Y. Li, C. Y. Yang and C. S. Hu, Electrochim. Acta, 2016, 211, 499 CrossRef CAS.
- Z. X. Yu, J. X. Song, M. L. Gordin, R. Yi, D. H. Tang and D. H. Wang, Adv. Sci., 2015, 2, 1400020 CrossRef PubMed.
- C. M. Subramaniyam, Z. Tai, N. Mahmood, D. Zhang, H. K. Liu, J. B. Goodenough and S. X. Dou, J. Mater. Chem. A, 2017, 5, 1925 CAS.
- L. Chen, G. M. Zhou, Z. B. Liu, X. M. Ma, J. Chen, Z. Y. Zhang, X. L. Ma, F. Li, H. M. Cheng and W. C. Ren, Adv. Mater., 2016, 28, 510 CrossRef CAS PubMed.
- L. Pan, X. D. Zhu, K. N. Sung, Y. T. Liu, X. M. Xie and X. Y. Ye, Nano Energy, 2016, 30, 347 CrossRef CAS.
- D. Zhao, B. B. Li, J. Y. Zhang, X. Li, D. B. Xiao, C. C. Fu, L. H. Zhang, Z. H. Li, J. Li and D. X. Cao, Nano Lett., 2017, 17, 3376 CrossRef CAS PubMed.
- H. W. Liu, Y. Q. Zou, L. Tao, Z. L. Ma, D. D. Liu, P. Zhou, H. B. Liu and S. Y. Wang, Small, 2017, 13, 1700758 CrossRef PubMed.
- D. M. Yuan, J. L. Cheng, G. X. Qu, X. D. Li, W. Ni, B. Wang and H. Liu, J. Power Sources, 2016, 301, 131 CrossRef CAS.
- N. Nitta, D. N. Lei, H. R. Jung, D. Gordon, E. B. Zhao, G. Gresham, J. Cai, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2016, 8, 25991 CAS.
- Z. W. Xu, Y. Zeng, L. Y. Wang, N. Li, C. Chen, C. Y. Li, J. Li, H. M. Lv, L. Y. Kuang and X. Tian, J. Power Sources, 2017, 356, 18 CrossRef CAS.
- Y. H. Du, Y. F. Tang and C. K. Chang, J. Electrochem. Soc., 2016, 163, A2938 CrossRef CAS.
- W. H. Li, Z. Z. Yang, Y. Jiang, Z. R. Yu, L. Gu and Y. Yu, Carbon, 2014, 78, 455 CrossRef CAS.
- G. M. Zhou, F. Li and H. M. Cheng, Energy Environ. Sci., 2014, 7, 1307 CAS.
- Y. H. Xu, Y. J. Zhu, F. D. Han, C. Luo and C. S. Wang, Adv. Energy Mater., 2015, 5, 1400753 CrossRef.
- X. Q. Xie, T. Makaryan, M. Q. Zhao, K. L. Van Aken, Y. Gogotsi and G. X. Wang, Adv. Energy Mater., 2016, 6, 1502161 CrossRef.
- H. R. An, Y. Li, Y. Gao, C. Cao, J. K. Han, Y. Y. Feng and W. Feng, Carbon, 2017, 116, 338 CrossRef CAS.
- R. Mukherjee, A. V. Thomas, A. Krishnamurthy and N. Koratkar, ACS Nano, 2012, 6, 7867 CrossRef CAS PubMed.
- C. Zhang, X. Wang, Q. F. Liang, X. Z. Liu, Q. H. Weng, J. W. Liu, Y. J. Yang, Z. H. Dai, K. J. Ding and Y. Bando, Nano Lett., 2016, 16, 2054 CrossRef CAS PubMed.
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759 CrossRef CAS PubMed.
- M. Iguchi, S. Yamanaka and A. Budhiono, J. Mater. Sci., 2000, 35, 261 CrossRef CAS.
- Z. Y. Wu, C. Li, H. W. Liang, J. F. Chen and S. H. Yu, Angew. Chem., 2013, 125, 2997 CrossRef.
- Y. Z. Wan, G. F. Zuo, F. Yu, Y. A. Huang, K. J. Ren and H. L. Luo, Surf. Coat. Technol., 2011, 205, 2938 CrossRef CAS.
- L. F. Chen, Z. H. Huang, H. W. Liang, W. T. Yao, Z. Y. Yu and S. H. Yu, Energy Environ. Sci., 2013, 6, 3331 CAS.
- L. X. Zhang, Z. H. Liu, G. L. Cui and L. Q. Chen, Prog. Polym. Sci., 2015, 43, 136 CrossRef CAS.
- R. E. Franklin, Acta Crystallogr., 1951, 4, 253 CrossRef CAS.
- J. M. Zaug, A. K. Soper and S. M. Clark, Nat. Mater., 2008, 7, 890 CrossRef CAS PubMed.
- V. Gomez-Serrano, J. Pastor-Villegas, A. Perez-Florindo, C. Duran-Valle and C. Valenzuela-Calahorro, J. Anal. Appl. Pyrolysis, 1996, 36, 71 CrossRef CAS.
- L. W. Daasch and D. C. Smith, Anal. Chem., 1951, 23, 853 CrossRef CAS.
- J. X. Song, Z. X. Yu, M. L. Gordin, X. L. Li, H. S. Peng and D. H. Wang, ACS Nano, 2015, 9, 11933 CrossRef CAS PubMed.
- Y. Kim, Y. Park, A. Choi, N. S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045 CrossRef CAS PubMed.
- H. W. Liang, Q. F. Guan, Z. Zhu, L. T. Song, H. B. Yao, X. Lei and S. H. Yu, NPG Asia Mater., 2012, 4, e19 CrossRef.
- Y. H. Liu, A. Y. Zhang, C. F. Shen, Q. Z. Liu, X. Cao, Y. Q. Ma, L. Chen, C. Lau, T. C. Chen, F. Wei and C. W. Zhou, ACS Nano, 2017, 11, 5530 CrossRef CAS PubMed.
- J. F. Ruan, T. Yuan, Y. P. Pang, X. B. Xu, J. H. Yang, W. B. Hu, C. Zhong, Z. F. Ma, X. X. Bi and S. Y. Zheng, ACS Appl. Mater. Interfaces, 2017, 9, 36261 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM and SEM images, digital photographs, the first three CV curves, cycle performance, and rate capabilities of PBC and P-PBC. See DOI: 10.1039/c8ra02370k |
| This journal is © The Royal Society of Chemistry 2018 |