
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLactobionic acid-functionalized polyethersulfone hollow fiber membranes promote HepG2 attachment and function
Surendra Kumar Vermaa,
Akshay Modi a,
Ashwin Dravida and
Jayesh Bellare*abc
a,
Ashwin Dravida and
Jayesh Bellare*abc
aDepartment of Chemical Engineering, Indian Institute of Technology Bombay, Mumbai, 400076, India. E-mail: jb@iitb.ac.in; Fax: +91-22-2572-6895; Tel: +91-22-2576-7207
bCentre for Research in Nanotechnology & Sciences, Indian Institute of Technology Bombay, Mumbai, 400076, India
cWadhwani Research Center for Bioengineering, Indian Institute of Technology Bombay, Mumbai, 400076, India
First published on 14th August 2018
Abstract
Surface modification of polyethersulfone hollow fibers, which are important in bio-artificial liver, is increasingly used to improve biocompatibility and promote the adhesion and proliferation of hepatocytes resulting in improved cell functionality. Hepatocytes are anchorage-dependent cells, and membrane surface modification enhances the hepatic cell adhesion and proliferation. Specific interaction of the asialoglycoprotein receptor on hepatocyte cell surfaces with a galactose moiety enhances the attachment of the cells on a biocompatible substrate. In this study, the outer surface of the polyethersulfone (P) hollow fiber membranes (HFMs) was chemically modified by covalent coupling with lactobionic acid (LBA). The energy dispersive X-ray spectrometry elemental mapping, attenuated total reflectance-Fourier transform infrared spectroscopy, and X-ray photoelectron spectroscopy confirmed the LBA-coupling on the outer surface of P-LBA HFMs. Hemocompatibility study indicated the suitability of the modified membranes with human blood. These membranes showed remarkably improved biocompatibility with human primary mesenchymal stem cells and HepG2 cells. Characteristic multi-cellular spheroids of HepG2 cells were observed under scanning electron and confocal microscopy. HepG2 cell functional activity was measured by quantifying the urea synthesis, albumin secretion and glucose consumption in the culture media, which indicated the improved HepG2 functions. These experimental results clearly suggest the potentiality of these LBA-modified P HFMs as a suitable biocompatible substrate for promoting HepG2 attachment and function leading to their application in bioreactors and bio-artificial liver devices.
1. Introduction
Both natural and synthetic biomaterials of different compositions are being explored for tissue engineering applications. Synthetic polymers are chosen because they can be easily chemically modified and mass-produced as per the desired application.1 Polyethersulfone (PES/P) is a nondegradable, biocompatible polymer membrane material, which is extensively being used in hemodialysis, ultrafiltration, gas separation and bioreactor technology.2–4 PES has excellent thermal stability and chemical resistance.5Surface properties of different materials including implanted devices, scaffolds, cell culture supports, and bioreactors control cell and host responses.6 Over the last few years, most of the advancement in the field of biomaterials has been focused on activating these different materials biologically, which would regulate cell behavior due to the interactions with specific cell membrane receptors via incorporation of ligand molecules.7 Membrane surface modification, involving both chemical modification and physical coating, enhances the hepatocytic cell adhesion and proliferation. Hepatocytes are known as anchorage-dependent cells, and their viability and differentiated function maintenance are highly sensitive to the extracellular matrix (ECM) environment.8 Very recently, the chitosan-modified composite polysulfone hollow fiber membranes (HFMs) showed enhanced attachment and proliferation of HepG2 cells exhibiting characteristic three dimensional multicellular spheroid morphology.9 In another study, PES HFMs were surface functionalized with glutaraldehyde-crosslinked gelatin, which supported the HepG2 cells attachment, growth, and proliferation.10 Galactose-derived Pluronic F68 (F68-gal) was adsorbed on polyvinylidene fluoride (PVDF) membrane promoted hepatocyte attachment and proliferation, which resulted in high albumin synthesis and P450 1A1 activity as compared to unmodified and collagen-coated PVDF membranes.11
Cell surface receptors of hepatocyte recognize and bind molecules with exposed galactose, N-acetylgalactosamine, or glucose residues.12 It is reported in the literature, that galactosylated alginate capsules binds with hepatocytes via the patch of asialoglycoprotein receptor (ASGPR). The galactose-modified substrate enhanced liver functions of hepatocytes compared with those in alginate capsules.13 Interaction between cells and extracellular matrix (ECM) permits cell attachment and provides regulatory signals to cells through adhesion.14 There are a variety of cell types that have been reported to adhere to PES, including endothelial cells, fibroblasts, osteoblasts, epithelial cells and keratinocytes.1,10 HFM based substrates are preferred because they provide three-dimensional microenvironment, immunoisolation, continuous flow, large capacity and scale up benefit over other systems.15
In the present study, lactobionic acid (LBA) as ASGPR ligand was covalently grafted on the outer surface of P HFMs to promote HepG2 cells adhesion and proliferation. LBA was covalently attached on the outer surface of HFMs by a facile method via amination of P HFMs. The modified HFMs are denoted as P-LBA HFMs, which were characterized by environmental scanning electron microscopy (ESEM), energy-dispersive X-ray spectroscopy (EDS) elemental mapping, attenuated total reflectance-Fourier transform infra-red (ATR-FTIR) spectroscopy, and X-ray photoelectron spectroscopy (XPS). The hemocompatibility of HFM samples was also evaluated using human blood. The developed membranes were evaluated for biocompatibility and cytocompatibility using human primary mesenchymal stem cells and HepG2 cells. HepG2 specific cell functional studies were performed to evaluate the cell activity.
2. Materials and methods
2.1. Materials
PES (Ultrason E 6020 P), and N-methyl-2-pyrrolidone (NMP) were procured from BASF (Germany), and Spectrochem (India), respectively, and used without any further purification. LBA was procured from Sigma-Aldrich (United States). N-Hydroxysuccinimide (NHS), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and chloromethyl methyl ether (CMME) were bought from Spectrochem Pvt. Ltd. (India). 2-Morpholine ethane sulphonic acid (MES) was purchased from Sisco Research Laboratories (India). Tin chloride (SnCl4) was procured from Sigma-Aldrich (United States). Hexane and ethylene diamine (EDA) were purchased from Merck Chemicals (India) and phosphate buffered saline (PBS) was bought from HiMedia Laboratories Pvt. Ltd., (India). Collagenase type IV and dispase were procured from Sigma-Aldrich (United States) and HiMedia Laboratories Pvt. Ltd., India respectively.BD Vacutainer® PLUS plastic plasma blood collection tubes containing 143 USP units sodium heparin (spray-coated) anticoagulant were procured from Becton, Dickinson and Company (United States). HepG2 cell lines were procured from National Centre for Cell Science (India). For cell culture, Dulbecco's modified eagle's medium (DMEM) (Gibco, Invitrogen, United States) containing 2 mM L-glutamine, supplemented with penicillin (100 units per mL), streptomycin (100 μg mL−1) (HiMedia Laboratories Pvt. Ltd., India) and 10% fetal calf serum (Invitrogen, United States) was used.
2.2. Development of P HFMs
P HFMs were developed as described in our previous study.3,16 Briefly, P was vacuum dried in oven for 24 h at 80 °C to remove the absorbed moisture and dissolved in NMP to prepare polymer dope solution. The dope solution was degassed to remove dissolved gas from the solution. Indigenously fabricated HFM spinning pilot plant was used for developing HFMs. The process parameters used for HFM spinning are listed in Table 1. The prepared fibers were left in water for a day to remove the residual solvent and used for further studies.| Ambient temperature (°C) | 25 |
| Relative humidity (%) | 50–60 |
| Dope solution composition | 25 wt% P |
| Bore solution composition | Deionized (DI) water |
| Dope and bore solution temperature (°C) | 25 |
| Dope flow rate (mL min−1) | 2 |
| Bore flow rate (mL min−1) | 2 |
| Air gap (cm) | 45 |
| Coagulation bath composition | DI water |
| Rinse bath composition | DI water |
| Coagulation bath temperature (°C) | 25 |
2.3. Surface functionalization of P HFMs
The surface functionalization of P HFMs was carried out in two steps: first step, amino groups were incorporated on outer surface of the PES HFMs, and second step, LBA were coupled with the animated P HFMs.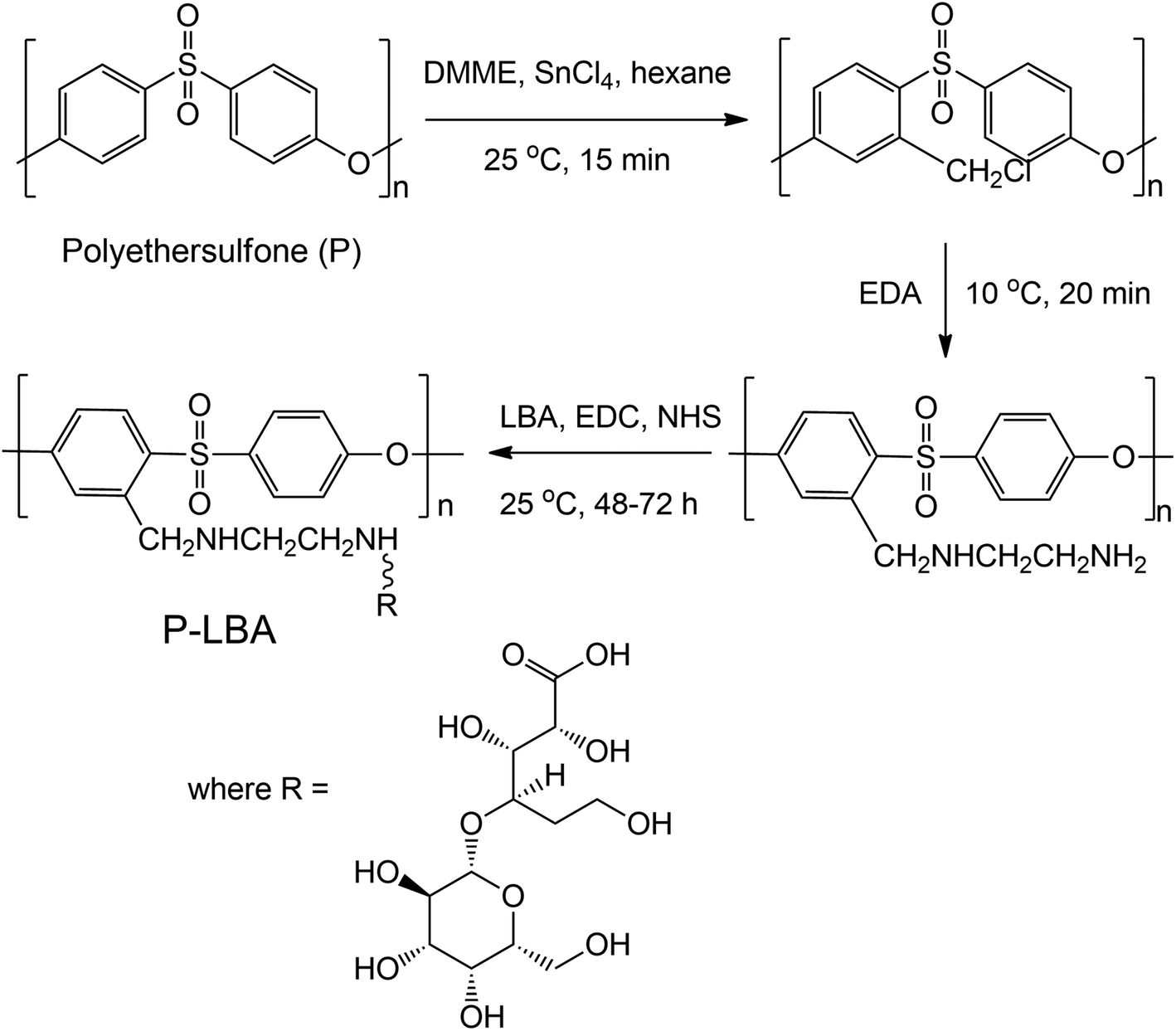 | ||
| Fig. 1 Reaction scheme for surface functionalization of P HFMs resulting in P-LBA HFMs. | ||
In this study, two samples were considered for the comparison purposes:
(1) P: plain HFMs.
(2) P-LBA: LBA-surface modified P HFMs.
2.4. Characterization of HFMs
The surface morphology of HFM samples (P and P-LBA) were observed by ESEM study. The HFM samples were then coated with gold/platinum by sputter coating using Auto fine coater JFC-1600 (JEOL, Japan) before observing under ESEM at 5–10 kV (Hitachi, S-3400 N, United Kingdom). EDS elemental mapping was also performed to map the elements present on the outer surface of P and P-LBA HFMs (X-Max 20 mm2 EDS system, Oxfords Instruments, United Kingdom). The surface modification of P-LBA HFMs was confirmed by ATR-FTIR (Bruker, Germany), and XPS (Kratos Analytical, United Kingdom) studies.In addition to the above-mentioned characterization, the hydrophilicity and roughness of the biomaterial surface are also important factors, which influence the cell attachment, and protein adsorption.19 Water contact angles were measured using goniometer, Digidrop GBX (GBX Instruments, Romans, France) to assess the wettability of the HFM samples. Membrane surface roughness is another factor, which affects the attachment of the cells on the membrane surface.20,21 The outer surface roughness was measured with the help of atomic force microscopy (AFM) (MFP-3 D-BIO, Asylum Research, USA). HFM samples were fixed on a cover slide with the help of adhesive tape. 5 × 5 μm area was scanned in the tapping mode in air. SiN probe cantilever having spring constant of 40 N m−1 was used for analysis. The average roughness (Rrms) of HFM samples was reported.
2.5. Evaluation of compatibility of HFMs with human blood
The compatibility of HFM samples with human blood was evaluated by measuring the hemolysis values. The protocol followed to perform hemolysis for HFM samples was described in the literature.21–23 Briefly, human blood was collected from a healthy donor in accordance with extant institutional guidelines and collected in BD Vacutainer® Plus plastic plasma tubes containing 143 USP units sodium heparin (spray-coated) anticoagulant. The collected blood sample was centrifuged at 1250 rpm for 15 min at 10 °C. After centrifugation, the supernatant containing platelet rich plasma was removed. The pellet containing erythrocytes were washed three times with normal saline solution (NSS) (0.9% w/v NaCl). The obtained hematocrit was incubated with HFM samples (4 pieces of 2 cm length) for 1 h at 37 °C and 5% carbon dioxide (CO2) in an incubator (Thermo Scientific™, United States). DI water and NSS were used as positive and negative controls, respectively. The samples were centrifuged at 1000 g for 5 min. The red blood cells lysis was quantified using UV-visible spectrophotometer (Molecular Devices, United States) at 542 nm. Hemolysis ratio (HR) was calculated using the equation:16
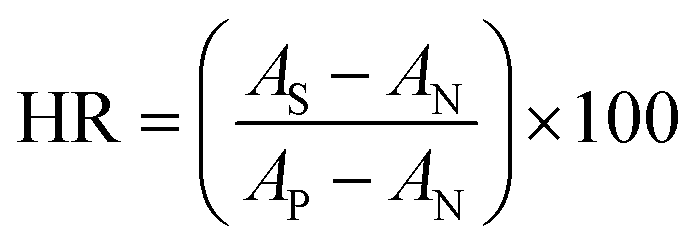 | (1) |
2.6. Study of human umbilical cord mesenchymal stem cells (hUMSCs) expansion on HFMs
Obtaining an adequate number of cells from a donor for transplantation is a major bottleneck to the success of stem cell therapy. A substrate, which can support the growth and expansion of cell especially human stem cell, is always desired.24,25 In this study, the HFM samples were studied for stem cell expansion. Human umbilical cords (hUCs) were obtained from a local maternity and health center after signing informed consents. UCs were collected in 50 mL falcon tube containing phosphate-buffered saline (PBS) supplemented with antibiotics, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin and transported to the laboratory.26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) on a magnetic orbital shaker at 37 °C. Further, a mixture of trypsin (0.05 wt%) and EDTA (0.02 wt%) was added and incubated on a magnetic orbital shaker for 15–20 min at 37 °C. The digested tissues were then filtered using sterile nylon filter and centrifuged at 1500 rpm for 10 min. Finally, the pellet was re-suspended, cultured in DMEM and incubated at 37 °C and 5% CO2. The culture media were replaced every two days alternatively.
:1 v/v) on a magnetic orbital shaker at 37 °C. Further, a mixture of trypsin (0.05 wt%) and EDTA (0.02 wt%) was added and incubated on a magnetic orbital shaker for 15–20 min at 37 °C. The digested tissues were then filtered using sterile nylon filter and centrifuged at 1500 rpm for 10 min. Finally, the pellet was re-suspended, cultured in DMEM and incubated at 37 °C and 5% CO2. The culture media were replaced every two days alternatively.At day 3 and 6, the samples were gently washed with PBS. The HFM samples with cells were evaluated for nutrient consumption and cell proliferation.
The quantitative evaluation of cell proliferation on HFM was assessed using Quant-iT PicoGreen ds DNA assay kit (Molecular probe, Invitrogen, United States). The HFM, seeded with cells were harvested at set time points. The samples were washed with PBS and dipped in deionized water to lyse the cells. Thereafter, the samples were frozen and thawed alternatively twice and centrifuged at 10000 rpm for 10 min. Then, 100 μL of PicoGreen working solution was added to supernatant of each sample, and left for incubation for 5 min. The concentration of DNA was measured using a fluorescence microplate reader (Spectramax M2e. Molecular devices, United States). The fluorescence readings were recorded at 480 nm excitation and 538 nm emission. The DNA amount was calculated by using the standard curve as prescribed by the manufacturers' kit. The HFM samples with cells were also evaluated for nutrient consumption (by determining the glucose levels in the culture media of different HFMs).
2.7. HepG2 cell culture study
HepG2 cells were cultured in DMEM containing 2 mM L glutamine, supplemented with penicillin (100 units per mL), streptomycin (100 mg mL−1) and 10% fetal calf serum at 37 °C in CO2 incubator. HFM samples with equal surface area, were used for cell seeding. Samples were sterilized by dipping in 70% ethanol followed by exposing to UV-light for 30 min. Excess of ethanol was removed by washing thoroughly with sterile PBS. 1 × 103 cells per cm2 were seeded on HFM samples. Cells were allowed to attach with these samples for 4 h, and then the HFM samples with cells were transferred into 12-well plate with fresh culture medium.21 At day 3 and 6, the samples were gently washed with PBS. The HFM samples with cells were evaluated for nutrient consumption, cell proliferation with characteristic morphology. HepG2 cell functional study was performed by evaluating the urea synthesis, albumin secretion and glucose consumption in the culture media.3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT)-based cytotoxicity test was performed to evaluate cytocompatibility of the HFM samples. After incubation of day 3 and day 6, 500 μL MTT was added to each well. After 4 h incubation at 37 °C, 500 μL dimethyl sulfoxide (DMSO) was added to each well to dissolve the formazan crystals. The optical density of the formazan solution was determined by a UV-visible spectrophotometer (Molecular Devices, United States) at 560 nm. In addition to this, DNA quantification was performed as described in the earlier Section 2.6.3.1.
Urea synthesis and albumin secretion are the liver cell specific functional marker.27 Spent cell culture media were evaluated for urea and albumin concentration at day 3 and day 6. The detection procedure was followed as per the estimation kit manufacturers' protocols. The glucose concentration in the spent medium was determined with a biochemical GOD/POD method using a kit procured from Coral Clinical Systems (India). In this, glucose is oxidised to gluconic acid and hydrogen peroxide in the presence of glucose oxidase. Further, hydrogen peroxide reacts with phenol and 4-aminoantipyrine by the catalytic action of peroxidase to form a red coloured quinoneimine dye complex. The glucose consumption was calculated at day 3 and day 6. This help to evaluate cell function and continuous proliferation.
3. Results and discussion
In this section, the results related to the physicochemical characterization, biocompatibility including hemocompatibility evaluation, and HepG2 functions are presented and discussed.3.1. HFM characterization
In this section, the results discussing the physicochemical characterization of HFMs by ESEM, EDS elemental mapping, ATR-FTIR, XPS, water contact angle, and AFM are presented. | ||
| Fig. 2 (A) ESEM micrographs of P and P-LBA HFMs showing the overview and cross-sectional view. Concentric HFMs with interconnected pores and fingers-like structures observed. (B) EDS elemental mapping of the outer surface of HFMs showed the presence of nitrogen in the P-LBA HFMs, which is an indicative of uniform surface modification via covalent coupling of LBA on aminated HFMs (P-LBA HFMs). | ||
Fig. 2B shows the EDS elemental mapping of both the HFM samples: P and P-LBA. The elemental map of N in P-LBA HFMs clearly indicates that the surface modification of HFMs was uniform across the length and width of the HFMs. Further, the change in intensity from left to right of an elemental map is due to the curvature of the HFMs.
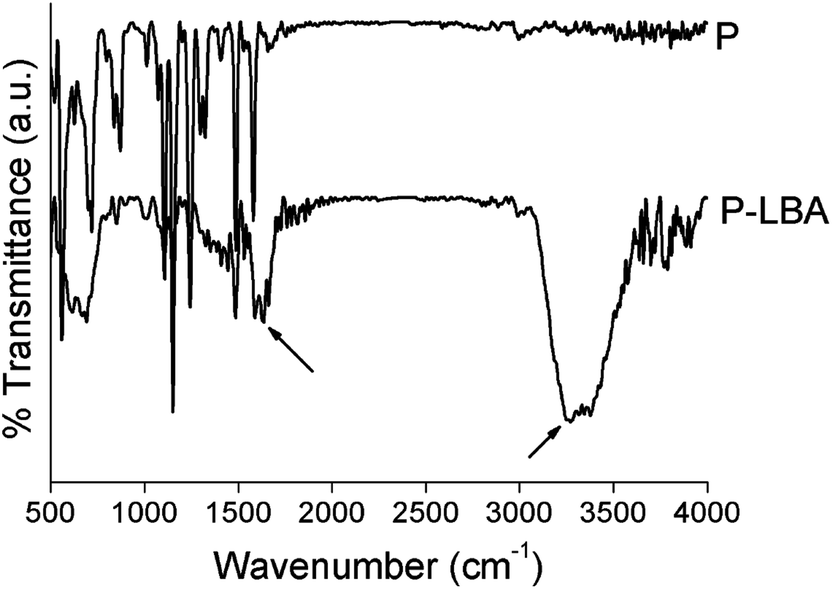 | ||
| Fig. 3 ATR-FTIR spectra of P and P-LBA HFMs. The presence of characteristic bands in the range 1580–1680 cm−1 corresponding to –NH2 and –CO–NH– groups, and 3200–3400 cm−1 corresponding to NH and –OH groups confirmed the surface modification on HFMs. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, respectively. For O 1s, the two peaks appeared at 532.5 eV and 534.3 eV, which represented CO and C–O–C/C–OH bonds, respectively. For N 1s, the peaks were recorded at 398.9, 400.2, and 403 eV, which corresponded to pyridinic, pyrrolic, and graphitic bonds, respectively. Furthermore, the mass composition (%) of C, N, O and S in P-LBA HFMs was 56.83 ± 0.69, 11.77 ± 0.64, 24.56 ± 0.46, and 6.83 ± 0.21. Thus, the XPS results clearly confirmed the presence of amide and LBA linkages in the P-LBA HFMs as discussed in the earlier results.
O bond, respectively. For O 1s, the two peaks appeared at 532.5 eV and 534.3 eV, which represented CO and C–O–C/C–OH bonds, respectively. For N 1s, the peaks were recorded at 398.9, 400.2, and 403 eV, which corresponded to pyridinic, pyrrolic, and graphitic bonds, respectively. Furthermore, the mass composition (%) of C, N, O and S in P-LBA HFMs was 56.83 ± 0.69, 11.77 ± 0.64, 24.56 ± 0.46, and 6.83 ± 0.21. Thus, the XPS results clearly confirmed the presence of amide and LBA linkages in the P-LBA HFMs as discussed in the earlier results.
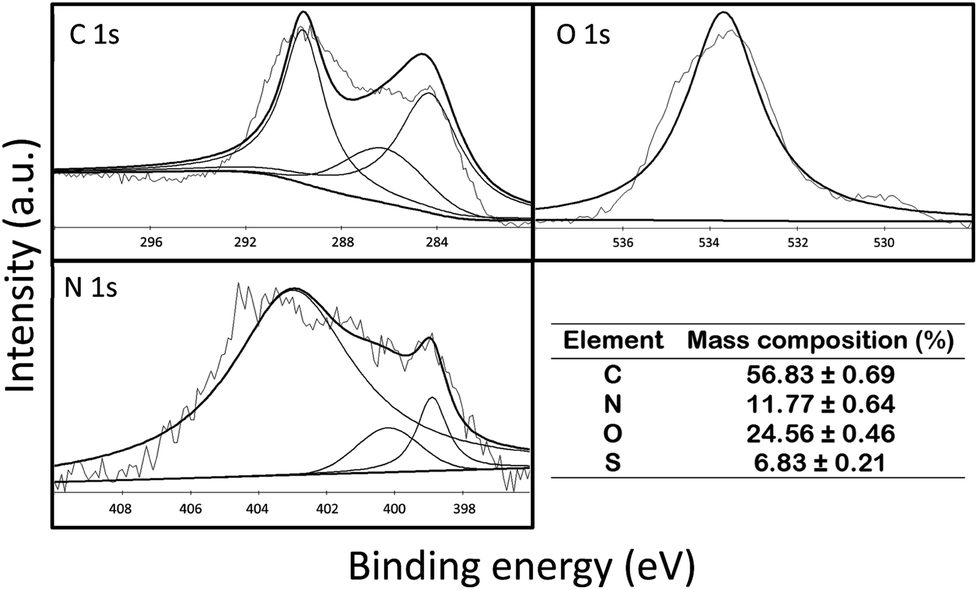 | ||
| Fig. 4 XPS spectra for C 1s, O 1s and N 1s for P-LBA HFMs. The appearance of the characteristic peaks confirmed the presence of amide and LBA linkages in the P-LBA HFMs. The mass composition (%) is also shown alongside the XPS spectra. | ||
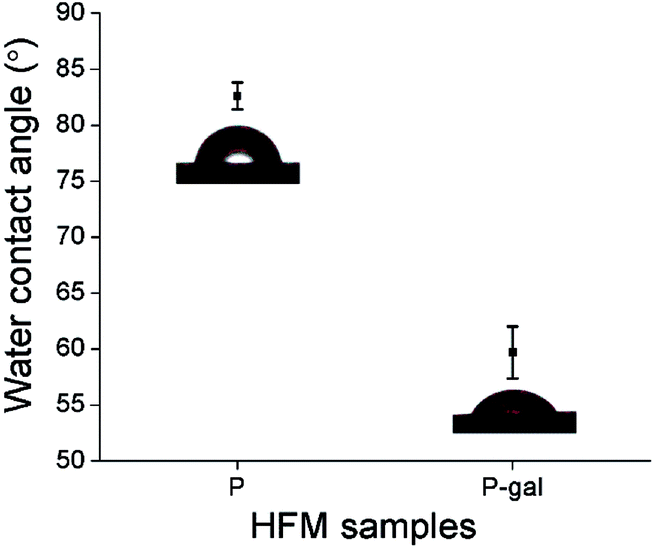 | ||
| Fig. 5 Water contact angles of P and P-LBA HFMs. Improvement in hydrophilicity of HFMs achieved with the surface modification. | ||
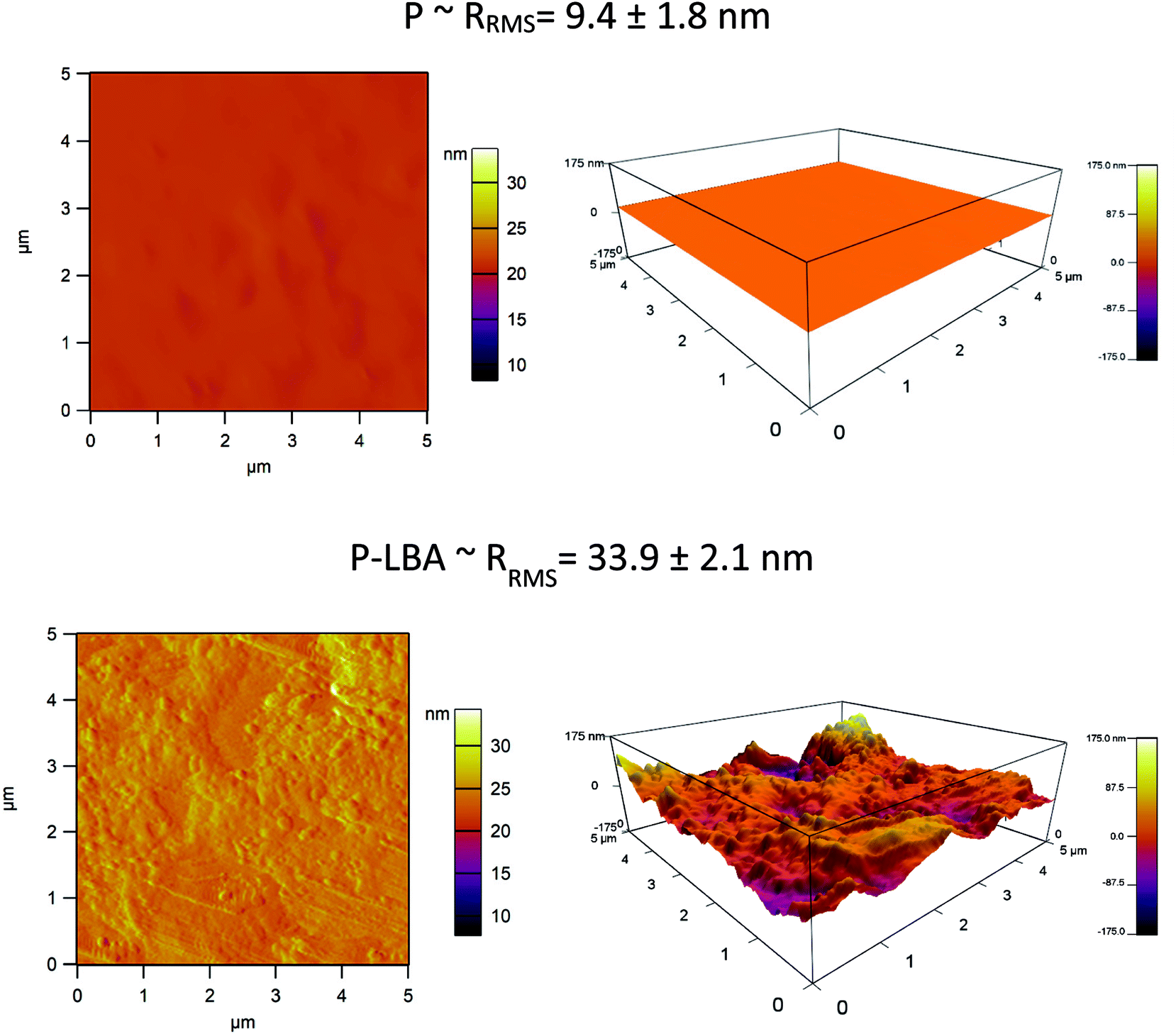 | ||
| Fig. 6 AFM images of P and P-LBA HFMs. The increased roughness on the outer surface of P-LBA HFMs enhanced the attachment of HepG2 cells. | ||
3.2. Biocompatibility evaluation of the developed HFMs
The HFM samples were evaluated for hemocompatibility and cytotoxicity. The related results are presented and discussed here.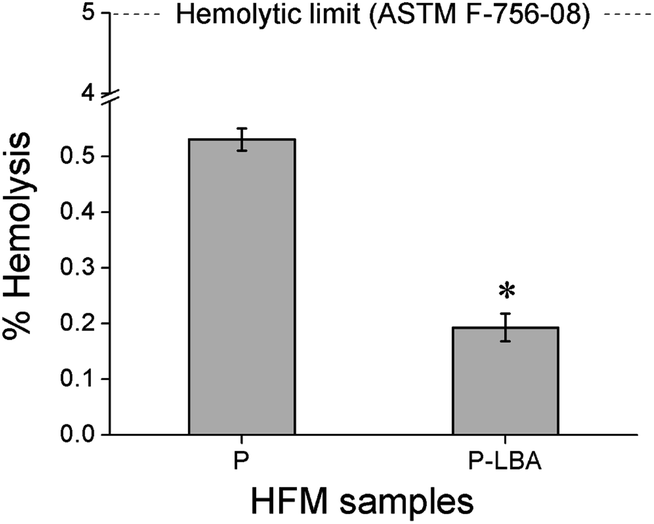 | ||
| Fig. 7 Bar charts showing % hemolysis values of P and P-LBA HFMs. Both fall within the hemolytic limit (5%) indicating the suitability for blood-contacting applications. | ||
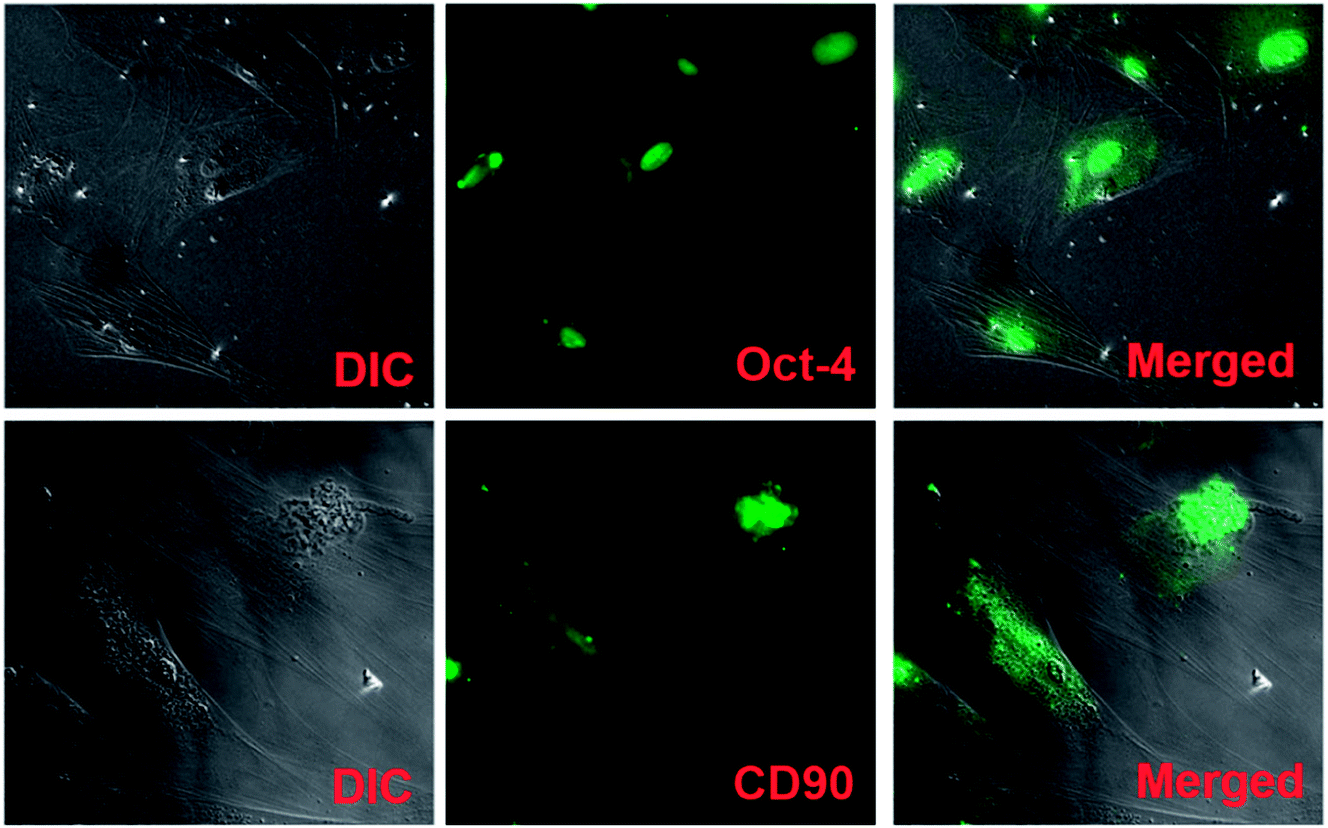 | ||
| Fig. 8 hUMSC characterization: DIC images show the fibroblast-like morphology of hUMSCs. Mesenchymal stem cell markers, Oct-4 and CD90, exhibit immunofluorescence in green color. Merged images show hUMSCs with fluorescence. | ||
3.2.2.1. hUMSCs viability and proliferation on HFMs. MTT and PicoGreen assays were performed at the set time points. MTT assay depicted the enhanced cell viability form day 3 to day 6 (Fig. 9A). PicoGreen double stranded DNA (dsDNA) reagent is an ultra-sensitive fluorescent nucleic acid stain for dsDNA quantification. PicoGreen assay showed the increased DNA amount with P-LBA HFMs, which confirmed the cell proliferation on HFMs (Fig. 9B). DNA content was measured to further increase in day 6 as compared to day 3 for both the HFM samples. The amount of DNA is proportional to cellular growth. Cell proliferation was observed more on HFMs, modified with LBA.
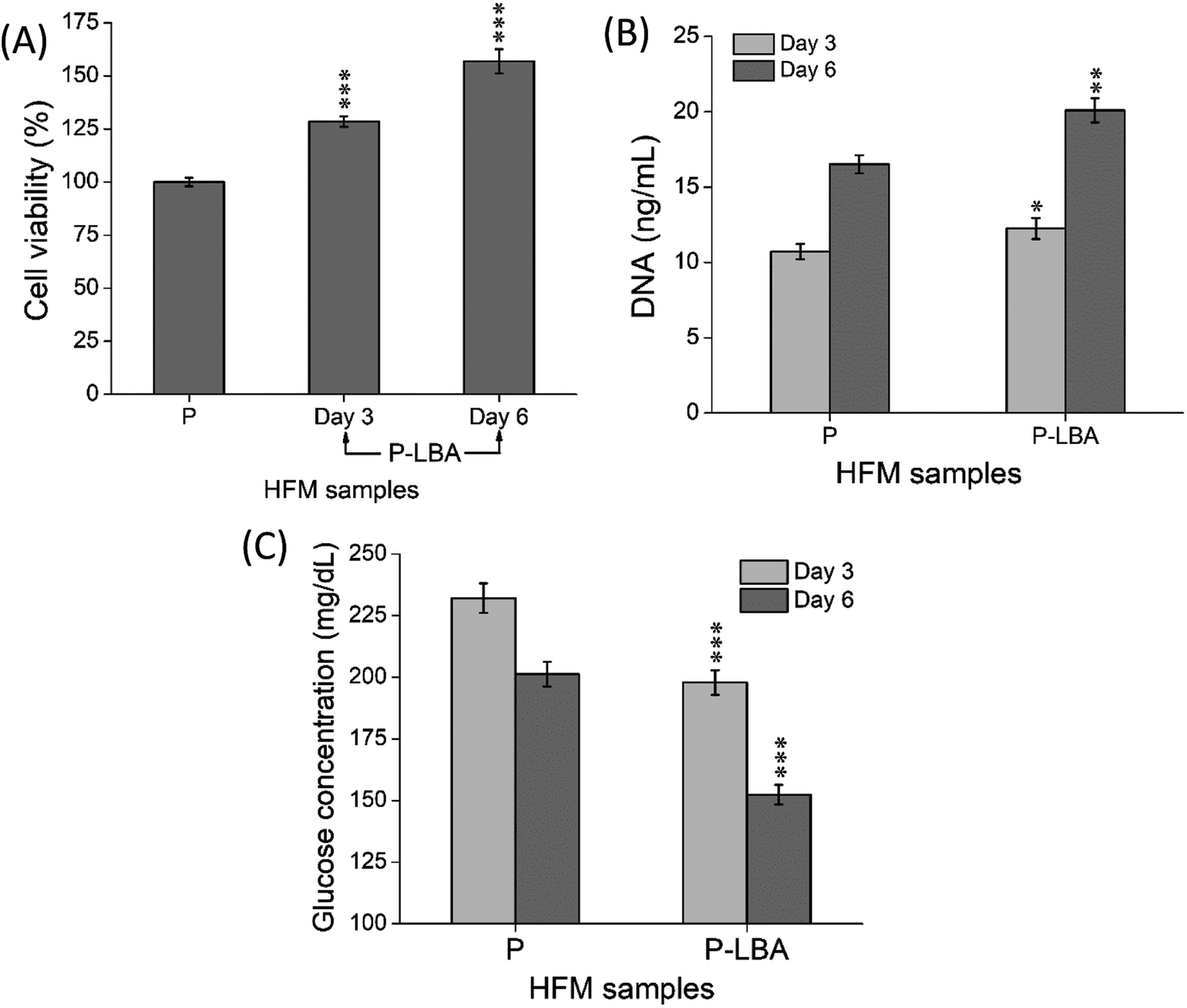 | ||
| Fig. 9 Quantitative evaluation of (A) % viability and (B) DNA content of cells attached on different HFMs. (C) Glucose levels in the spent cell culture media of different HFMs. The results indicated the suitability of P-LBA HFMs for the hUMCSs attachment and proliferation. Values are expressed as mean ± SD (n = 3); *p < 0.05, **p < 0.01, ***p < 0.001 versus P HFMs. | ||
Furthermore, glucose levels were also measured in the culture media of different HFMs, which are shown in Fig. 9C. In general, the lower glucose levels in the spent culture medium are desirable. In this study, the glucose levels associated with P-LBA HFMs were found to be lower than that of the P HFMs on both the set time points. Thus, the results of MTT cell viability, PicoGreen DNA quantification, and glucose consumption assays confirmed the increase in the cell viability and proliferation.
3.3. HepG2 cell culture study
It has been known that apart from energy source, carbohydrates can serve as structural components of the natural product as a key element in various molecular recognition processes, including bacterial and viral infections, cell adhesion, differentiation, development, regulation and signal transduction events.8,34 Carbohydrate recognizing receptors found on the cell surface including hepatocytes, alveolar macrophages, and L1210 mouse leukemia cell are specifically bound to galactose/N-acetyl-b-galactosamine,35 a-mannose36 and a-fucose,37 respectively. ASGPR recognizes galactose as a specific ligand, which was discovered and characterized in mammalian liver.38 In this part of study, HepG2 cell adhesion, proliferation and morphology study were performed with the help of confocal microscopy. The results of the cell function studies were also presented and discussed.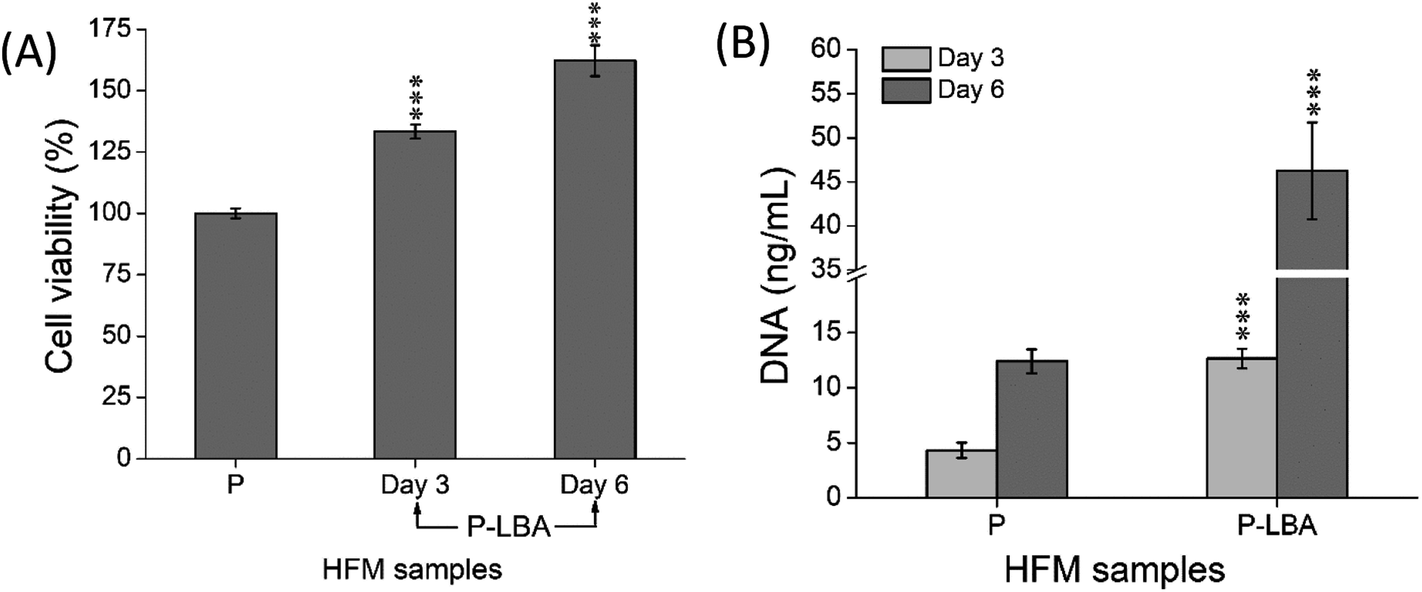 | ||
| Fig. 10 Quantitative evaluation of (A) % viability and (B) DNA content of HepG2 cells attached on different HFMs. The results indicated the suitability of P-LBA HFMs for the cell attachment and proliferation. Values are expressed as mean ± SD (n = 3); ***p < 0.001 versus P HFMs. | ||
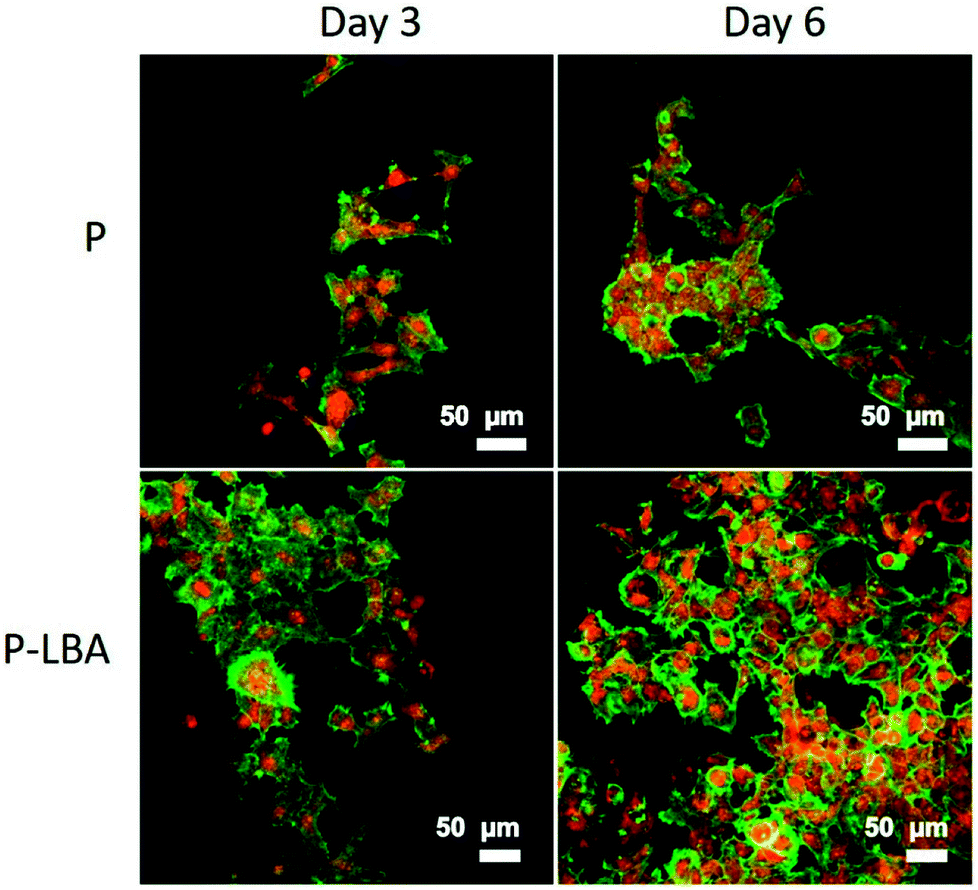 | ||
| Fig. 11 Confocal micrographs showing HepG2 cell growth on P and P-LBA HFMs: enhanced HepG2 cell attachment and proliferation observed on P-LBA HFMs. Nuclei was stained with PI (red), and actins with FITC-ph (green). | ||
Urea synthesis and albumin secretion are the key markers of HepG2 liver cell functional evaluation, which was measured in the spent media, on days 3, and 6. Enhanced urea and albumin was quantified with P-LBA HFMs (Fig. 12A and B). The binding of galactosylated alginate with hepatocytes via the patch of ASGPR improved liver functions of hepatocytes.13 Furthermore, glucose is the primary source of energy in living systems. It is an important factor for cells growth and viability.40 High glucose consumption was measured with P-LBA HFMs (Fig. 12C). LBA-modification promoted more cells to attach on the outer surface of P-LBA HFMs, resulting in higher consumption of glucose as compared to P-HFMs.
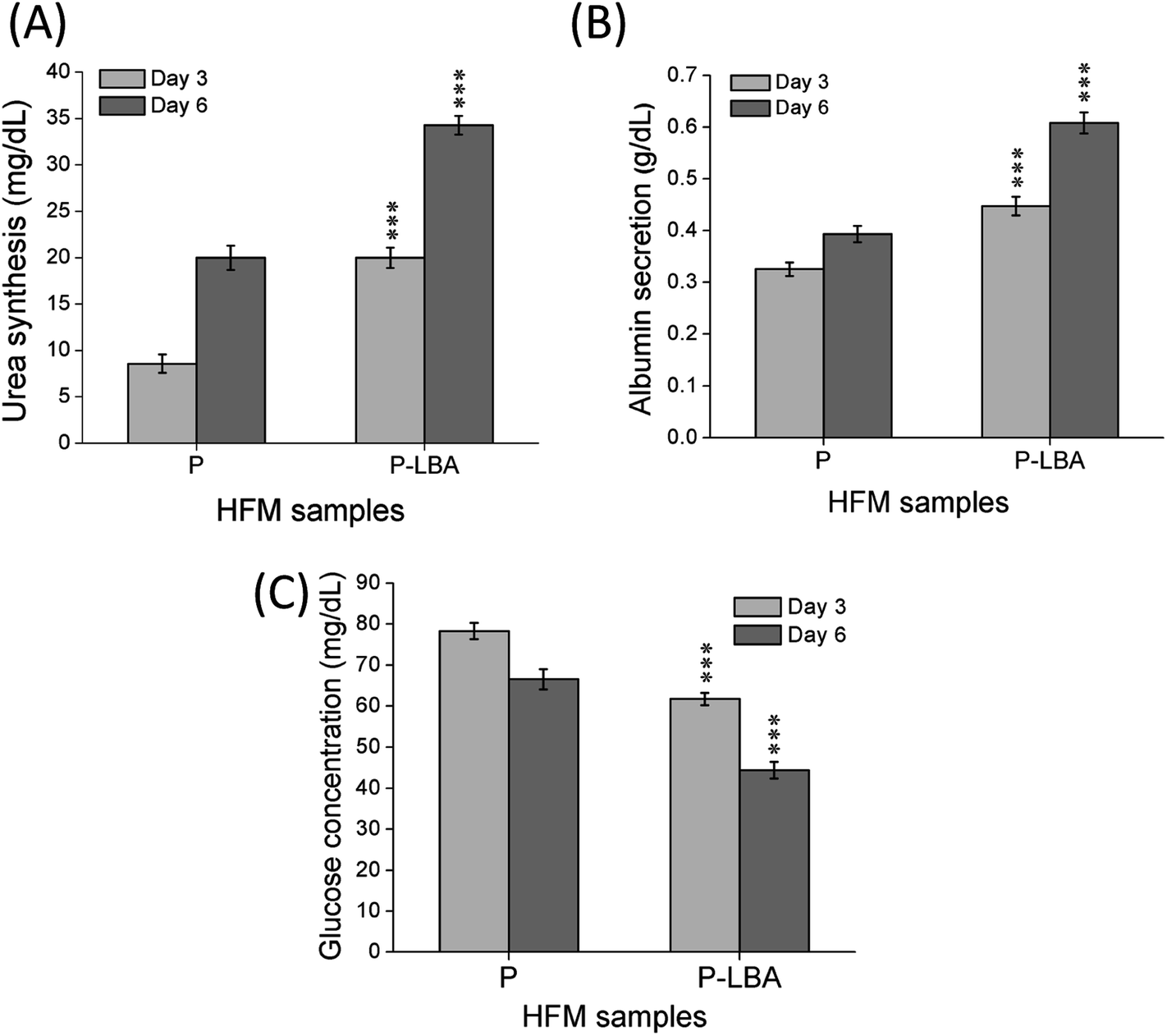 | ||
| Fig. 12 Quantitative determination of (A) urea synthesis, (B) albumin secretion, and (C) glucose consumption in the spent medium of HFM samples (P and P-LBA) at day 3 and day 6. Higher values of urea and albumin, and lower value of glucose on both the day were measured with P-LBA HFMs, which confirmed the high attachment and proliferation of cells on P-LBA HFMs as observed in the microscopy studies. Values are expressed as mean ± SD (n = 3); ***p < 0.001 versus P HFMs. | ||
4. Conclusions
In this study, LBA was covalently coupled with P HFMs. First of all, P HFMs were prepared by phase inversion process with the help of our indigenously developed HFM spinning pilot plant. The outer surface of HFMs was modified with the LBA as a ASGPR ligand. The functional modification was confirmed by ATR-FTIR, EDS elemental mapping, and XPS analysis. The surface modification improved hydrophilicity as exhibited by water contact angle measurements, and also surface roughness, which is desirable for cells attachment and proliferation. P-LBA HFMs were found to be homocompatible with human blood as evaluated by hemolysis. Cytocompatibility was evaluated with human primary mesenchymal stem cells and HepG2 liver cells, which was found to be improved. Enhanced attachment and proliferation of both the cells was observed. The urea synthesis, albumin secretion and glucose consumption analysis showed the active functionality of the attached HepG2 cells on the P-LBA HFMs. Thus, in this study, the P-LBA HFMs, developed with LBA-modification, may open up the possibility to use it for various applications including (1) HFM membrane material for bio-artificial liver development, (2) stem cell expansion, (3) drug testing bioreactor, and (4) mass cell culture.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the help of Dr Rohit Teotia in the hUMSCs culture studies. The authors are thankful to Centre for Research in Nanotechnology and Science (CRNTS), National Centre for Photovoltaic Research and Education (NCPRE), and Central Instrumentation Facility, Indian Institute of Technology Bombay, India for providing the access to various characterization facilities used in this study. The authors acknowledge research fellowship grants from the U.G.C. (S. K. V.), M.H.R.D. (A. M). The authors also acknowledge the Wadhwani Research Centre for Bioengineering, IIT Bombay, India for the research grants.References
- R. Unger, Q. Huang, K. Peters, D. Protzer, D. Paul and C. Kirkpatrick, Biomaterials, 2005, 26, 1877–1884 CrossRef PubMed.
- K.-V. Peinemann and S. P. Nunes, Membrane Technology, Volume 1: Membranes for Life Sciences, John Wiley & Sons, 2011 Search PubMed.
- S. K. Verma, A. Modi and J. Bellare, Biomater. Sci., 2018, 6, 280–291 RSC.
- S. K. Verma, A. Modi, A. K. Singh, R. Teotia and J. Bellare, J. Biomed. Mater. Res., Part B, 2018, 106, 1286–1298 CrossRef PubMed.
- B. Su, S. Sun and C. Zhao, in Progress in Hemodialysis-From Emergent Biotechnology to Clinical Practice, InTech, 2011 Search PubMed.
- J. M. Anderson, Annu. Rev. Mater. Res., 2001, 31, 81–110 CrossRef.
- B. D. Ratner, J. Biomed. Mater. Res., Part A, 1993, 27, 837–850 CrossRef PubMed.
- C. Cho, S. Seo, I. Park, S. Kim, T. Kim, T. Hoshiba, I. Harada and T. Akaike, Biomaterials, 2006, 27, 576–585 CrossRef PubMed.
- R. S. Teotia, D. Kalita, A. K. Singh, S. K. Verma, S. S. Kadam and J. R. Bellare, ACS Biomater. Sci. Eng., 2015, 1, 372–381 CrossRef.
- S. K. Verma, A. Modi, A. K. Singh, R. Teotia, S. Kadam and J. Bellare, Colloids Surf., B, 2018, 164, 358–369 CrossRef PubMed.
- H.-F. Lu, W. S. Lim, J. Wang, Z.-Q. Tang, P.-C. Zhang, K. W. Leong, S. M. Chia, H. Yu and H.-Q. Mao, Biomaterials, 2003, 24, 4893–4903 CrossRef PubMed.
- E. F. Neufeld and G. Ashwell, in The biochemistry of glycoproteins and proteoglycans, Springer, 1980, pp. 241–266 Search PubMed.
- J. Yang, M. Goto, H. Ise, C.-S. Cho and T. Akaike, Biomaterials, 2002, 23, 471–479 CrossRef PubMed.
- H. K. Kleinman, R. J. Klebe and G. R. Martin, J. Cell Biol., 1981, 88, 473–485 CrossRef PubMed.
- J. Lee, M. J. Cuddihy and N. A. Kotov, Tissue Eng., Part B, 2008, 14, 61–86 CrossRef PubMed.
- G. J. Dahe, R. S. Teotia, S. S. Kadam and J. R. Bellare, Biomaterials, 2011, 32, 352–365 CrossRef PubMed.
- A. Higuchi, K. Shirano, M. Harashima, B. O. Yoon, M. Hara, M. Hattori and K. Imamura, Biomaterials, 2002, 23, 2659–2666 CrossRef PubMed.
- S. K. Vashist, Diagnostics, 2012, 2, 23–33 CrossRef PubMed.
- D. Vaikkath, R. Anitha, B. Sumathy and P. D. Nair, Colloids Surf., B, 2016, 141, 112–119 CrossRef PubMed.
- T.-W. Chung, D.-Z. Liu, S.-Y. Wang and S.-S. Wang, Biomaterials, 2003, 24, 4655–4661 CrossRef PubMed.
- A. Modi, S. K. Verma and J. Bellare, J. Colloid Interface Sci., 2017, 504, 86–100 CrossRef PubMed.
- A. Modi, S. K. Verma and J. Bellare, Mater. Sci. Eng., C, 2018, 91, 524–540 CrossRef PubMed.
- A. Modi, S. K. Verma and J. Bellare, Colloids Surf., B, 2018, 167, 457–467 CrossRef PubMed.
- O. Lindvall, Z. Kokaia and A. Martinez-Serrano, Nat. Med., 2004, 10, S42 CrossRef PubMed.
- L. T. Lock and E. S. Tzanakakis, Tissue Eng., 2007, 13, 1399–1412 CrossRef PubMed.
- R. S. Teotia, S. Kadam, A. K. Singh, S. K. Verma, A. Bahulekar, S. Kanetkar and J. Bellare, Mater. Sci. Eng., C, 2017, 77, 857–866 CrossRef PubMed.
- B. Lee, Y. Baek, M. Lee, D. H. Jeong, H. H. Lee, J. Yoon and Y. H. Kim, Nat. Commun., 2015, 6, 7109 CrossRef PubMed.
- R. Lou, W. Yu, Y. Song, Y. Ren, H. Zheng, X. Guo, Y. Lin, G. Pan, X. Wang and X. Ma, Carbohydr. Polym., 2017, 155, 456–465 CrossRef PubMed.
- A. Modi, S. K. Verma and J. Bellare, J. Colloid Interface Sci., 2018, 514, 750–759 CrossRef PubMed.
- R. Teotia, S. K. Verma, D. Kalita, A. K. Singh, G. Dahe and J. Bellare, J. Mater. Sci., 2017, 52, 12513–12523 CrossRef.
- M. Amirilargani, A. Sabetghadam and T. Mohammadi, Polym. Adv. Technol., 2012, 23, 398–407 CrossRef.
- C. H. Kim, M. S. Khil, H. Y. Kim, H. U. Lee and K. Y. Jahng, J. Biomed. Mater. Res., Part B, 2006, 78, 283–290 CrossRef PubMed.
- U. Nekanti, L. Mohanty, P. Venugopal, S. Balasubramanian, S. Totey and M. Ta, Stem Cell Res., 2010, 5, 244–254 CrossRef PubMed.
- A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef PubMed.
- M. Sarkar, J. Liao, E. A. Kabat, T. Tanabe and G. Ashwell, J. Biol. Chem., 1979, 254, 3170–3174 Search PubMed.
- P. Stahl, P. H. Schlesinger, E. Sigardson, J. S. Rodman and Y. Lee, Cell, 1980, 19, 207–215 CrossRef PubMed.
- K. Y. Chen, R. H. Kramer and E. Canellakis, Biochim. Biophys. Acta, Biomembr., 1978, 507, 107–118 CrossRef.
- G. Ashwell and A. G. Morell, Advances in Enzymology and Related Areas of Molecular Biology, 2006, vol. 41, pp. 99–128 Search PubMed.
- S. Zhang, T. Liu, L. Chen, M. Ren, B. Zhang, Z. Wang and Y. Wang, J. Mater. Sci.: Mater. Med., 2012, 23, 2001–2011 CrossRef PubMed.
- B. Deorosan and E. A. Nauman, Stem Cells Int., 2011, 2011 DOI:10.4061/2011/429187 , 12 pages.
| This journal is © The Royal Society of Chemistry 2018 |