
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: SNHG3 promotes proliferation and invasion by regulating the miR-101/ZEB1 axis in breast cancer
Liang Chang†
 *a,
Zhuang Hu†a,
Zhenyu Zhoua and
Hui Zhangb
*a,
Zhuang Hu†a,
Zhenyu Zhoua and
Hui Zhangb
aDepartment of Thyroid Breast Surgery, Huaihe Hospital of Henan University, No. 115, Ximen Street, Gulou District, Kaifeng, 475000, China. E-mail: changliang0221@sina.com; Tel: +86-0371-23906894
bDepartment of Gastroenterology, Huaihe Hospital of Henan University, Kaifeng, 475000, China
First published on 24th April 2018
Abstract
Background: Dysregulated lncRNA expression contributes to the pathogenesis of human tumors via the lncRNAs functioning as oncogenes or tumor suppressors. Small nucleolar RNA host gene 3 (SNHG3) was demonstrated to be upregulated in breast cancer cells. However, the detailed roles and molecular mechanism of SNHG3 in breast cancer are largely unknown. Methods: The expression of SNHG3, miR-101, and zinc finger E-box-binding protein 1 (ZEB1) in breast cancer tissues and cells was detected using qRT-PCR. The effects of SNHG3 on cell proliferation and invasion were evaluated using MTT, EdU, and cell invasion assays. The protein levels of Ki-67, proliferating cell nuclear antigen (PCNA), matrix metalloproteinase MMP-2, and MMP-9 were analyzed using western blot analysis. A luciferase reporter assay and RNA immunoprecipitation (RIP) were performed to explore the interaction between SNHG3, ZEB1 and miR-101. A subcellular fractionation assay was used to detect the subcellular location of SNHG3. Xenograft tumor experiments were conducted to verify the role and mechanism of SNHG3 in breast cancer in vivo. Results: SNHG3 expression was upregulated in breast cancer tissues and correlated with poor prognosis. SNHG3 knockdown suppressed breast cancer cell proliferation and invasion, which was further demonstrated by high levels of proliferation marker proteins Ki-67/PCNA and metastasis-related proteins MMP-2/MMP-9. Additionally, SNHG3 was located in the cytoplasm of breast cancer cells. SNHG3 functioned as a molecular sponge for miR-101 in breast cancer cells. miR-101 was downregulated in breast cancer tissues and negatively correlated with SNHG3 expression. Moreover, ZEB1, a target of miR-101, was positively regulated by SNHG3 in breast cancer cells. ZEB1 mRNA expression was upregulated in breast cancer tissues and positively correlated with SNHG3 expression. Mechanistically, SNHG3 knockdown suppressed cell proliferation and invasion by upregulation of miR-101 and downregulation of ZEB1 expression in breast cancer cells in vitro and in vivo. Conclusion: SNHG3 promoted proliferation and invasion by regulating the miR-101/ZEB1 axis in breast cancer.
1. Introduction
Breast cancer continues to be the most frequently diagnosed malignancy and it leads to high rates of cancer-related mortality in females worldwide, with an estimated 25% of new cancer cases diagnosed in 2015 being breast cancer.1,2 There were approximately 234![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 190 new diagnosed cases and 40730 deaths in the United States in 2015.2 Despite the advances in early diagnosis and comprehensive therapy for breast cancer patients in recent years, the overall prognosis of breast cancer is still not satisfactory.3 Recurrence and distant metastasis still remain as the present major obstacles in the treatment of breast cancer.4 Currently, the exploration of the underlying mechanisms involved in the initiation and development of breast cancer is the subject of extensive research.
190 new diagnosed cases and 40730 deaths in the United States in 2015.2 Despite the advances in early diagnosis and comprehensive therapy for breast cancer patients in recent years, the overall prognosis of breast cancer is still not satisfactory.3 Recurrence and distant metastasis still remain as the present major obstacles in the treatment of breast cancer.4 Currently, the exploration of the underlying mechanisms involved in the initiation and development of breast cancer is the subject of extensive research.
Recently, mounting evidence has highlighted the crucial roles of noncoding RNAs (ncRNAs), including the well-known microRNA (miRNA) and the recently acknowledged long noncoding RNA (lncRNA), in the pathogenesis and progression of various tumors.5,6 The well-known miRNAs, which are approximately 20–24 nucleotides in length, are important post-transcriptional regulators that directly bind to the 3′-untranslated region (3′-UTR) of target genes.7 Dysregulation of miRNAs has been robustly implicated in human cancer tumorigenesis and progression.8–10 More recently, miR-101 was reported to be downregulated in several types of malignancy and functioned as a tumor suppressor in tumors, including those of breast cancer.11,12
lncRNAs are a class of ncRNAs longer than 200 nucleotides in length with limited or no protein coding capacity and recent studies have demonstrated that dysregulated lncRNA expression contributes to the pathogenesis of human tumors via the lncRNAs functioning as oncogenes or tumor suppressors.13 Due to their involvement in diverse biological processes including development, differentiation and carcinogenesis,14 lncRNAs have received increasing attention. Small nucleolar RNA host gene 3 (SNHG3; GenBank Accession no. AJ006835), located at 1q35.3, is a recently identified lncRNA. Previously, SNHG3 was demonstrated to be upregulated in breast cancer cells.15 However, the detailed roles and the molecular mechanism of SNHG3 in breast cancer are largely unknown. Numerous studies have proposed a competing endogenous RNA (ceRNA) hypothesis that lncRNAs suppress the expression and biological functions of miRNAs by functioning as ceRNAs or molecular sponges for miRNAs, thereby modulating the derepression of miRNA targets.16 For example, SNHG6 was reported to act as an oncogene in gastric cancer cells through regulation of miR-101-3p/ZEB1 at a post-transcriptional level.17 Notably, our bioinformatics analysis revealed that SNHG3 contained a sequence complementary to the seed region in the 3′-UTR of miR-101. Therefore, we hypothesized that SNHG3 could function as a molecular sponge for miR-101 in breast cancer.
In the present study, we investigated the expression and functional roles of SNHG3 in breast cancer cells, as well as the underlying mechanism of SNHG3 involved in the progression of breast cancer in vitro and in vivo.
2. Materials and methods
2.1. Patient samples
The study was approved by the Institutional Review Board of the Huaihe Hospital of Henan University, and informed consent was obtained from all subjects in the study. Our study was approved by the Ethics Committee of the Huaihe Hospital of Henan University in accordance with the Declaration of Helsinki principles. 42 paired breast cancer tissue samples and adjacent normal samples were collected from patients who underwent surgical resection between November 2014 and November 2016 at the Huaihe Hospital of Henan University. All specimens were snap-frozen in liquid nitrogen and stored at −80 °C immediately after resection. None of the patients received any preoperative radiotherapy or chemotherapy. Histological diagnosis of each case was independently confirmed by two pathologists based on WHO classification. The tumor stage was classified according to the American Joint Committee on Cancer (AJCC) tumor-lymph node-metastasis (TNM) classification system.18 In these 42 breast patients, 34 patients had lymph node metastasis (N1: n = 14, N2: n = 11, and N3: n = 9), while the other patients (n = 8) had no lymph node metastasis (N0 patients). Meanwhile, 31 patients had distant metastasis, while 11 patients had no distant metastasis.2.2. Cell culture and transfection
Human breast cancer cell lines (MCF-7, MDA-MB-231, and MDA-MB-453) and the normal breast epithelial cell line MCF-10A were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA) and antibiotics (100 U ml−1 penicillin and 100 μg ml−1 streptomycin sulfate) (Invitrogen, Carlsbad, CA, USA) at 37 °C in a humidified incubator with 5% CO2. Cells at 80% confluence were harvested and used in our experiments.siRNAs against SNHG3 (si-SNHG3#1 and si-SNHG3#2), siRNA scrambled control (si-con), pcDNA-SNHG3 (SNHG3), pcDNA-zinc finger E-box-binding protein 1 (ZEB1), pcDNA empty vector (Vector), miR-101 mimic (miR-101), miRNA scrambled control (miR-con), miR-101 antagomir (anti-miR-101), and antagomir control (anti-miR-con) were obtained from GenePharma (Shanghai, China). The full length of SNHG3 fragments containing the mutated miR-101 binding sites was synthesized (RiboBio Co., Ltd., Guangzhou, China) and cloned into pcDNA vector (Promega, Madison, WI, USA) to construct pcDNA-SNHG3-MUT (mutant SNHG3). Cells were plated at a density of 1 × 106 cells per ml and transfected with these oligonucleotides or plasmids using Lipofectamine 2000 (Invitrogen). Cells were harvested at 48 h post-transfection for the next experiments.
2.3. Construction of lentivirus
shRNAs targeting the sequence of SNHG3 (sh-SNHG3) or scrambled control (sh-con) were cloned into pFH-L vectors (Shanghai Hollybio, Shanghai, China) containing green fluorescent protein (GFP) reporter. Briefly, lenti-sh-SNHG3 plasmids were transfected into HEK293T cells, along with pCMVΔR8.92 and pVSVG-I helper as packaging vectors (Shanghai Hollybio, Shanghai, China). The supernatant containing the lentivirus expressing sh-SNHG3 (lenti-sh-SNHG3) or sh-con (lenti-sh-con) was harvested via purification and precipitation at 72 h post-transfection. Lentiviruses containing miR-101 antagomirs (lenti-miR-101) or antagomir scrambled control (lenti-miR-con) were purchased from GenePharma.2.4. Quantitative real-time PCR
Total RNA was extracted from the tissue samples and cells using TRIzol reagent (Invitrogen) and the RNA concentration was measured using a NanoDrop 2000/2000c (Thermo Fisher Scientific, Inc.). Total RNA was reversely transcribed into cDNA using an EasyScript First-Strand cDNA Synthesis SuperMix kit (TransGen Biotech, Inc., Beijing, China). The expression of miR-101 was examined using a TaqMan MicroRNA Assay kit (Thermo Fisher Scientific, Inc.) and SNHG3 and ZEB1 mRNA expression was detected using a SYBR Premix Ex Taq kit (Thermo Fisher Scientific, Inc.) on a Roche Lightcycler 480 real-time PCR system (Roche Diagnostics, Basel, Switzerland), respectively. GAPDH and U6 small nuclear RNA (snRNA) were used as the internal references for lncRNA, mRNA and miRNA, respectively. The relative fold change of gene expressions was calculated using the 2−ΔΔCt method.2.5. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Following 48 h of transfection, MCF-7 and MDA-MB-231 cells were seeded in a 96-well plate at a density of 5000 cells per well and then incubated for 0, 24, 48 or 72 h. Then, 10 μl MTT solution (5 mg ml−1; Beyotime, Haimen, China) was added to each well, followed by incubation for another 4 h. Thereafter, the supernatant was removed and 150 μl dimethyl sulfoxide (DMSO) was added to dissolve the formazan crystals. Absorbance at a wavelength of 570 nm was measured using a Benchmark Plus™ microplate spectrometer (Model 680; Bio-Rad Laboratories, Inc., Hercules, CA, USA).2.6. 5-Ethynyl-2-deoxyuridine (EdU) proliferation assay
Cell proliferation was also evaluated using an EdU proliferation assay (RiboBio, Nanjing, China). Briefly, MCF-7 and MDA-MB-231 cells at 48 h post-transfection (5000 cells per well) were seeded into 96-well plates. Following incubation for 24 h, the cells were then incubated with 50 μM EdU for 2 h, fixed with 4% paraformaldehyde for 30 min, and permeabilized with 0.5% Triton X-100 for 10 min. After washing with PBS three times, the cells were incubated with 100 μl 1× Alexa-Fluor 594 reaction cocktail for 30 min at room temperature and then stained with 100 μl Hoechst 33342 for 30 min. Images were visualized using a fluorescence microscope (Nikon, Tokyo, Japan) and the numbers of proliferative cells (EdU-positive) in three random fields of view per slide were examined under a confocal laser scanning microscope (FV300, Olympus, Tokyo, Japan).2.7. Cell invasion assay
Transwell chambers (8.0 μm pore size, Corning Inc., Corning, NY, USA) coated with Matrigel (BD Biosciences, San Jose, CA, USA) were used to assess cell invasion potential according to the manufacturer’s instructions. Briefly, 3 × 105 transfected MCF-7 and MDA-MB-231 cells were seeded into the upper chamber with 200 μl serum-free medium, while 500 μl complete medium supplemented with 10% FBS as a chemoattractant was added to the lower chamber. Following incubation for 24 h at 37 °C, the cells in the upper chamber were removed. The cells that invaded the lower chamber were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet solution for 30 min, and counted in five randomly selected fields using a microscope (Olympus).2.8. Subcellular fractionation
To determine the cellular localization of SNHG3, the nuclear fraction was isolated from cytoplasm using a Nuclear/Cytosol Fractionation Kit (Biovision, San Francisco Bay, CA, USA). Then the expression patterns of SNHG3, U6 and GAPDH in nuclear and cytoplasm fractions were respectively determined using a qRT-PCR assay. U6 and GAPDH were used as fractionation indicators.2.9. Luciferase reporter assay
SNHG3 fragments containing the predicted wild-type (WT) or mutated (MUT) miR-101 binding sites were amplified by PCR and cloned downstream of firefly luciferase gene in pGL3 plasmids (Promega), namely SNHG3-WT and SNHG3-MUT. Similarly, the 3′-UTR sequence of ZEB1 containing the predicted miR-101 binding sites was synthesized and inserted into pGL3 plasmids, named ZEB1 3′-UTR. For the luciferase activity assay, MCF-7 cells were seeded in 24-well plates and cotransfected with 200 ng SNHG3-WT or SNHG3-MUT, 50 ng of renilla luciferase reporter vector (pRL-TK; Promega) and miR-101, anti-miR-101, or matched controls, or cotransfected with ZEB1 3′-UTR and miR-con, miR-101, miR-101 + Vector, miR-101 + ZEB1, anti-miR-con, anti-miR-101, anti-miR-101 + si-con, or anti-miR-101 + si-SNHG3#1 using Lipofectamine 2000 (Invitrogen). Cells were collected at 48 h post-transfection and the luciferase activity was determined using the Dual-Luciferase Reporter Assay system (Promega). The firefly luciferase activity was normalized against the renilla luciferase activity.2.10. RNA immunoprecipitation (RIP)
According to the manufacturer’s protocol, the RIP assay was performed using a Magna RIP RNA-binding protein immunoprecipitation kit (Millipore, Billerica, MA, USA). MCF-7 cells at 80% confluence were scraped off and lysed in the RIP lysis buffer. Afterwards, 100 μl of whole cell extract was incubated with RIP buffer containing magnetic beads coated with human anti-argonaute2 (Ago2) antibody (Millipore) and immunoglobulin G (IgG) (Millipore) as a negative control. Samples were digested with proteinase K and then immunoprecipitated RNA was isolated. The coprecipitated RNAs were subjected to qRT-PCR for the detection of the enrichment of SNHG3 and miR-101.2.11. Western blot
Total proteins were extracted from the cultured cells using RIPA buffer (Sigma) in the presence of PMSF (Beyotime) on ice. Equal amounts of protein samples were subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). The membranes were blocked in 5% nonfat milk dissolved in Tris-buffered saline (TBS) containing 0.1% Tween-20 for 2 h at room temperature and then incubated at 4 °C overnight with diluted primary antibodies against matrix metalloproteinase (MMP)-2 (1:500; Abcam, Cambridge, MA, USA), MMP-9 (1:1000; Abcam), Ki-67 (1:1000; Abcam), proliferating cell nuclear antigen (PCNA) (1:2000; Abcam), ZEB1 (1:1000; Abcam) or GAPDH (1:1000; Abcam), followed by incubation with horseradish peroxidase-conjugated secondary antibody (1:1000; Abcam) for 2 h at room temperature. GAPDH was used as an internal control. Immunoreactive bands were developed using an enhanced chemiluminescence kit (Amersham Pharmacia, Piscataway, NJ, USA).
2.12. Xenograft tumor model
All animal procedures were approved by the Ethics Committee of the Huaihe Hospital of Henan University and performed following the guidelines for the Care and Use of Laboratory Animals. Female BALB/c nude mice aged 4 weeks were purchased from the Shanghai Experimental Animal Center of the Chinese Academy of Sciences (Shanghai, China). MCF-7 cells were seeded in a 24-well plate and infected with lenti-sh-SNHG3 or lenti-sh-con at a multiplicity of infection (MOI) of 20. After infection, lenti-anti-miR-con or lenti-anti-miR-101 was transduced in lenti-sh-SNHG3 or lenti-sh-con stably transfected MCF-7 cells. Stably transfected MCF-7 cells were collected and suspended in 100 μl of PBS at a concentration of 1 × 107 cells per ml and then subcutaneously implanted into the posterior flank of the same nude BALB/c mice (n = 4). The tumor volumes were measured using a Vernier scale every 7 days for 42 days and calculated using the formula: volume = (length × width2)/2. The mice were killed 42 days after injection and the tumors were dissected and weighed. Total RNA and protein were extracted for the detection of specific RNA and protein levels.2.13. Statistical analysis
All results were presented as mean ± standard deviation (SD) from three independent experiments. All statistical analyses were performed using GraphPad Prism V5.0 software. The significant differences between two or more groups were analyzed using Student’s t-test or one-way analysis of variance (ANOVA). *P < 0.05, **P < 0.01, and ***P < 0.001 were considered to be statistically significant.3. Results
3.1. SNHG3 was aberrantly upregulated in breast cancer patients and correlated with poor prognosis
To determine the functional role of SNHG3 in breast cancer, SNHG3 expression in 42 paired breast cancer tissue samples and the adjacent normal samples was detected using qRT-PCR. The results showed that SNHG3 expression was significantly higher in breast cancer tissues than in matched normal tissues (Fig. 1A). Moreover, we found that SNHG3 expression showed a strong correlation with breast cancer lymph node metastasis (Fig. 1B) and distant metastasis (Fig. 1C). The overall survival, calculated using Kaplan–Meier curves and a log-rank test, showed that breast cancer patients with high expression of SNHG3 had poor prognosis compared to those with low SNHG3 levels (Fig. 1D). Collectively, these results demonstrated that upregulated SNHG3 was closely correlated with poor prognosis for breast cancer patients.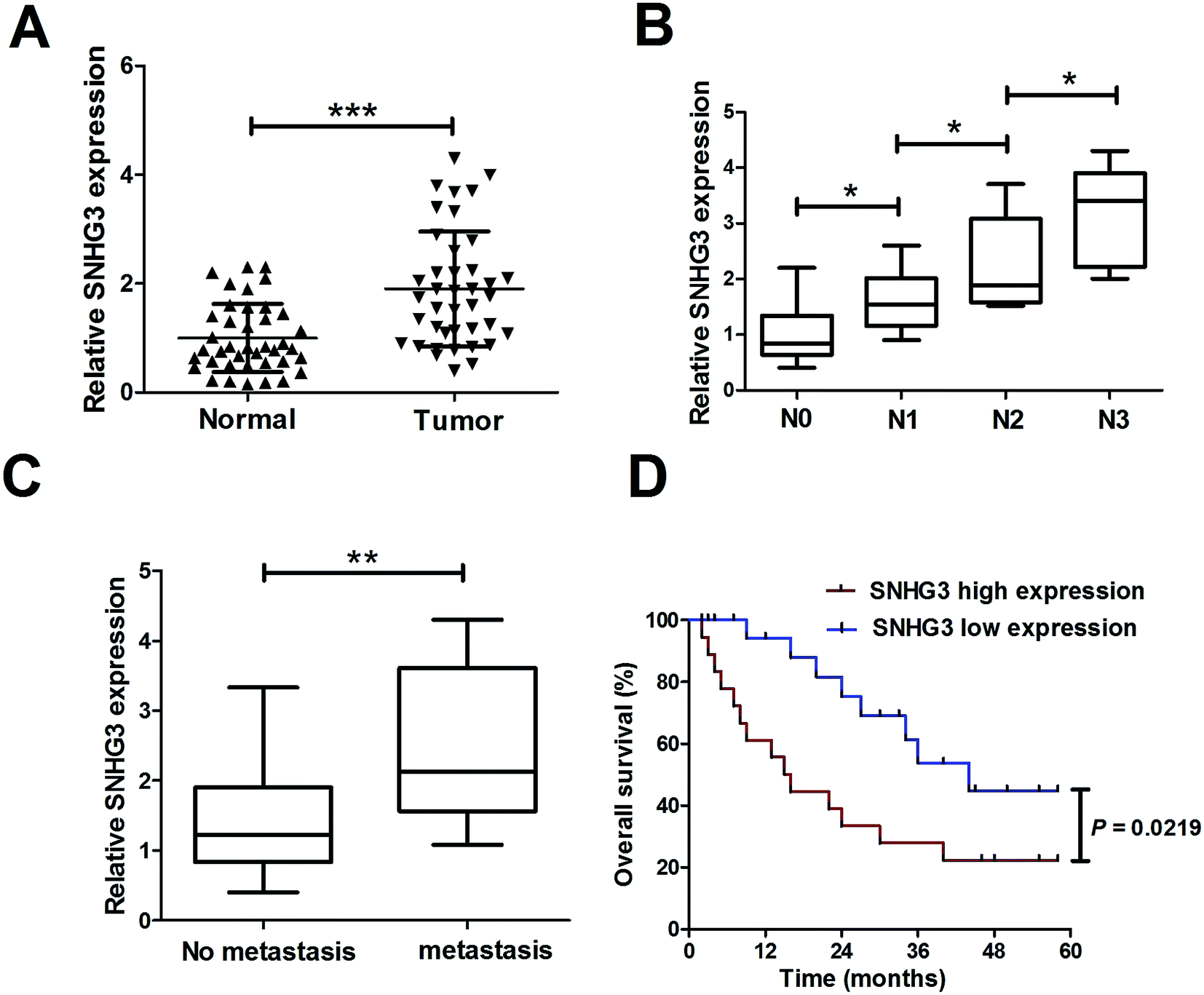 | ||
| Fig. 1 Upregulated SNHG3 was closely correlated with poor prognosis for breast cancer patients. (A) The expression of SNHG3 in 42 paired breast cancer tissues and matched adjacent normal tissues was detected using qRT-PCR. (B) The expression of SNHG3 in different advanced pathological stages N0, N1, N2 and N3. (C) The expression of SNHG3 in 31 breast cancer patients with distant metastasis and 11 breast cancer patients with no distant metastasis. (D) Kaplan–Meier curves and a log-rank test of overall survival for all breast cancer patients. *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
3.2. SNHG3 silencing suppressed cell proliferation and invasion in breast cancer cells
The expression of SNHG3 in breast cancer cells was further evaluated and the results demonstrated that SNHG3 expression was robustly upregulated in different metastatic potential breast cancer cells (MCF-7, MDA-MB-453 and MDA-MB-231) compared with that in the normal breast epithelial cell line MCF-10A (Fig. 2A). Among these cells, the metastatic breast cancer cells with high expression (MDA-MB-231) and the metastatic breast cancer cells with low expression (MCF-7) were selected for further analyses.19 Loss-of-function approaches were performed to address the functional role of SNHG3 in breast cancer progression. MTT and EdU proliferation assays were applied to assess cell proliferation changes in MCF-7 and MDA-MB-231 cells transfected with si-SNHG3#1, si-SNHG3#2 or si-con. As compared with the si-con group, si-SNHG3#1- or si-SNHG3#2-transfected MCF-7 (Fig. 2C and E) and MDA-MB-231 (Fig. 2D and F) cells exhibited an obvious suppression of cell proliferation, as demonstrated by the MTT assay and the EdU proliferation assay. Consistently with this, the protein levels of proliferation marker proteins Ki-67 and PCNA were distinctly hindered by transfection of si-SNHG3#1 or si-SNHG3#2 in MCF-7 (Fig. 2G) and MDA-MB-231 (Fig. 2H) cells. In addition, a cell invasion assay showed decreased cell invasion ability after si-SNHG3#1 or si-SNHG3#2 transfection in MCF-7 (Fig. 2I) and MDA-MB-231 (Fig. 2J) cells compared with si-con-transfected cells. Moreover, SNHG3 knockdown by si-SNHG3#1 or si-SNHG3#2 evidently reduced the levels of metastasis-related proteins including MMP-2 and MMP-9 in MCF-7 (Fig. 2K) and MDA-MB-231 (Fig. 2L) cells. These results implied that SNHG3 silencing suppressed cell proliferation and invasion in breast cancer cells.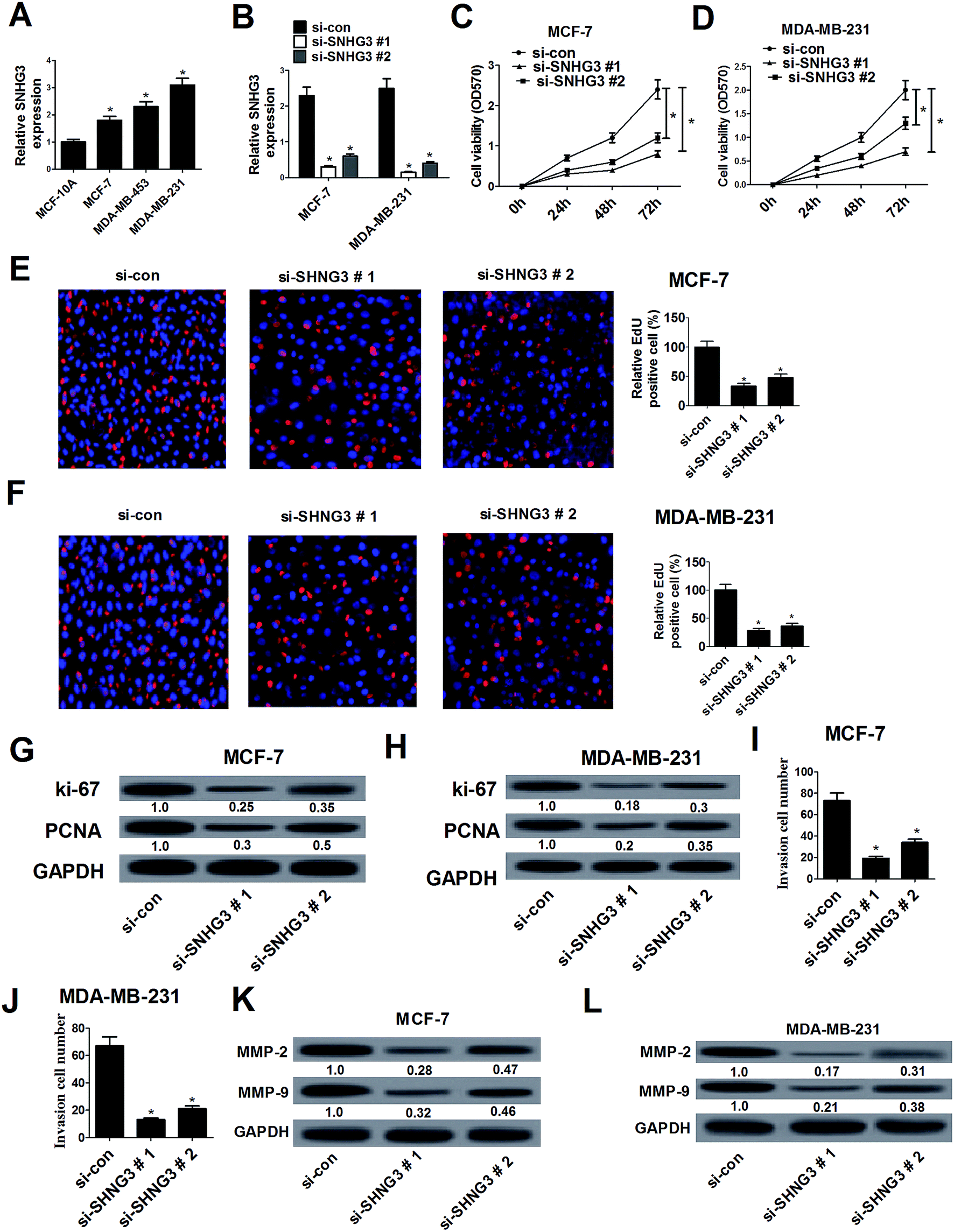 | ||
| Fig. 2 SNHG3 silencing suppressed cell proliferation and invasion in breast cancer cells. (A) The expression level of SNHG3 in breast cancer cells (MCF-7, MDA-MB-453 and MDA-MB-231) and the normal breast epithelial cell line MCF-10A was estimated using qRT-PCR. (B) The expression level of SNHG3 in MCF-7 and MDA-MB-231 cells transfected with si-SNHG3#1, si-SNHG3#2 or si-con was evaluated using qRT-PCR. The MTT assay (C and D) and EdU proliferation assay (E and F) were performed to detect cell proliferation at 24 h, 48 h, and 72 h in MCF-7 and MDA-MB-231 cells transfected with si-SNHG3#1, si-SNHG3#2 or si-con. Western blot analysis was conducted to evaluate the protein levels of Ki-67 and PCNA in MCF-7 (G) and MDA-MB-231 (H) cells transfected with si-SNHG3#1, si-SNHG3#2 or si-con. Cell invasion ability was examined using the cell invasion assay in MCF-7 (I) and MDA-MB-231 (J) cells transfected with si-SNHG3#1, si-SNHG3#2 or si-con. The protein levels of MMP-2 and MMP-9 in MCF-7 (K) and MDA-MB-231 (L) cells transfected with si-SNHG3#1, si-SNHG3#2 or si-con were determined using western blot analysis. *P < 0.05. | ||
3.3. SNHG3 functioned as a molecular sponge for miR-101 in breast cancer cells
As mentioned before, recent studies have documented that lncRNAs could function as ceRNAs or molecular sponges to regulate miRNA expression and function.20 To delineate the molecular mechanism by which SNHG3 exerts its oncogenic role in breast cancer cells, several bioinformatics tools (http://www.microRNA.org and miRBase) were applied to analyze the potential targets of SNHG3. Bioinformatics tool analysis revealed that SNHG3 contained the complementary binding sites for the seed region of miR-101, as shown in Fig. 3A. Additionally, SNHG3 was analyzed for nucleic–cytoplasmic compartmentalization by subcellular fractionation of MCF-7 cells and we found that SNHG3 expression was specifically enriched in the cytoplasm versus the nucleus, which indicated that SNHG3 was mainly localized in the cytoplasm and exerted its function at the post-transcriptional level. The luciferase reporter assay showed that miR-101 overexpression remarkably reduced the luciferase activity of SNHG3-WT, but not SNHG3-MUT, in MCF-7 cells (Fig. 3C). By contrast, inhibition of miR-101 apparently increased the luciferase activity of SNHG3-WT in MCF-7 cells, but had no obvious effect on SNHG3-MUT (Fig. 3D). In addition, it is well characterized that miRNAs exert their gene silencing function via binding to Ago2, a major component of the RNA-induced silencing complex (RISC). To confirm whether SNHG3 physically interacts with the RISC complex and hence regulates the function of miRNAs, an anti-Ago2 RIP assay was conducted in MCF-7 cell extracts. The results showed that SNHG3 and miR-101 were preferentially enriched in Ago2-coating beads compared with in the IgG control group (Fig. 3E). Furthermore, qRT-PCR showed that SNHG3 expression was significantly increased in either SNHG3 or mutant SNHG3-transfected MCF-7 cells (Fig. 3F). However, miR-101 expression was markedly diminished by transfection of SNHG3, but showed no obvious change in the mutant SNHG3-introduced group in MCF-7 cells (Fig. 3G). Moreover, si-SNHG3#1 transfection dramatically enhanced miR-101 expression in MCF-7 cells (Fig. 3H). We found that miR-101 expression was abnormally downregulated in breast cancer tissues with respect to that in the corresponding normal tissues (Fig. 3I), and negatively correlated with the expression of SNHG3 in breast cancer cells (Fig. 3J). Moreover, miR-101 expression was significantly correlated with breast cancer lymph node metastasis (Fig. 3K) and distant metastasis (Fig. 3L). Furthermore, we found that breast cancer patients with low expression of miR-101 showed poor prognosis compared to those with high expression of miR-101 (Fig. 3M). Collectively, these results demonstrated that SNHG3 repressed miR-101 expression in breast cancer cells by functioning as a ceRNA.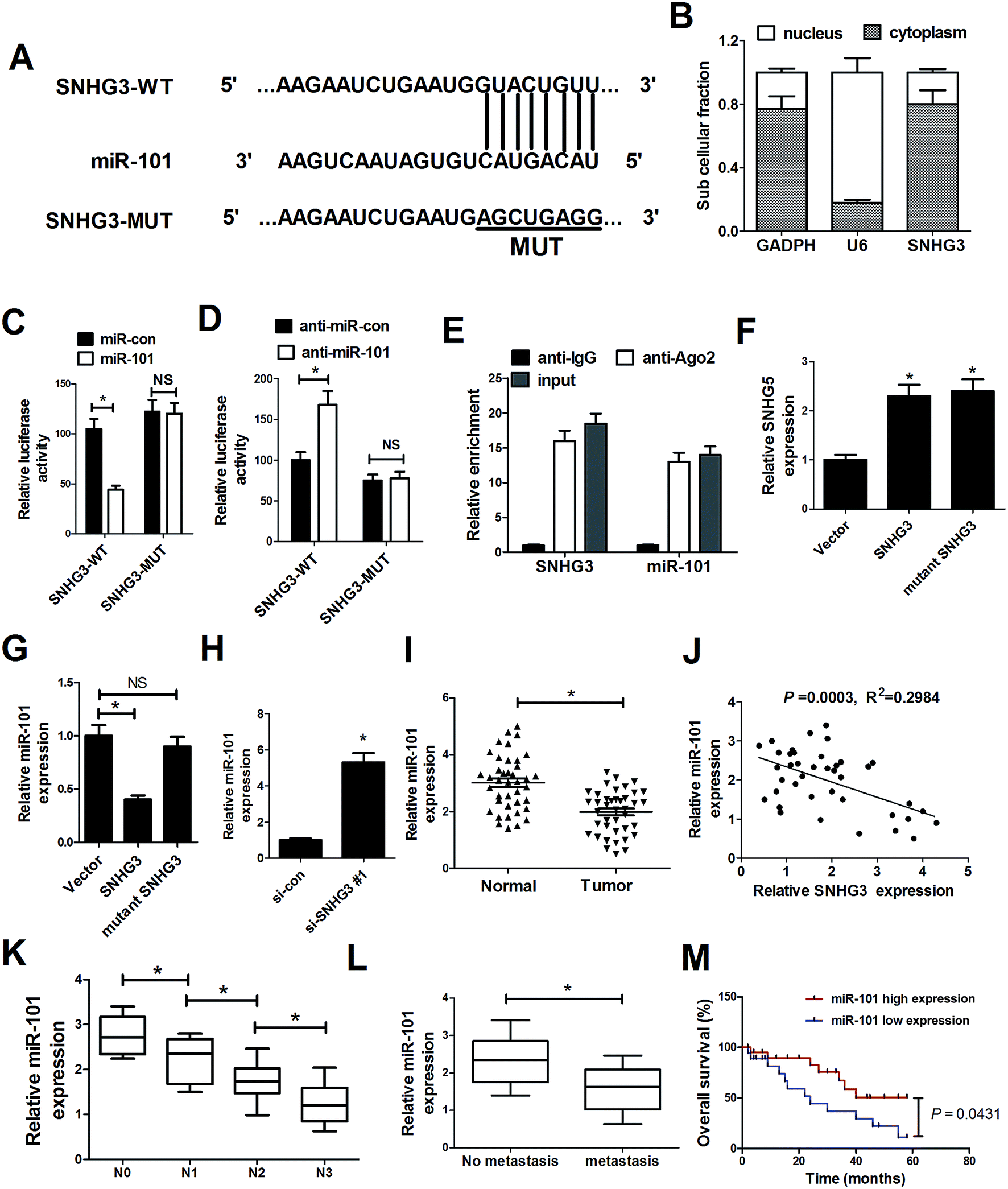 | ||
| Fig. 3 SNHG3 functioned as a molecular sponge for miR-101 in breast cancer cells. (A) The luciferase reporter plasmids containing the predicted wild-type or mutated miR-101 in SNHG3. (B) Subcellular fractionation for SNHG3 in MCF-7 cells, where GAPDH and U6 were used as the controls. (C and D) A luciferase reporter assay was conducted to measure the luciferase activity in MCF-7 cells cotransfected with SNHG3-WT or SNHG3-MUT and miR-101, anti-miR-101, or matched controls. (E) Association between SNHG3 and miR-101 with Ago2 antibody. An anti-Ago2 RIP assay was performed in MCF-7 cell extract and the expressions of SNHG3 and miR-101 were detected using qRT-PCR. qRT-PCR was performed to detect the expressions of SNHG3 (F) and miR-101 (G and H) in MCF-7 cells transfected with SNHG3, mutant SNHG3, Vector, si-SNHG3#1, or si-con. (I) miR-101 expression in 42 paired breast cancer tissues and matched normal tissues. (J) The correlation between SNHG3 and miR-101 expression in breast cancer tissues. (K) The expression of miR-101 in different advanced pathological stages N0, N1, N2 and N3. (L) The expression of miR-101 in 31 breast cancer patients with distant metastasis and 11 breast cancer patients with no distant metastasis. (M) Kaplan–Meier curves and a log-rank test of overall survival for all breast cancer patients. *P < 0.05. | ||
3.4. ZEB1, a target of miR-101, was positively regulated by SNHG3 in breast cancer cells
In a previous study, ZEB1, a well-known transcriptional regulator that induces epithelial–mesenchymal transition (EMT), was identified as a target of miR-101 in breast cancer.21 Therefore, we hypothesized whether the SNHG3/miR-101 axis could regulate the progression of breast cancer via ZEB1. qRT-PCR results demonstrated that ectopic expression of miR-101 and SNHG3 silencing significantly reduced the protein level of ZEB1 in breast cancer cells, while suppression of miR-101 and forced expression of SNHG3 dramatically enhanced the ZEB1 level in MCF-7 cells. The subsequent luciferase reporter assay showed that miR-101 mimics drastically decreased the luciferase activity of ZEB1 3′-UTR reporter plasmids compared to that with the miR-con group, while cotransfection with SNHG3 and miR-101 evidently restored the luciferase activity of ZEB1 3′-UTR reporter plasmids (Fig. 4C). In contrast, cotransfection of MCF-7 cells with anti-miR-101 and ZEB1 3′-UTR vector produced a higher level of luciferase activity, which was conspicuously reversed by additional transfection with si-SNHG3#1 (Fig. 4D). Additionally, we found that ZEB1 mRNA expression was markedly upregulated in breast cancer tissues with respect to the adjacent normal tissues (Fig. 4E). Moreover, a positive correlation between SNHG3 and ZEB1 mRNA expression in breast cancer was observed (Fig. 4F). In addition, ZEB1 expression was dramatically correlated with breast cancer lymph node metastasis (Fig. 4G) and distant metastasis (Fig. 4H). Furthermore, we found that breast cancer patients with low expression of ZEB1 showed poor prognosis compared to those with high expression of ZEB1 (Fig. 4I). Collectively, these findings suggested that ZEB1 was a target of miR-101 and was positively regulated by SNHG3.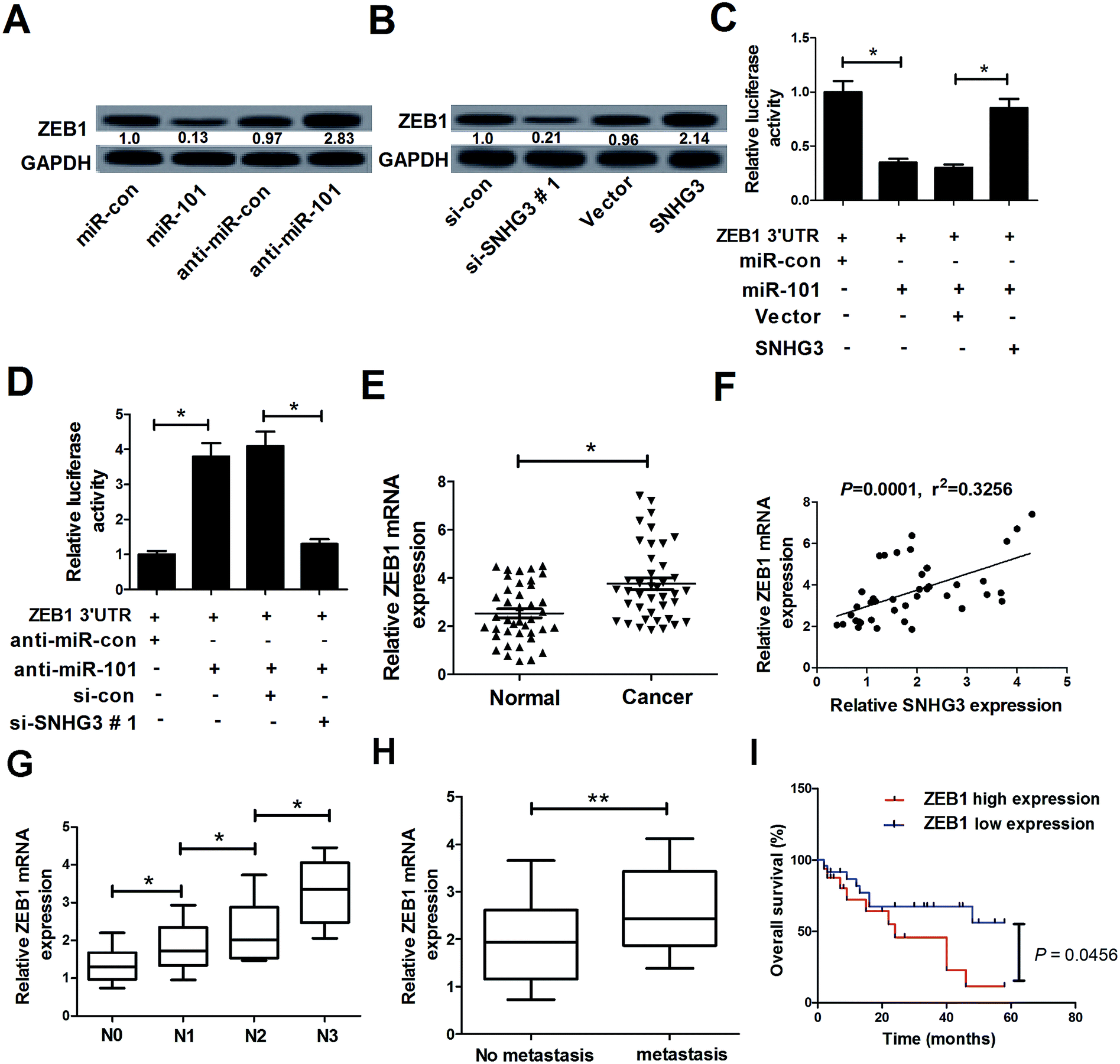 | ||
| Fig. 4 The interaction between SNHG3, ZEB1 and miR-101 in breast cancer cells. (A) Western blot analysis of the ZEB1 level in MCF-7 cells transfected with miR-101, anti-miR-101, or matched controls. (B) Western blot analysis of the ZEB1 level in MCF-7 cells introduced to si-SNHG3#1, SNHG3, or the respective controls. (C) MCF-7 cells were cotransfected with ZEB1 3′-UTR and miR-101, miR-con, miR-101 + Vector, or miR-101 + SNHG3. A luciferase reporter assay was performed to detect the luciferase activity at 48 h post-transfection. (D) MCF-7 cells were cotransfected with ZEB1 3′-UTR and anti-miR-101, anti-miR-con, anti-miR-101 + si-con, or anti-miR-101 + si-SNHG3. A luciferase reporter assay was performed to measure the luciferase activity at 48 h post-transfection. (E) ZEB1 mRNA expression in 42 paired breast cancer tissues and adjacent normal tissues. (F) The correlation between SNHG3 and ZEB1 mRNA expression in breast cancer tissues. (G) The expression of ZEB1 in different advanced pathological stages N0, N1, N2 and N3. (H) The expression of ZEB1 in 31 breast cancer patients with distant metastasis and 11 breast cancer patients with no distant metastasis. (I) Kaplan–Meier curves and a log-rank test of overall survival for all breast cancer patients. *P < 0.05, **P < 0.01. | ||
3.5. SNHG3 knockdown suppressed cell proliferation and invasion by upregulation of miR-101 and downregulation of ZEB1 expression in breast cancer cells in vitro
To further assess whether SNHG3 affected breast cancer progression by regulating the expression of miR-101 and ZEB1, MCF-7 cells were cotransfected with si-SNHG3#1 or si-con, along with anti-miR-101, anti-miR-con, ZEB1, or Vector. Western blot analysis demonstrated that downregulation of miR-101 (Fig. 5A) and overexpression of ZEB1 (Fig. 5B) significantly reversed SNHG3 silencing-mediated repression of the ZEB1 protein level in MCF-7 cells. The MTT assay revealed that inhibition of miR-101 (Fig. 5C) and forced expression of ZEB1 (Fig. 5D) distinctly restored the inhibitory effect on cell proliferation mediated by SNHG3 knockdown in MCF-7 cells. Moreover, the protein levels of Ki-67 and PCNA were dramatically augmented following transfection of anti-miR-101 or ZEB1 in MCF-7 cell, and this increase could be partially abated by SNHG3 knockdown (Fig. 5E). As illustrated by the cell invasion assay, miR-101 antagomirs and ectopic expression of ZEB1 prominently enhanced the cell invasion capability in MCF-7 cells, which was strikingly undermined by SNHG3 knockdown (Fig. 5F). Likewise, western blot analysis revealed that SNHG3 knockdown obviously attenuated the increase of the protein levels of MMP-2 and MMP-9 in MCF-7 cells caused by anti-miR-101 and ZEB1 overexpression (Fig. 5G). All of these data indicated that SNHG3 knockdown suppressed cell proliferation and invasion by upregulation of miR-101 and downregulation of ZEB1 expression in breast cancer cells in vitro.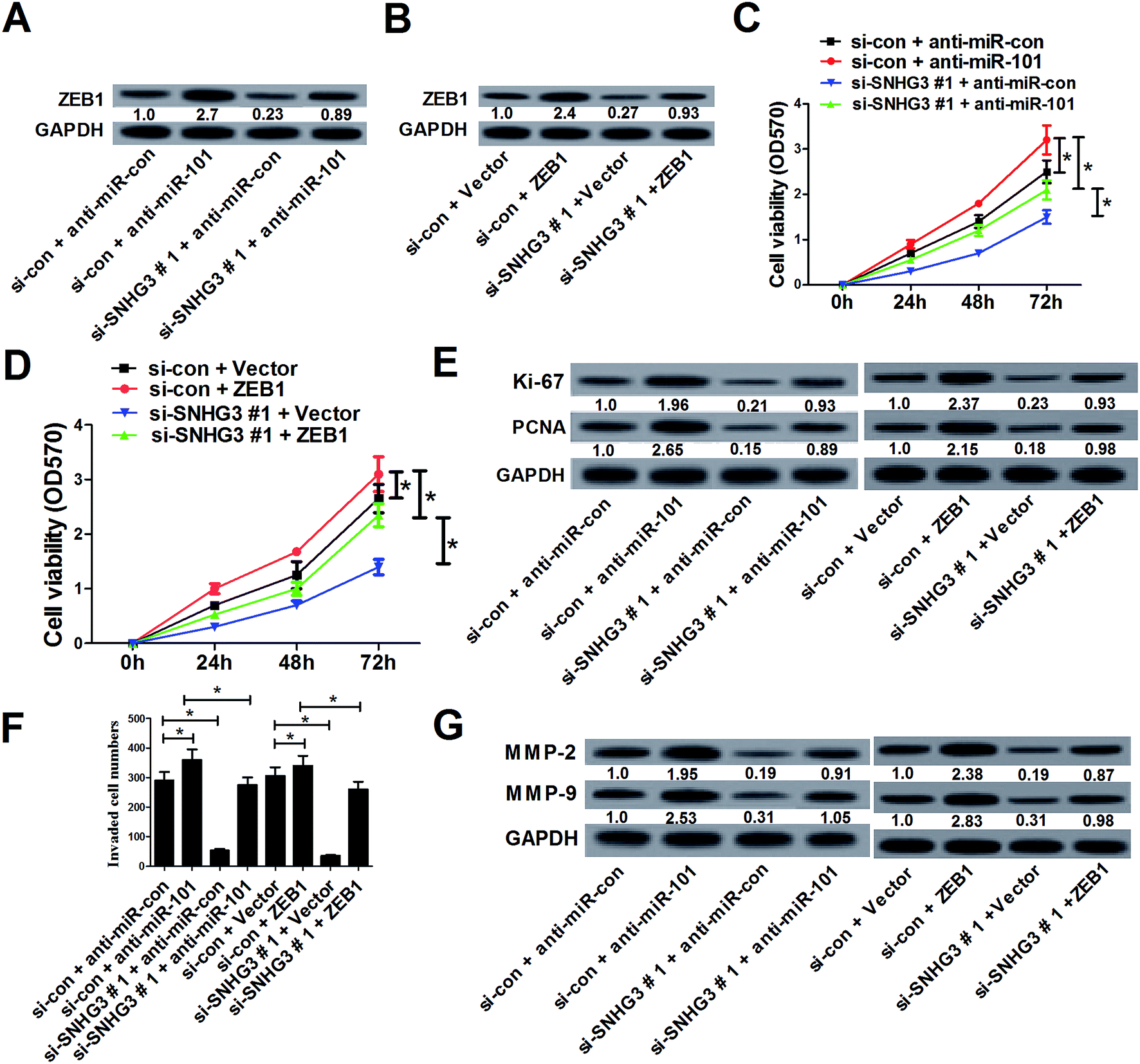 | ||
| Fig. 5 SNHG3 knockdown suppressed cell proliferation and invasion by upregulation of miR-101 and downregulation of ZEB1 expression in breast cancer cells in vitro. MCF-7 cells were cotransfected with si-SNHG3#1 or si-con, along with anti-miR-101, anti-miR-con, ZEB1, or Vector, and incubated for 48 h. (A and B) The protein level of ZEB1 in transfected MCF-7 cells. (C and D) Cell proliferation was estimated using an MTT assay in transfected MCF-7 cells. (E) The protein levels of Ki-67 and PCNA in transfected MCF-7 cells. (F) The cell invasion capability was evaluated using a cell invasion assay in transfected MCF-7 cells. (G) The protein levels of MMP-2 and MMP-9 in transfected MCF-7 cells. *P < 0.05. | ||
3.6. SNHG3 knockdown suppressed breast cancer tumor growth and invasion in vivo by upregulating miR-101 and downregulating ZEB1
To confirm whether SNHG3 could affect breast cancer development in vivo, MCF-7 cells stably transfected with lenti-sh-SNHG3 or lenti-sh-con combined with lenti-anti-miR-con or lenti-anti-miR-101 were subcutaneously injected into nude mice. As shown in Fig. 6A, the tumor growth was significantly inhibited by lenti-anti-miR-101 compared with that in the lenti-anti-miR-con group in breast cancer, which was drastically reduced by SNHG3 knockdown. Moreover, SNHG3 knockdown strikingly mitigated a miR-101 inhibition-mediated increase of tumor weight (Fig. 6B). Additionally, we found that lenti-sh-SNHG3 injection led to a marked decrease of SNHG3 expression, and an evident increase of miR-101 expression in xenografted tumor samples (Fig. 6C). Thereafter, we analyzed the expression patterns of ZEB1, Ki-67, PCNA, MMP-2 and MMP-9 in these xenografted tumor samples from nude mice. Consistently with the in vitro results, downregulation of miR-101 remarkably promoted the protein levels of ZEB1, Ki-67, PCNA, MMP-2 and MMP-9 in xenografted tumors, which was significantly attenuated by SNHG3 knockdown (Fig. 6D). These results substantiated that SNHG3 knockdown suppressed breast cancer tumor growth and invasion in vivo by upregulating miR-101 and downregulating ZEB1.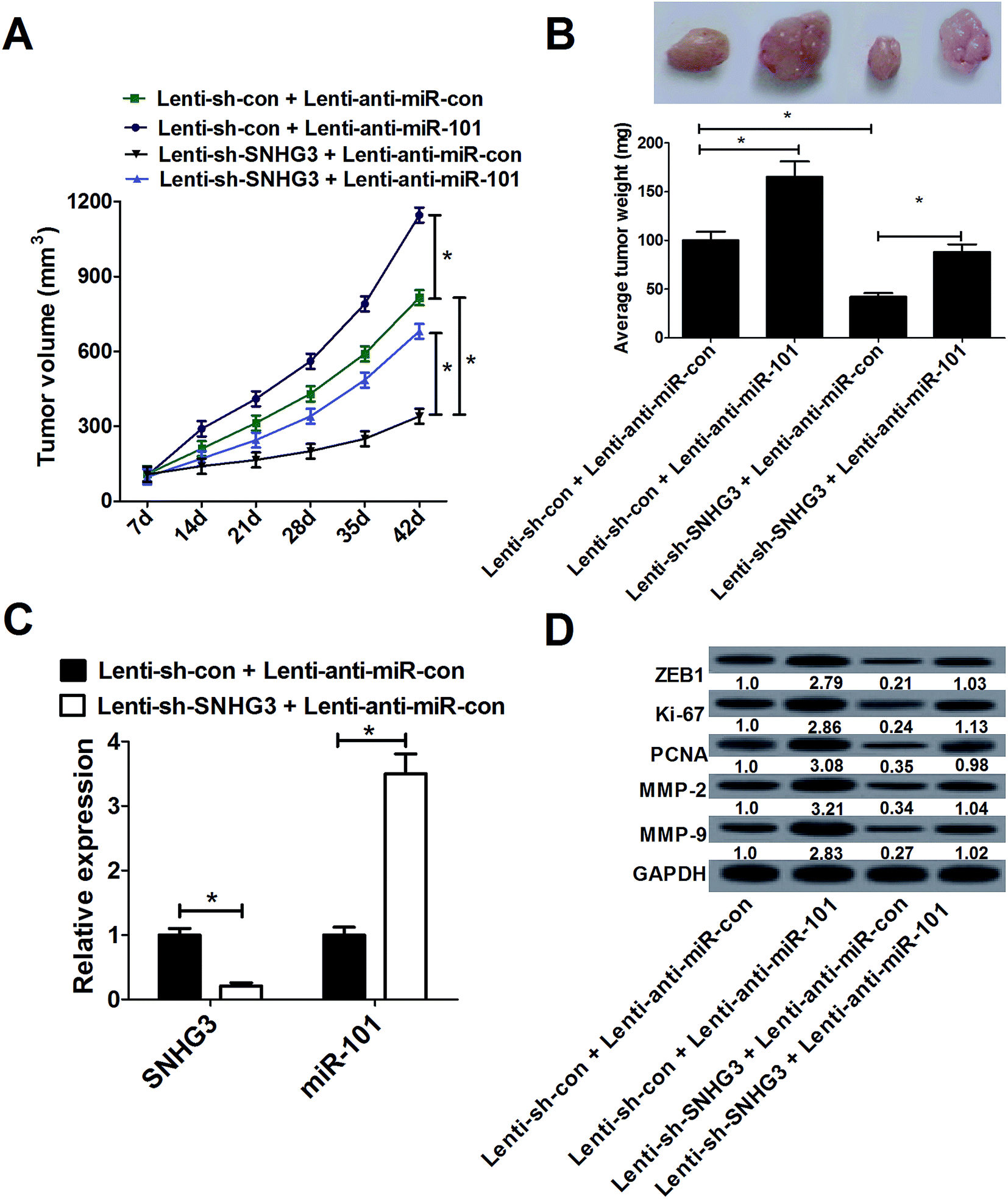 | ||
| Fig. 6 SNHG3 knockdown suppressed breast cancer tumor growth and invasion in vivo by upregulating miR-101 and downregulating ZEB1. Lenti-anti-miR-con or lenti-anti-miR-101 was transduced in lenti-sh-SNHG3 or lenti-sh-con stably transfected MCF-7 cells and then MCF-7 cells were subcutaneously injected into nude mice. (A) Tumor volume was measured every 7 days for 42 days. (B) Forty-two days after injection, the mice were sacrificed and the tumor tissues were isolated and weighed. (C) The expressions of SNHG3 and miR-101 in xenografted tumors. (D) The protein levels of ZEB1, Ki-67, PCNA, MMP-2 and MMP-9 in xenografted tumors were detected using western blot analysis. *P < 0.05. | ||
4. Discussion
Recently, emerging evidence has indicated that dysregulation of lncRNAs plays a significant role in cancer pathogenesis, and targeted therapies applying lncRNAs as a novel diagnostic and therapeutic tool have received increasing attention, such as therapies using SNHG15,22 MIAT,23 and PTENP1.24 In our study, we demonstrated that SNHG3 was aberrantly upregulated in breast cancer patients and this correlated with poor prognosis. SNHG3 knockdown suppressed breast cancer cell proliferation and invasion in vitro and in vivo. Mechanistically, SNHG3 functioned as a ceRNA behaving as a sponge for miR-101 to modulate ZEB1 expression in breast cancer cells. To our knowledge, the current study is the first that provides evidence of the functional role and molecular mechanism of SNHG3 in breast cancer.As a novel identified lncRNA, there had been limited investigations reporting the role of SNHG3 in malignant tumors up to now. It had been identified that SNHG3 was highly expressed in hepatocellular carcinoma and was associated with malignant status and poor prognosis in hepatocellular carcinoma patients.25 Moreover, SNHG3 was also upregulated in colorectal cancer (CRC), which predicted poor prognosis for patients with CRC, and promoted the malignant development of CRC.25 Importantly, SNHG3 was previously identified as an oncogene in breast cancer.15 However, the detailed roles and molecular mechanism of SNHG3 in breast cancer remain largely unclear. Our study implied that SNHG3 was upregulated in breast cancer tissues and cells, in accordance with a previous study.15 Moreover, we found that upregulation of SNHG3 was correlated with poor prognosis in breast cancer patients. Functional analysis revealed that SNHG3 knockdown suppressed cell proliferation and invasion in breast cancer cells. This viewpoint was further demonstrated by the high levels of proliferation marker proteins Ki-67/PCNA and metastasis-related proteins MMP-2/MMP-9 in breast cancer cells. These results indicated that SNHG3 knockdown restrained cell proliferation and invasion of breast cancer cells.
Recently, the interaction between lncRNAs and miRNAs has attracted more and more attention in research into the pathological mechanism of cancer. Mounting evidence showed that lncRNAs can serve as ceRNAs or molecular sponges and directly interact with miRNAs in order to suppress the inhibitory effect on downstream target mRNA.27 For example, upregulation of lncRNA urothelial carcinoma associated 1 (UCA1) could promote glioma cell proliferation and migration through interaction with miR-182 via miR-182-dependent inhibitor of apoptosis-stimulating protein of p53 (iASPP) regulation.28 SNHG16 induced breast cancer cell migration by competitively binding miR-98 with E2F transcription factor 5 protein (E2F5).29 lncRNA-X-inactive specific transcript (lncRNA XIST) inhibited cell proliferation and cell mobility by competitively binding to miR-21-5p and upregulating programmed cell death 4 (PDCD4) in osteosarcoma (OS).30 Of note, it was reported that SNHG3 functioned as a ceRNA behaving as a sponge for miR-182-5p, thus leading to the release of c-Myc from miR-182-5p and modulating the expression of c-Myc in colorectal cancer.26
To further explore the underlying molecular mechanism by which SNHG3 regulated breast cancer, we made a prediction and found that miR-101 was a potential target of SNHG3. miR-101 is a highly conserved miRNA in many species, which is located at chromosomes 1p31.3 and 9p24.1.31 A wide array of documents unveiled that miR-101 functioned as a tumor suppressor, and was frequently underexpressed in various types of cancer, such as lung cancer,32 glioblastoma,33 and breast cancer.12 In our study, we identified using a luciferase reporter assay and RIP that SNHG3 directly interacted with miR-101 and suppressed its expression in breast cancer in vitro and in vivo. In parallel, we confirmed that miR-101 was downregulated in breast cancer tissues and negatively correlated with SNHG3 expression in breast cancer patients. Additionally, in agreement with the previous observations,25 our results demonstrated that SNHG3 was distributed in the cytoplasm of breast cancer cells. Moreover, we further found that ZEB1, a well-known target of miR-101,21,34–36 was positively regulated by SNHG3 in breast cancer cells. ZEB1, a member of the ZEB family, has been shown to induce EMT by repression of E-cadherin and promotion of mesenchymal markers, and consequently contributes to cancer progression by facilitating tumor cell migration and invasion.37 Our study demonstrated that ZEB1 mRNA was upregulated in breast cancer tissues, in accordance with the previous studies,38,39 and positively correlated with SNHG3 expression. Rescue experiments demonstrated that SNHG3 knockdown suppressed cell proliferation and invasion by upregulation of miR-101 and downregulation of ZEB1 expression in breast cancer in vitro and in vivo, suggesting that SNHG3 promoted breast cancer cell proliferation and apoptosis by regulating the miR-101/ZEB1 axis.
5. Conclusion
In conclusion, we provided evidence that SNHG3 knockdown suppressed cell proliferation and invasion by upregulation of miR-101 and downregulation of ZEB1 expression in breast cancer in vitro and in vivo, which contributed to our understanding of the pathogenesis and development of breast cancer. Therefore, our study may be important in facilitating the development of lncRNA-directed therapy against breast cancer.Conflicts of interest
The authors declare that they have no competing interests.References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- B. A. Kohler, R. L. Sherman, N. Howlader, A. Jemal, A. B. Ryerson, K. A. Henry, F. P. Boscoe, K. A. Cronin, A. Lake, A. M. Noone, S. J. Henley, C. R. Eheman, R. N. Anderson and L. Penberthy, J. Natl. Cancer Inst., 2015, 107, djv048 CrossRef PubMed.
- M. Martin, S. Lopez-Tarruella and Y. J. Gilarranz, Breast, 2016, 28, 161–166 CrossRef PubMed.
- D. F. Alonso, G. V. Ripoll, J. Garona, N. B. Iannucci and D. E. Gomez, Curr. Pharm. Biotechnol., 2011, 12, 1974–1980 CAS.
- X. Shi, M. Sun, H. Liu, Y. Yao and Y. Song, Cancer Lett., 2013, 339, 159–166 CrossRef CAS PubMed.
- T. Nagano and P. Fraser, Cell, 2011, 145, 178–181 CrossRef CAS.
- A. Grigoriev and N. M. Bonini, Aging, 2014, 6, 705–706 CAS.
- O. Slaby, R. Laga and O. Sedlacek, Biochem. J., 2017, 474, 4219–4251 CrossRef CAS PubMed.
- G. A. Galin and C. M. Croce, Nat. Rev. Cancer, 2006, 11, 857–866 Search PubMed.
- E. van Schooneveld, H. Wildiers, I. Vergote, P. B. Vermeulen, L. Y. Dirix and S. J. Van Laere, Breast Cancer Res., 2015, 17, 21 CrossRef PubMed.
- J. Wang, H. Zeng, H. Li, T. Chen, L. Wang, K. Zhang, J. Chen, R. Wang, Q. Li and S. Wang, Cell. Physiol. Biochem., 2017, 43, 717–732 CrossRef CAS PubMed.
- J. T. Li, L. T. Jia, N. N. Liu, X. S. Zhu, Q. Q. Liu, X. L. Wang, F. Yu, Y. L. Liu, A. G. Yang and C. F. Gao, Oncotarget, 2015, 6, 30818–30830 Search PubMed.
- S. W. Cheetham, F. Gruhl, J. S. Mattick and M. E. Dinger, Br. J. Cancer, 2013, 108, 2419–2425 CrossRef CAS PubMed.
- C. P. Ponting, P. L. Oliver and W. Reik, Cell, 2009, 136, 629–641 CrossRef CAS PubMed.
- S. Xu, D. Kong, Q. Chen, Y. Ping and D. Pang, Mol. Cancer, 2017, 16, 129 CrossRef PubMed.
- P. Paci, T. Colombo and L. Farina, BMC Syst. Biol., 2014, 8, 83 CrossRef PubMed.
- K. Yan, J. Tian, W. Shi, H. Xia and Y. Zhu, Cell. Physiol. Biochem., 2017, 3, 999–1012 CrossRef PubMed.
- S. E. Singletary, C. Allred, P. Ashley, L. W. Bassett, D. Berry, K. I. Bland, P. I. Borgen, G. M. Clark, S. B. Edge, D. F. Hayes, L. L. Hughes, R. V. Hutter, M. Morrow, D. L. Page, A. Recht, R. L. Theriault, A. Thor, D. L. Weaver, H. S. Wieand and F. L. Greene, Surg. Clin. North Am., 2003, 83, 803–819 CrossRef PubMed.
- O. Isbilen, N. Rizaner and E. Volkan, BMC Complementary Altern. Med., 2018, 1, 30 CrossRef PubMed.
- L. Salmena, L. Poliseno, Y. Tay, L. Kats and P. P. Pandolfi, Cell, 2011, 146, 353–358 CrossRef CAS PubMed.
- K. Chandra Mangalhara, S. Manvati, S. K. Saini, K. Ponnusamy, G. Agarwal, S. K. Abraham and R. N. K. Bamezai, Cancer Lett., 2017, 391, 59–73 CrossRef CAS PubMed.
- Q. Kong and M. Qiu, Biochem. Biophys. Res. Commun., 2017, 495, 1594–1600 CrossRef PubMed.
- T. Luan, X. Zhang, S. Wang, Y. Song, S. Zhou, J. Lin, W. An, W. Yuan, Y. Yang, H. Cai, Q. Zhang and L. Wang, Oncotarget, 2017, 8, 76153–76164 Search PubMed.
- S. Chen, Y. Wang, J. H. Zhang, Q. J. Xia, Q. Sun, Z. K. Li, J. G. Zhang, M. S. Tang and M. S. Dong, Oncol. Lett., 2017, 14, 4659–4662 CrossRef PubMed.
- T. Zhang, C. Cao, D. Wu and L. Liu, Tumor Biol., 2016, 37, 2379–2385 CrossRef CAS PubMed.
- W. Huang, Y. Tian, S. Dong, Y. Cha, J. Li, X. Guo and X. Yuan, Oncol. Rep., 2017, 38, 1402–1410 CrossRef PubMed.
- A. Sahu, U. Singhal and A. M. Chinnaiyan, Trends Cancer, 2015, 1, 93–109 CrossRef PubMed.
- Z. He, Y. Wang, G. Huang, Q. Wang, D. Zhao and L. Chen, Arch. Biochem. Biophys., 2017, 623–624, 1–8 CrossRef CAS PubMed.
- C. Cai, Q. Huo, X. Wang, B. Chen and Q. Yang, Biochem. Biophys. Res. Commun., 2017, 485, 272–278 CrossRef CAS PubMed.
- R. Zhang and T. Xia, Internet J. Oncol., 2017, 51, 1460–1470 CrossRef PubMed.
- T. Gui and K. Shen, Cancer Epidemiol., 2012, 36, 537–540 CrossRef CAS PubMed.
- J. Zhang, J. Liu, Y. Liu, W. Wu, X. Li, Y. Wu, H. Chen, K. Zhang and L. Gu, Cancer Epidemiol., 2015, 74, 215–221 CAS.
- L. Xiaoping, Y. Zhibin, L. Wenjuan, W. Zeyou, X. Gang, L. Zhaohui, Z. Ying, W. Minghua and L. Guiyuan, Cell Death Dis., 2013, 4, e675 CrossRef CAS PubMed.
- B. Wu, D. Lei, L. Wang, X. Yang, S. Jia, Z. Yang, C. Shan, X. Yang, C. Zhang and B. Lu, Am. J. Cancer Res., 2016, 6, 1396–1407 CAS.
- S. Zhang, W. Wang, G. Liu, S. Xie, Q. Li, Y. Li and Z. Lin, Biomed. Pharmacother., 2017, 95, 711–720 CrossRef CAS PubMed.
- F. Guo, D. Cogdell, L. Hu, D. Yang, A. K. Sood, F. Xue and W. Zhang, Oncol. Rep., 2014, 31, 2021–2028 CrossRef CAS PubMed.
- H. Peinado, D. Olmeda and A. Cano, Nat. Rev. Cancer, 2007, 7, 415–428 CrossRef CAS PubMed.
- Z. Ma, Y. Li, J. Xu, Q. Ren, J. Yao and X. Tian, IUBMB Life, 2016, 68, 394–402 CrossRef CAS PubMed.
- Q. Zou, E. Zhou, F. Xu, D. Zhang, W. Yi and J. Yao, J. Cell. Biochem., 2017, 119, 2189–2199 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |