DOI:
10.1039/C8RA02056F
(Paper)
RSC Adv., 2018,
8, 15762-15772
One-step fabrication of carbonaceous solid acid derived from lignosulfonate for the synthesis of biobased furan derivatives
Received
8th March 2018
, Accepted 21st April 2018
First published on 26th April 2018
Abstract
An eco-friendly and low-cost lignosulfonate-based acidic carbonaceous catalyst (LS-SO3H) was effectively fabricated using the sulfite pulping by-product of sodium lignosulfonate as a precursor by facile one-step simultaneous carbonization and sulfonation, and employed for the synthesis of promising biofuel furan derivatives from biorenewable feedstocks. The catalyst preparation conditions significantly affected the preparation and properties of LS-SO3H. A relatively high catalyst preparation yield (40.4%) with strong –SO3H density (1.33 mmol g−1) were achieved when the lignosulfonate was treated in concentrated H2SO4 solution at 120 °C for 6 h. The preparation yield of LS-SO3H was nearly twice as much as that of one-step prepared catalyst using alkaline lignin (another technical lignin from pulping) as a precursor. The as-prepared LS-SO3H had similar textural characteristics to the frequently-used two-step prepared carbonaceous catalyst involving pyrolysis carbonization and sulfonation. LS-SO3H was found to show good catalytic activity for the synthesis of 5-ethoxymethylfurfural (EMF) in ethanol medium, affording around 86%, 57% and 47% yields from 5-hydroxymethylfurfural (HMF), fructose and inulin, respectively. Also, a high HMF yield of 83% could be obtained from fructose when DMSO was replaced as reaction medium. The used LS-SO3H was readily recovered by filtration, and remained active in recycle runs.
1. Introduction
With the diminishing fossil fuels and worsening environmental quality, the development of alternative fuels and chemicals from renewable biomass resources has been receiving much needed attention.1 As a key bio-based platform compound for fuels and plastic, the synthesis of 5-hydroxymethylfurfural (HMF) from biomass-derived carbohydrates has been broadly investigated.2 Recently, 5-ethoxymethylfurfural (EMF), as one of the derivatives of HMF, has received considerable attention purporting its potential use as an alternative biofuel owing to its relatively high energy density (8.7 kW h L−1).3 It is known that these furan derivatives can be directly produced from one-pot conversion of carbohydrates catalyzed by acid catalyst. Presently, solid acid catalysts such as protonated zeolites,4 ion-exchange resins,5 sulfonated metal oxides,6 heteropoly acid salts,7 metal–organic frameworks,8 and carbon-based acids9 are widely applied in these reactions because they are less corrosive than liquid acids, form less pollutants after reaction and can be readily recycled.3,10 In spite of significant advances from the persistent studies, the main obstacles for practical application include the complex preparation procedures of the most solid acids and expensive catalyst cost. Consequently, the development of easy-prepared and low-cost solid acid catalysts for the transformation of carbohydrates into furan derivatives such as EMF is still a challenge.
Lignin is the second most abundant lignocellulosic biomass fraction after cellulose and the richest natural aromatic organic polymer. At an industrial scale, technical lignin is generally produced as a by-product of lignocellulosic biomass utilization in pulp mills and it is traditionally divided into two distinct classes, namely alkaline lignins (include kraft lignin and soda lignin) and lignosulfonate.11 Of late years, lignin has attracted increasing attention as an underutilized renewable material for the preparation of high-value polymeric materials due to its highly phenolic and aliphatic hydroxyl groups,12 as well as the production of liquid fuels and chemicals.13 On the other hand, lignin has higher carbon content than carbohydrates that benefits the economics in carbonaceous material production. Recently, lignin has been proposed as a superior carrier material for the fabrication of carbonaceous solid acid catalyst, and the related reports were summarized in Table 1.14 Beyond doubt, using lignin as a precursor of solid acid catalyst not only provides an inexpensive and biorenewable catalyst source but also supplies a complementary pathway for the resource utilization of lignin as a part of biorefinery. It can be found from literatures that alkaline lignins were usually used as lignin-based solid acid catalyst materials, and the prepared catalysts were active in the synthesis of biodiesel, cellulose hydrolysis, and sugar dehydration. Nevertheless, it is a fact that almost all alkaline lignins are burned to generate energy for conferring to the pulp mills self-sufficiency, only a small amount is currently extracted for practical applications.15 As for the preparation of lignin-based solid acid catalysts, the previous report method generally involves two steps: pyrolysis carbonization or solvothermal carbonization of lignin and the subsequent sulfonation of the resulting carbon material. In particular, acid hydrolysis residual lignin was found to synthesize acidic carbonaceous catalyst by one-step simultaneous carbonization and sulfonation in concentrated sulfuric acid or sulfuryl chloride solutions.14b,d Presently, there were also some reports about the one-step preparation of carbonaceous solid acid in concentrated sulfonic acid solutions by using cellulose, glucose, or cellulosic biomass as the carrier material.16 The one-step method for the fabrication of carbonaceous solid acid is highly facile with the promising potential for actual application.
Table 1 Preparation method, properties and application of lignin-based acidic carbonaceous catalysts in previous reports14
| Catalyst |
Carrier material |
Preparation method |
BET surface area, m2 g−1 |
|
Densities of acid groups, mmol g−1 |
Application |
| Total acid |
–SO3H |
|
| Solid acid catalyst |
Kraft lignin |
Impregnation: 85% H3PO4, 1 h |
54.8 |
— |
1.30 |
Esterification of oleic acid and conversion of jatropha oil to biodiesel |
|
| Pyrolysis carbonization: 400 °C, 1 h |
|
| Sulfonation: H2SO4, 200 °C, 2 h |
|
| Lignin-derived carbonaceous catalyst |
Lignin |
Sulfonation: N2, H2SO4, 150 °C, 1 h |
4.7 |
1.71 |
0.86 |
Synthesis of biodiesel from acidified soybean soapstock |
|
| Amorphous carbon-based sulfonated catalyst |
Dealkaline lignin |
Solvothermal carbonization: H2O, 225 °C, 20 h |
<2 |
1.21 |
0.87 |
HMF production from inulin |
|
| Sulfonation: H2SO4, 150 °C, 12 h |
|
| Lignin-based solid acid |
Acid hydrolysis lignin |
Sulfonation: sulfuryl chloride, 50 °C, 5 h |
12.1 |
2.22 |
0.37 |
Esterification of acetic acid and hydration of 2,3-dimethyl-2-butene |
|
| Magnetic lignin-derived carbonaceous catalyst |
Enzymatic hydrolysis lignin |
Impregnation: 10 mmol L−1 FeCl3, 5 h |
234.6 |
1.95 |
0.77 |
Dehydration of fructose into HMF |
|
| Pyrolysis carbonization: 400 °C, 1 h |
|
| Sulfonation: H2SO4, 150 °C, 10 h |
|
| One-dimensional solid acid catalysts |
Lignin-based activated carbon fibers |
Sulfonation: H2SO4, 150 °C, 20 h |
475.0 |
0.88 |
0.56 |
Hydrolysis cellulose to glucose and nano-cellulose |
|
| Hydrothermal treatment: 100 °C, 20 h |
|
| Carbonaceous acids |
Dealkaline lignin |
Solvothermal carbonization: subcritical ethanol, 180 °C, 1 h |
2.7 |
5.35 |
1.06 |
Produce biodiesel from actual plant oils |
|
| Pyrolysis carbonization: 400 °C, 2 h |
|
| Sulfonation: H2SO4, 150 °C, 10 h |
|
| Lignin-derived solid acids |
Alkali lignin |
Impregnation: 40% H3PO4, 6 h |
488.4 |
3.52 |
0.88 |
Hydrolysis of microcrystalline cellulose into reducing sugar |
|
| Pyrolysis carbonization: 450 °C, 3 h |
|
| Sulfonation: H2SO4, 180 °C, 12 h |
|
Lignosulfonate is one of technical lignin, which was generally produced by the sulfite pulping process. It is found in large quantities with a world production of around one million tonnes of solids per year.17 Currently, lignosulfonate was mainly used for preparing of concrete admixtures, oil well dispersants, dyestuff, coal water slurry dispersants, and industrial binders.18 Herein, we first proposed to make use of lignosulfonate as a precursor to synthesize a novel carbonaceous solid acid catalyst by facile one-step simultaneous carbonization and sulfonation. Effect factors of catalyst preparation were explored, and the physicochemical properties of as-prepared catalysts were well characterized using a variety of techniques. The catalytic activities of as-prepared catalyst were evaluated for the one-pot conversion of biorenewable carbohydrates to furan derivatives including EMF and HMF.
2. Experimental section
2.1. Materials
Glucose, fructose, sucrose, inulin, HMF, ethyl levulinate (EL), NaOH, NaHCO3, NaCl, and N,N-dimethylformamide (DMF) were purchased from Aladdin Reagent (Shanghai, China). EMF was obtained from Sigma-Aldrich (Shanghai, China). Ethyl glucoside (EGO) and ethyl fructoside (EFO) were supplied by Chemsynlab Pharmaceutical Science & Technology Co., Ltd (Beijing, China). Ethanol, dimethylsulfoxide (DMSO), and tetrahydrofuran (THF) were obtained from Fengchuan Chemical Technology Co., Ltd (Tianjin, China). H2SO4 (98%) and HCl (38%) were from Kelong Chemical Reagent Factory (Chengdu, China) and Chuandong Chemical Co., Ltd (Chongqing, China), respectively. Sodium lignosulfonate (LS) was provided by Jiangmen Sugarcane Chemical Factory Co., Ltd (Guangdong, China). Alkaline lignin (AL) that is from kraft pulping process was supplied by Cool Chemical Science and Technology Co., Ltd (Beijing, China).
2.2. Catalyst preparation
A typical one-step fabrication procedure of lignosulfonate-based acidic carbonaceous catalyst (labeled as LS-SO3H) was as follows: 10 g LS powder and 100 mL concentrated H2SO4 were mixed into 250 mL three-necked round-bottomed flask. The flask was immersed in an oil-bath controlled at 120 °C and the mixture was continuously stirred at 500 rpm for the reaction. After 6 h, the mixture was cooled to room temperature, filtered and washed with hot deionized water (80 °C) to dislodge the physically absorbed sulfate ions until the washed water became neutral. Finally, the resulting precipitated solid was dried in a vacuum oven at 80 °C for 12 h to obtain the prepared LS-SO3H. For comparison, another lignin-based acidic carbonaceous catalyst using AL instead of LS as the carrier material was prepared with the same one-step procedure as above, which was denoted as AL-SO3H. In addition, to find out the differences of textural and catalytic properties, other lignosulfonate-based carbonaceous catalysts were also prepared based on a known and frequently-used two-step process including pyrolysis carbonization and sulfonation for the preparation of carbonaceous solid acids.14g,19 Concretely, LS powder was first carbonized at 200–500 °C for 1 h under N2 gas flow in a tube furnace to produce the lignosulfonate-derived amorphous carbon (labeled as LSC). Subsequently, the obtained LSC was sulfonated at 120 °C for 6 h to introduce –SO3H groups. The next operations were the same as the preparation of LS-SO3H. Finally, a series of lignosulfonate-based carbonaceous catalysts (LSC-SO3H) were obtained and labeled as LSC200-SO3H, LSC300-SO3H, LSC400-SO3H, and LSC500-SO3H according to the carbonization temperature of 200 °C, 300 °C, 400 °C, and 500 °C, respectively.
2.3. Catalyst characterization
Surface morphologies were obtained by using a Hitachi S4800 scanning electron microscope (SEM). Powder X-ray diffraction (XRD) patterns were performed on a Bruker D8 advanced D8 X-ray diffractometer with a Cu Kα radiation source, and the scan range was 10–80° with a step of 0.02°. The functional groups of samples were determined by a Nicolet 5700 Fourier transform infrared (FT-IR) spectrometer in the wavenumber range of 400–4000 cm−1. X-ray photoelectron spectroscopy (XPS) measurements were conducted on a Thermo ESCALAB 250XI system with an Al Kα radiation source. Thermo gravimetric analyzer (TGA) was made on a TA Instruments Q500 TGA under N2 atmosphere at a constant heating rate of 10 °C min−1 in a temperature range of 30–800 °C. The N2 adsorption–desorption isotherms were obtained by JW-BK 112 surface analyzer at liquid nitrogen temperature. The surface areas of samples were estimated by using the method of Brunauer–Emmett–Teller (BET). The densities of total acids, –SO3H, –COOH, and phenolic –OH were measured by acid–base titration based on the previous reported procedures.20
2.4. Typical procedure for the conversion of carbohydrate into furan derivatives
In a typical synthesis of EMF from fructose, 1.08 g fructose and 30 mL ethanol solution contains 0.2 g catalyst were loaded into 50 mL cylindrical stainless steel pressurized reactor. The sealed reactor was placed into a preheated oil-bath for the reaction and stirred magnetically at 800 rpm. After running a given time, the reactor was took out and placed into cool water to terminate the reaction. Then, the liquid products and solid acid catalyst were separated by filtration. The obtained filtrate was diluted and analyzed by high-performance liquid chromatography (HPLC) and gas chromatography (GC) analysis, respectively. To test the catalyst reusability, the LS-SO3H catalyst was collected by filtration after reaction. The recovered catalyst was washed several times with ethanol, and dried in a vacuum oven at 80 °C for 24 h. Then a new reaction with the recovered catalyst under the identical conditions was started as a new cycle.
2.5. Product analysis
The analysis of EMF and HMF were conducted on a Shimadzu HPLC (LC-10AVP) with a UV detector. Samples were separated by a Copsil C18 column (250 × 4.6 mm) at 30 °C with a detection wavelength of 280 nm. The samples were eluted using the mixture solution of methanol and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) with the flow rate of 1.0 mL min−1. A Copsil NH2 column (250 × 4.6 mm) at 30 °C coupled with a refractive index detector was used to determine sugars, EFO and EGO. The samples were eluted at 1.0 mL min−1 with the mixture solution of acetonitrile and water (7:3 v/v). EL was analyzed by GC on an Agilent 6890 instrument with a capillary DB-5 column (30 m × 0.32 mm × 0.25 μm) equipped with a flame-ionization detector. Operating conditions were as follow: N2 carrier gas with a flow rate of 1.0 mL min−1, injection port temperature of 250 °C, oven temperature of 200 °C, and detector temperature of 250 °C. The amounts of detected substances were calculated according to the external standard curves constructed as their authentic standards. The yields of products were defined as the molar ratio of the obtained products to the potential hexose monomer in carbohydrates for the feed.
:1 v/v) with the flow rate of 1.0 mL min−1. A Copsil NH2 column (250 × 4.6 mm) at 30 °C coupled with a refractive index detector was used to determine sugars, EFO and EGO. The samples were eluted at 1.0 mL min−1 with the mixture solution of acetonitrile and water (7:3 v/v). EL was analyzed by GC on an Agilent 6890 instrument with a capillary DB-5 column (30 m × 0.32 mm × 0.25 μm) equipped with a flame-ionization detector. Operating conditions were as follow: N2 carrier gas with a flow rate of 1.0 mL min−1, injection port temperature of 250 °C, oven temperature of 200 °C, and detector temperature of 250 °C. The amounts of detected substances were calculated according to the external standard curves constructed as their authentic standards. The yields of products were defined as the molar ratio of the obtained products to the potential hexose monomer in carbohydrates for the feed.
3. Results and discussion
3.1. Catalyst screening in the conversion of fructose to EMF
Initially, various lignin-based acidic carbonaceous catalysts that prepared by one-step sulfonation or two-step treatment (i.e., pyrolysis carbonization and sulfonation) of the technical lignins (i.e., ligninsulfonate and alkaline lignin) were investigated for the conversion of fructose in ethanol medium at 110 °C for 15 h. The results of catalyst preparation yield, fructose conversion and the yield of typical products (EMF, HMF, EL, EFO, and EGO) are depicted in Table 2. The technical lignins directly as the catalyst did not lead effectively to the conversion of fructose to EMF, and there were no obvious difference with no catalyst (Table 2, Entries 2–3). However, fructose was consumed with the conversion of around 70%. Two main reaction products including EFO and EGO were detected, which are the products of the etherification and isomerization of fructose, respectively.21 On the contrary, the SO3H-functionalized lignin-based catalysts except LSC500-SO3H exhibited catalytic activity in the synthesis of EMF, suggesting that –SO3H groups played a key role in the reaction process. Among these as-prepared catalysts, one-step prepared LS-SO3H and AL-SO3H catalysts were found to show similarly remarkable catalytic performance for the conversion of fructose, giving EMF yields of above 50%. However, it is important to note that AL was readily dissolved in concentrated H2SO4 and serious blebbing phenomenon was observed in the one-step sulfonation process for AL-SO3H preparation, thus lead to a relative low catalyst preparation yield of 22.3%. Fortunately, the catalyst preparation yield of LS-SO3H (40.4%) was obtained almost twice as much as that of AL-SO3H. This could potentially be linked to the introduction of sulfonate groups in LS from sulfite pulping process. Based on this arresting superiority, LS was emerged as a preferred carrier material for the one-step fabrication of acidic carbonaceous catalyst. For comparison, other LS-based carbonaceous catalysts that prepared by a frequently-used two-step process including pyrolysis carbonization and sulfonation were investigated. Although the catalyst preparation yield of LSC-SO3H was similar to that of LS-SO3H, the yields of EMF and total detected products (EMF, HMF and EL) were found to be lower. Especially, the prepared catalyst at elevated carbonization temperature (LSC500-SO3H) was inactive for the reaction. Considering the preparation and properties of catalyst, the one-step prepared LS-SO3H catalyst was well characterized and applied for the synthesis of furans in the subsequent explorations.
Table 2 Catalyst preparation yield of various lignin-based carbonaceous catalysts and their catalytic performance for the conversion of fructose in ethanol mediuma
| Entry |
Catalyst |
CP yieldb, wt% |
Conversion, % |
Product yield, % |
| HMF |
EMF |
EL |
EFO |
EGO |
| Reaction conditions: 1.08 g fructose, 0.2 g catalyst, 30 mL ethanol, 110 °C, 15 h. CP yield (representing catalyst preparation yield) was calculated as the ratio of the weight of obtained resulting sample to the weight of initial material for catalyst preparation. 0.133 mmol H2SO4 (H+ amount of loaded H2SO4 is consistent with –SO3H amount of 0.2 g LS-SO3H). |
| 1 |
— |
— |
88.5 |
1.5 |
0 |
0.2 |
34.2 |
24.3 |
| 2 |
LS |
— |
71.2 |
0.2 |
0.2 |
0 |
28.1 |
28.7 |
| 3 |
AL |
— |
69.9 |
0.3 |
0 |
0 |
25.6 |
21.1 |
| 4 |
LS-SO3H |
40.4 |
99.4 |
3.0 |
53.4 |
11.8 |
0 |
0.2 |
| 5 |
AL-SO3H |
22.3 |
99.5 |
7.4 |
55.9 |
8.1 |
0 |
0.5 |
| 6 |
LSC |
61.7 |
68.4 |
0.5 |
1.3 |
0 |
23.0 |
22.6 |
| 7 |
LSC200-SO3H |
40.8 |
97.5 |
4.2 |
43.8 |
5.9 |
1.3 |
1.4 |
| 8 |
LSC300-SO3H |
41.6 |
97.0 |
6.3 |
45.4 |
6.8 |
0 |
0.5 |
| 9 |
LSC400-SO3H |
39.6 |
96.5 |
6.1 |
42.9 |
6.0 |
0 |
0.4 |
| 10 |
LSC500-SO3H |
41.8 |
69.2 |
0.1 |
0.2 |
0 |
20.4 |
17.8 |
| 11 |
H2SO4c |
— |
99.1 |
8.4 |
49.8 |
7.3 |
0 |
0.7 |
3.2. Catalysts characterization
The structural parameters and acid densities of carbonaceous materials derived from LS and AL are listed in Table 3. It can be seen that all samples had small BET surface areas (0.8–5.4 m2 g−1). It was considered as devoid of porous structures due to incomplete carbonization, and a great quantity of hydrogen and oxygen atoms still reserved without the formation of porous particles.22 The measurement of acid densities showed that the catalyst preparation method evidently affects the acid densities. LS-SO3H exhibited higher densities of –SO3H (1.33 mmol g−1) and –COOH (3.32 mmol g−1) than LSC-SO3H. Nevertheless, the acid densities of LS-SO3H was slightly lower than AL-SO3H. Combined with the results of catalytic reaction in Table 2, it can be concluded that the –SO3H density of carbonaceous catalyst is a sensitive indicator for the production of EMF. Fructose could not be converted effectively into EMF only by –COOH and phenolic –OH groups without a certain amount of –SO3H groups, as reflected by the results of LSC and LSC500-SO3H. However, when H2SO4 was selected as the catalyst (H+ amount of loaded H2SO4 in keeping with –SO3H amount in used LS-SO3H), the EMF yield became lower than that for LS-SO3H. It follows that the catalytic activity of carbonaceous catalyst for the synthesis of EMF was probably attributed to the synergic effect of –SO3H, –COOH and phenolic –OH groups.
Table 3 Physicochemical properties of various lignin-based carbonaceous catalysts
| Catalyst |
BET surface area, m2 g−1 |
Densities of acid groups, mmol g−1 |
| Total acid |
–SO3H |
–COOH |
Phenolic –OH |
| Recovered LS-SO3H catalyst after the third recycle run. |
| LS |
1.4 |
1.28 |
0.23 |
0.63 |
0.42 |
| AL |
1.1 |
— |
— |
— |
— |
| LS-SO3H |
2.0 |
5.51 |
1.33 |
3.32 |
0.86 |
| LS-SO3Ha |
1.4 |
4.23 |
0.82 |
2.66 |
0.75 |
| AL-SO3H |
5.4 |
5.89 |
1.41 |
3.44 |
1.04 |
| LSC |
0.8 |
2.13 |
— |
0.74 |
1.39 |
| LSC200-SO3H |
2.2 |
5.57 |
1.08 |
3.05 |
1.44 |
| LSC300-SO3H |
2.8 |
5.35 |
1.11 |
3.09 |
1.15 |
| LSC400-SO3H |
3.3 |
4.58 |
0.83 |
2.46 |
1.29 |
| LSC500-SO3H |
3.9 |
1.25 |
0.05 |
0.31 |
0.79 |
As can be observed from SEM images in Fig. 1 that all samples had an anomalous structure and covered by some irregular particles. LSC (Fig. 1b) and LS-SO3H (Fig. 1c) had relatively smooth surface topography with a particle size of 20–40 μm. After sulfonation for LSC, the surface of LSC-SO3H was etched to be rough and had layered structures (Fig. 1d). This phenomenon was even more distinct for AL-SO3H (Fig. 1f). The former could be attributed to the release of the volatile substances and decomposition of sulfonate groups in LS from the combination of pyrolysis carbonization and sulfonation process, and the latter could be due to the release of more volatile substances in AL from the strong acid attack during the sulfonation process.14h,23 This may be the reason for the low catalyst preparation yield and relatively high BET surface area of AL-SO3H.
 |
| | Fig. 1 SEM images of the LS (a), LSC (b), LS-SO3H (c), LSC300-SO3H (d), AL (e), and AL-SO3H (f). | |
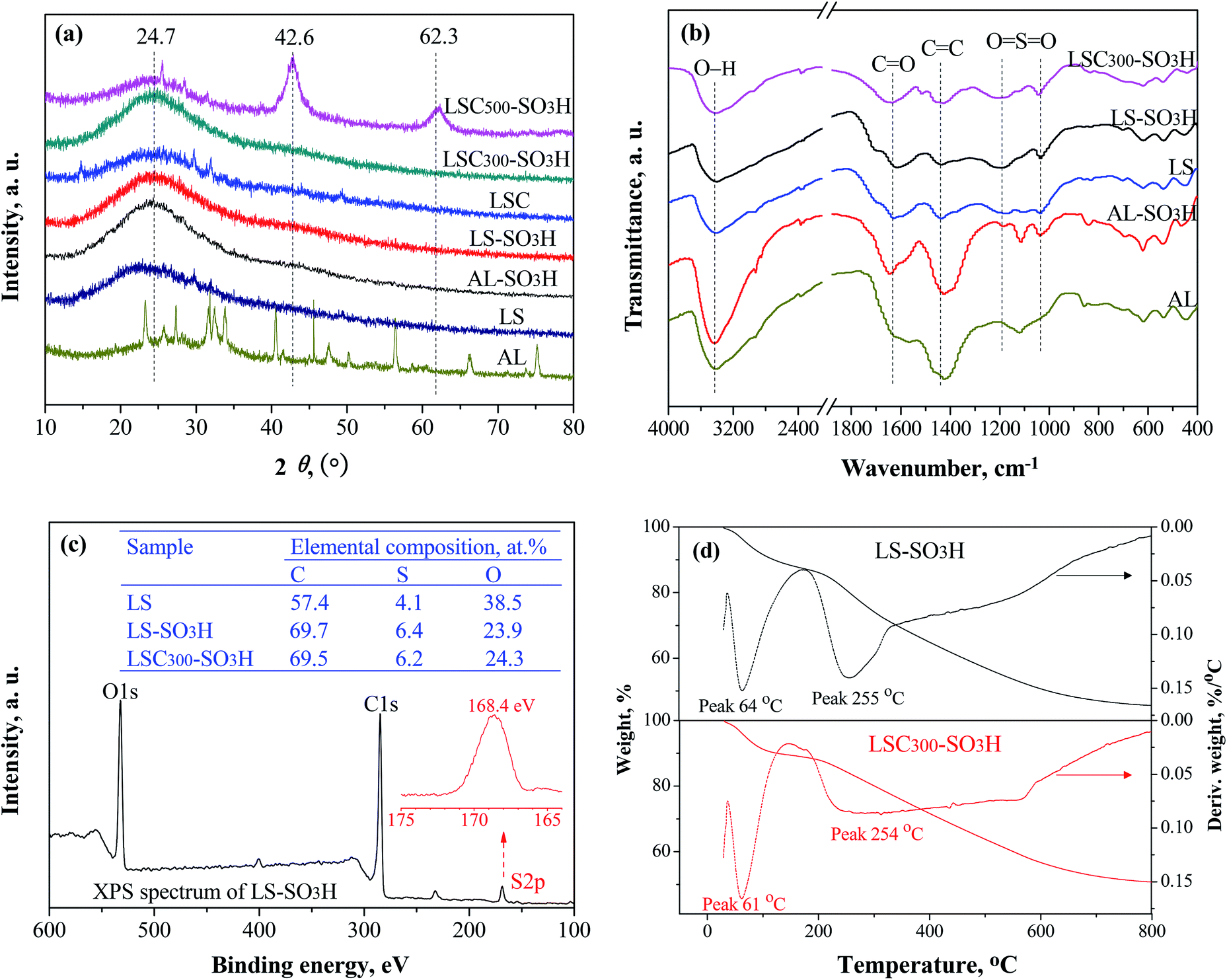
XRD patterns of the samples are given in Fig. 2a. All carbonaceous materials exhibit a broad and weak diffraction peak (2θ = 20–25°), which is typical of amorphous carbon consisting of aromatic carbon sheets oriented in a random fashion.24 The diffraction pattern of LS was similar to that of the carbonaceous materials. However, a closer examination reveals that the diffraction peak of LS appears at the 2θ of 22.4°, which may be due to remnant cellulose contained in the LS.25 AL appears many distinct diffraction peaks, and these peaks disappeared after sulfonation (AL-SO3H), presumably because AL includes some sugars composition26 and they were destroyed during the sulfonation process. Beyond that, there was a highly similar texture property between the one-step prepared catalyst (AL-SO3H, LS-SO3H) and the two-step obtained catalyst carbonized at 300 °C (LSC300-SO3H). It suggests that amorphous structure can be successfully formed by one-step sulfonation process of ligninsulfonate. In addition, two new diffraction peaks at 2θ = 42° and 62° were observed by increasing the carbonization temperature from 300 to 500 °C (LSC500-SO3H) due to stacking of carbon sheets.27
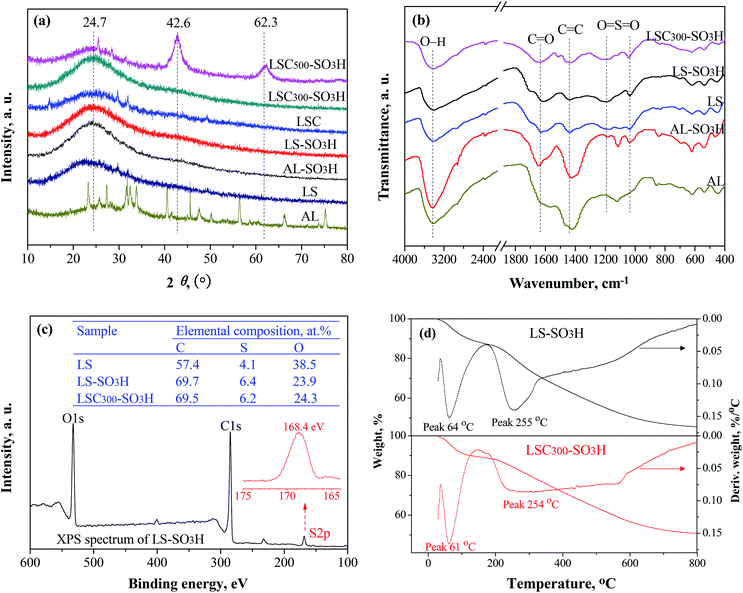 |
| | Fig. 2 XRD patterns (a), FT-IR spectra (b), XPS analyses (c), and TGA curves (d) of the samples. | |
FT-IR spectra of the samples from Fig. 2b showed that the intensities of two absorption peaks at 1040 cm−1 and 1190 cm−1 from O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO symmetric and asymmetric stretching vibrations enhanced after sulfonation,28 meaning that –SO3H groups were loaded on catalyst surface successfully. The absorptions at 1440 cm−1, 1640 cm−1 and 3430 cm−1 are contributed by CC, CO and O–H stretching vibration in aromatic carbons, –COOH and phenolic –OH groups, respectively.29 The intensities of CO and O–H peaks increased after carbonization and sulfonation. There were no obvious differences in characteristic bands between LS-SO3H and LSC300-SO3H. However, AL and AL-SO3H had a very strong CC absorption at 1440 cm−1, indicating abundant aromatic carbons exist in AL. In short, all prepared lignin-based carbonaceous catalysts probably composed of polycyclic aromatic carbon sheets bearing –SO3H, –COOH, and phenolic –OH groups.
SO symmetric and asymmetric stretching vibrations enhanced after sulfonation,28 meaning that –SO3H groups were loaded on catalyst surface successfully. The absorptions at 1440 cm−1, 1640 cm−1 and 3430 cm−1 are contributed by CC, CO and O–H stretching vibration in aromatic carbons, –COOH and phenolic –OH groups, respectively.29 The intensities of CO and O–H peaks increased after carbonization and sulfonation. There were no obvious differences in characteristic bands between LS-SO3H and LSC300-SO3H. However, AL and AL-SO3H had a very strong CC absorption at 1440 cm−1, indicating abundant aromatic carbons exist in AL. In short, all prepared lignin-based carbonaceous catalysts probably composed of polycyclic aromatic carbon sheets bearing –SO3H, –COOH, and phenolic –OH groups.
Fig. 2c presents the elemental compositions of LS, LS-SO3H and LSC300-SO3H determined by XPS analysis. The 4.1% S for LS is due to the existence of sulfonate groups in LS. After sulfonation, the S content had visibly increased, meaning that a certain amount of –SO3H groups was introduced into the LS-SO3H and LSC300-SO3H. The S content of LS-SO3H was similar to that of LSC300-SO3H. This observation is consistent with the result of FT-IR spectra. The single S2p peak at 168.4 eV from XPS spectrum of LS-SO3H (Fig. 2c) was attributed to the contribution of –SO3H groups. The C content was distinctly enlarged after carbonization and sulfonation. There was an almost C content between LS-SO3H and LSC300-SO3H, indicating that the similar carbonization level can be achieved by one-step directly sulfonation of LS.
As can be seen from TGA spectra in Fig. 2d that the LS-SO3H and LSC300-SO3H had similar thermal stability. A weight loss of ca. 10% was detected below 100 °C on account of the removal of adsorbed water and volatile components. Further, a sharp weight loss was observed from 220 to 300 °C, which is caused by the decomposition of –SO3H groups.23,30 When the temperature over 300 °C, a continuous weight loss may be attributed to further condensation of amorphous carbon.31 From a reaction temperature perspective, the lignin-based acidic carbonaceous catalysts can be fully qualified for the reaction of this study.
3.3. Effect of catalyst preparation conditions on the properties of LS-SO3H
The LS-SO3H catalyst was directly prepared by one-step treatment of LS in concentrated H2SO4. Here, the influences of preparation conditions on the catalyst characteristics and the conversion of fructose into EMF were evaluated. As can be seen from Table 4, the yield of EMF increased first and after that it kept an approximate constant with the increase of H2SO4 dosage. The change rule of EMF yield were in good consistent with –SO3H density. However, with increasing of H2SO4 dosage, the catalyst preparation yield continued to diminish. The sulfonation temperature had significantly affected –SO3H density and EMF yield. The formation of EMF was favored at low sulfonation temperature. A possible explanation is that the elevated temperature may lead to the collapse of carbon skeleton structure to affect the introduction of –SO3H group.32 But unfortunately, hardly any LS-SO3H catalyst could be obtained by separation when sulfonation temperature below 120 °C. The effect of sulfonation time (4–12 h) showed that the catalytic activity of LS-SO3H catalyst was not fundamentally changed with the prolonging of time, and the catalyst preparation yield had risen notably from 4 to 6 h. From the above, the sulfonation conditions significantly affected the preparation and properties of LS-SO3H catalyst. Taking the preparation yield and catalysis of LS-SO3H into considerations, the right catalyst preparation process was as follows: 1 g LS was sulfonated at 120 °C for 6 h with 10 mL concentrated H2SO4.
Table 4 Effects of preparation conditions of LS-SO3H catalyst on the catalyst characteristics and the conversion of fructose to EMF
| Entry |
Catalyst preparation conditionsa |
CP yieldb, wt% |
–SO3H density, mmol g−1 |
Conversion, % |
Product yieldc, % |
| H2SO4 dosage, mL |
Temperature, °C |
Time, h |
HMF |
EMF |
EL |
| 1 g ligninsulfonate was used for sulfonation. CP yield represents catalyst preparation yield. Reaction conditions: 1.08 g fructose, 0.2 g LS-SO3H, 30 mL ethanol, 110 °C, 15 h. |
| 1 |
5 |
160 |
4 |
38.9 |
0.92 |
98.8 |
9.6 |
37.7 |
4.2 |
| 2 |
10 |
160 |
4 |
37.1 |
1.11 |
98.3 |
7.7 |
45.7 |
5.8 |
| 3 |
15 |
160 |
4 |
34.7 |
1.05 |
97.6 |
8.8 |
44.8 |
4.6 |
| 4 |
20 |
160 |
4 |
25.8 |
1.09 |
97.6 |
7.4 |
45.6 |
6.2 |
| 5 |
25 |
160 |
4 |
19.6 |
1.13 |
97.7 |
5.6 |
46.8 |
7.4 |
| 6 |
30 |
160 |
4 |
14.3 |
1.07 |
98.8 |
8.7 |
45.3 |
4.2 |
| 7 |
10 |
120 |
4 |
33.8 |
1.31 |
99.0 |
4.1 |
53.5 |
9.1 |
| 8 |
10 |
140 |
4 |
35.4 |
1.14 |
96.3 |
8.9 |
46.3 |
4.9 |
| 9 |
10 |
180 |
4 |
37.9 |
0.83 |
90.1 |
14.2 |
29.0 |
3.0 |
| 10 |
10 |
200 |
4 |
39.2 |
0.76 |
91.6 |
13.1 |
23.4 |
2.6 |
| 11 |
10 |
120 |
6 |
40.4 |
1.33 |
99.6 |
3.0 |
53.4 |
11.8 |
| 12 |
10 |
120 |
8 |
40.7 |
1.40 |
99.2 |
6.0 |
55.2 |
5.5 |
| 13 |
10 |
120 |
10 |
41.1 |
1.39 |
99.6 |
3.7 |
55.1 |
11.1 |
| 14 |
10 |
120 |
12 |
41.6 |
1.35 |
99.4 |
7.4 |
53.8 |
3.7 |
3.4. Effect of reaction parameters on EMF synthesis from fructose
To achieve the highest possible yield of EMF, the effects of reaction parameters including reaction temperature and catalyst dosage on the conversion of fructose into EMF were studied as a function of reaction time. Fig. 3 exhibits the effect of reaction temperature (90–130 °C) on the yield of products. Rising temperature can greatly increase the reaction rate, and the yield of EMF increased rapidly in the initial reaction stage. However, when temperatures exceed 110 °C, the yield of EMF first increased and then decreased with the time continued, and the yield of HMF was significantly reduced. Instead, the production of EL was favored when elevating temperature and prolonging time. These findings showed that HMF and EMF are instable at elevated temperature, which is readily further degraded to form stabilized EL.33 Hence, the elevation of temperature is also unfavorable for the extent of reaction, and the optimized temperature was set to 110 °C in this reaction.
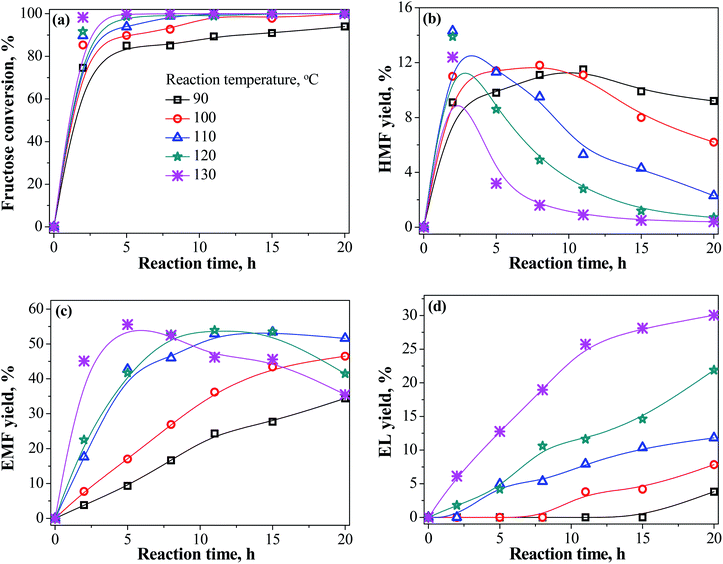 |
| | Fig. 3 Effect of reaction temperature on the yield of products (EMF, HMF, and EL) from fructose conversion in ethanol medium as a function of time. Reaction conditions: 1.08 g fructose, 0.2 g LS-SO3H, 30 mL ethanol. | |
Fig. 4 presents the influence of catalyst dosage on the conversion of fructose to EMF. Fructose was almost entirely consumed within a short reaction time of 2 h for all test catalyst dosages. When the catalyst dosage was 0.05 g, the yield of EMF increased slowly and a relatively high HMF yield of 17.7% could be obtained after 20 h. With increasing the catalyst dosage, the increased total number of available acid sites result in a faster reaction rate to facilitate the conversion of fructose to EMF. The catalyst dosage had a similar influence rule as the temperature for fructose conversion and the yield of products. The highest EMF yield of 57.3% was achieved at 110 °C for 11 h with the catalyst loading of 0.4 g. However, the equilibrium yield of EMF showed no remarkable difference between the catalyst loading of 0.4 and 0.2 g. Therefore, low catalyst loading of 0.2 g was used in the next experiments.
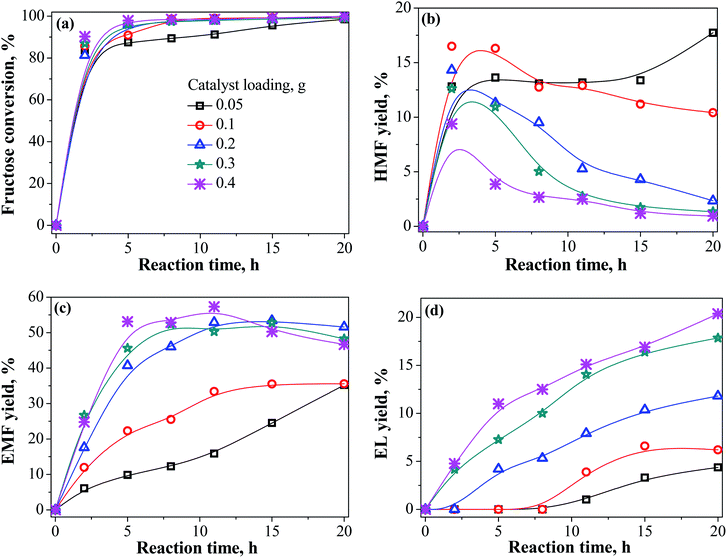 |
| | Fig. 4 Effect of catalyst loading on the yield of products (EMF, HMF, and EL) from fructose conversion in ethanol medium as a function of time. Reaction conditions: 1.08 g fructose, 30 mL ethanol, 110 °C. | |
3.5. Reusability of catalyst
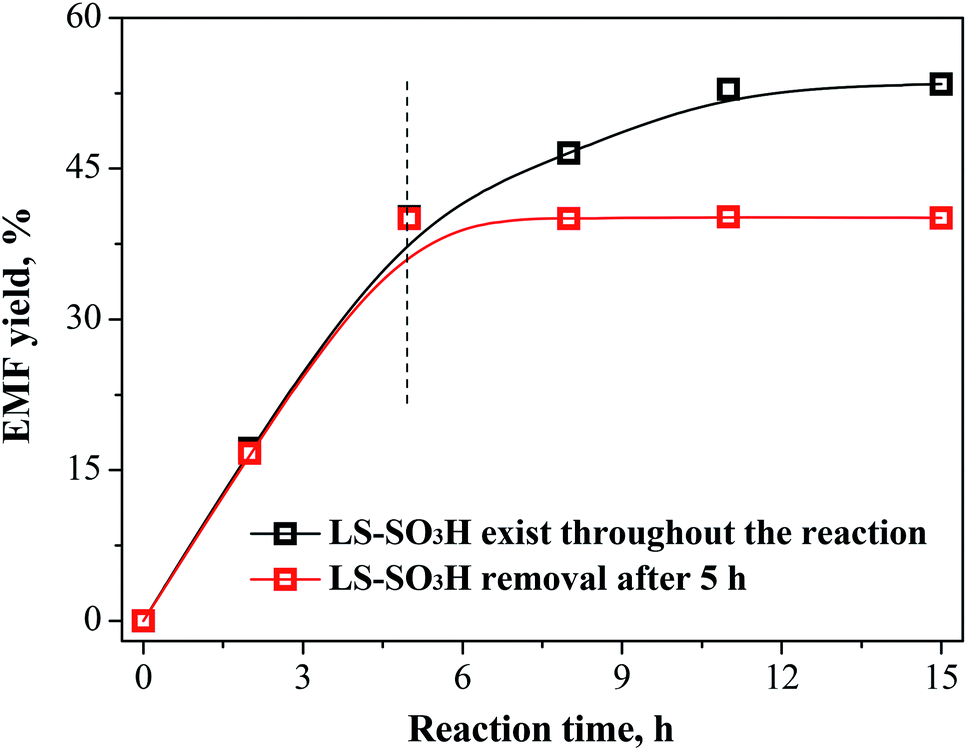
The recycle experiments of LS-SO3H catalyst were carried out for the conversion of fructose in ethanol medium to synthesize EMF. The used LS-SO3H catalyst could be sufficiently separated from the resulting reaction mixture by filtration. After cleaning and drying, it was directly employed in a new experiment under identical reaction conditions. As seen from Fig. 5, the recovered LS-SO3H remained catalytic active for each recycle run, although the EMF yield decreased from 53.4% (Run 1) to 48.8% (Run 2) and 34.3% (Run 3). To explore the stability of acid groups on the catalyst, the density of acid groups on the used catalyst after the third recycle run was measured (Table 3), and it was found that the –SO3H density on the used catalyst (0.82 mmol g−1) declined compared with the fresh catalyst (1.33 mmol g−1). It suggests that the shedding of functional groups from the LS-SO3H might be occurred. However, a verification experiment revealed in Fig. 6 that the yield of EMF remained constant over the reaction time when the LS-SO3H catalyst was removed after 5 h, implying that no active groups exist in the homogeneous system for the reaction. The decreases of –SO3H density and catalytic activity of the recovered LS-SO3H did not major caused by the leaching of acid groups. On the other hand, the catalyst recovery rate, which is defined as the weight ratio of the recovered catalyst to the initial catalyst load in the feed, was determined to be 110% after the first run. This is likely due to the adsorption and deposition of humin products on the surface of catalyst,34 thus resulting in the partly shielding of surface acid sites and the decrease of catalytic activity for the recovered LS-SO3H. Fortunately, the slightly-deactivated catalyst could be easily regenerated by simple treatment in concentrated H2SO4 solution at 120 °C for 1 h, and a similar high EMF yield of 56.8% was regained in a subsequent recycling experiment (Fig. 5, Run 4).
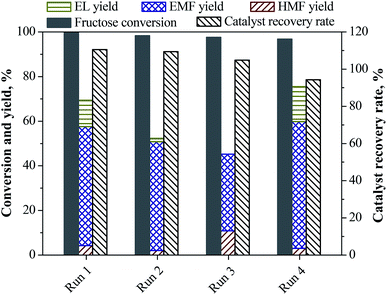 |
| | Fig. 5 Reusability of the LS-SO3H catalyst on the conversion of fructose into EMF. Reaction conditions: 1.08 g fructose, 0.2 g LS-SO3H, 30 mL ethanol, 110 °C, 15 h. The recovered catalyst was used without any pretreatment in the first three runs, and the fourth used catalyst was regenerated at 120 °C for 1 h in concentrated H2SO4 solution. Catalyst recovery rate was defined as the weight ratio of the recovered catalyst to the initial catalyst load in the first run. | |
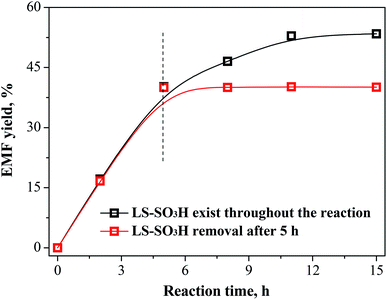 |
| | Fig. 6 The experiment results for the synthesis of EMF from fructose after the removal of LS-SO3H at 5 h. Reaction conditions: 1.08 g fructose, 0.2 g LS-SO3H, 30 mL ethanol, 110 °C. | |
3.6. Synthesis of EMF from various biorenewable feedstocks
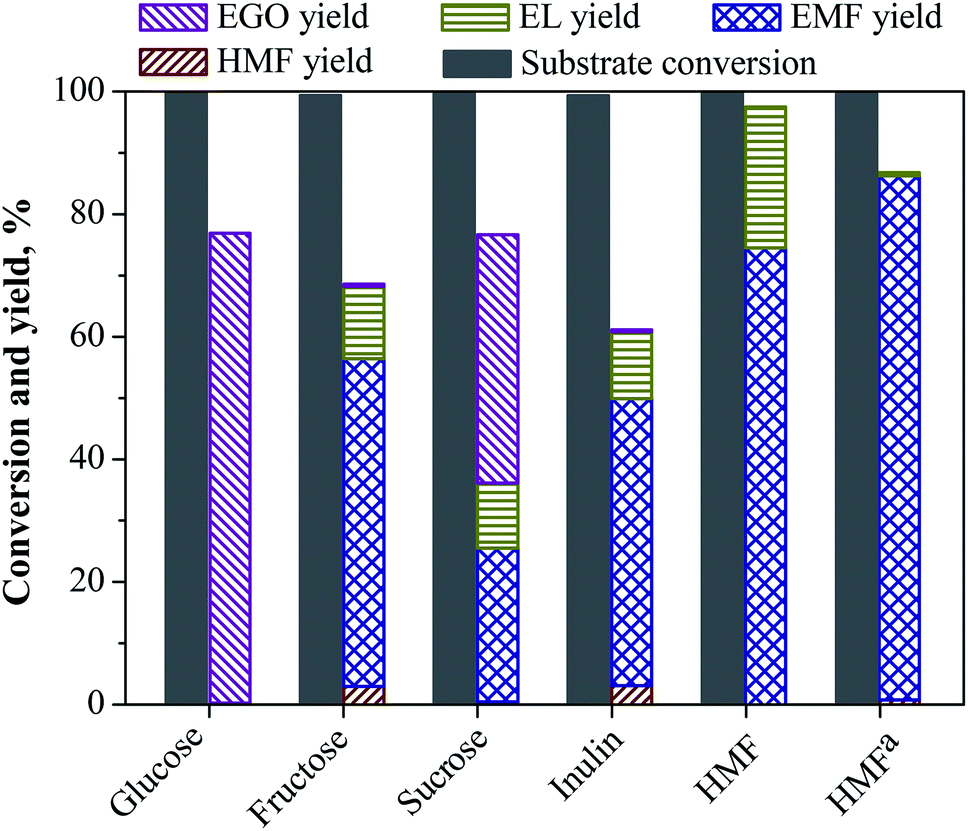
To explore the application scope of the catalyst system for the production of EMF catalyzed by LS-SO3H, other biorenewable feedstocks such as HMF, inulin, sucrose and glucose were subjected as the starting material, and the results are shown in Fig. 7. The conversion of HMF gave 74.5% of EMF yield, along with 22% yield of EL at 110 °C for 15 h, indicating that HMF was easily translated into EMF by etherification reaction. Further, an improved EMF yield of 85.5% could be achieved under milder conditions at 80 °C for 11 h. When glucose was subjected to our reaction system, the reactant was almost entirely converted. However, hardly any furans products (i.e., EMF, and HMF) were observed, and instead ethyl glucoside (EGO) with a high yield of 77% yield was formed. This result clearly indicates that the LS-SO3H catalyst did not work for the isomerization of glucose to fructose, and EGO was obtained preferentially from the direct etherification of glucose and fairly stable in the reaction system. For sucrose, a moderate yield of EMF (25%) and EGO (41%) could be achieved, it follows that the conversion of sucrose followed the accumulation of the reaction pathways of fructose and glucose. In this case, the EMF was formed from the fructose unit in sucrose and the EGO was formed from the glucose unit in sucrose. Interestingly, when inulin was used as the substrate, a good EMF yield of 46.8% could also be obtained, although it was slightly lower than that from fructose (53.4%). Consequently, these results suggest that the LS-SO3H catalyst could not only catalyze the dehydration of fructose and the etherification of HMF, but also facilitate the hydrolysis of inulin into fructose.
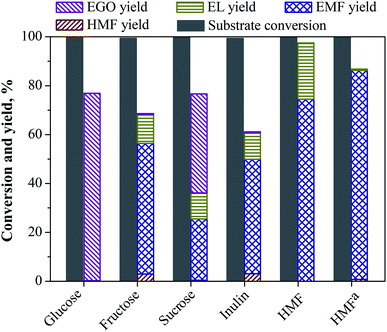 |
| | Fig. 7 Synthesis of EMF using various biorenewable feedstocks in ethanol medium over LS-SO3H. Reaction conditions: 1.08 g substrate, 0.2 g LS-SO3H, 30 mL ethanol, 110 °C, 15 h. aThe reaction was performed at 80 °C for 11 h. | |
3.7. Synthesis of HMF from fructose
The catalytic activity of LS-SO3H catalyst was further evaluated for the dehydration of fructose into HMF as far as possible, and various solvents including DMF, THF and DMSO were used as the reaction medium. As shown in Table 5, the HMF could be produced from fructose in these solvents media catalyzed by LS-SO3H. A high HMF yield of 83.1% was obtained in the presence of DMSO at 140 °C for 3 h, which was comparable with the previous obtained by carbonaceous solid acid catalysts.14e,35 In addition, a good yield of HMF was also achieved at lower temperature of 80 °C after 16 h. By contrast, HMF was formed in traces without catalyst. It follows that the LS-SO3H can also act as an efficient catalyst for the conversion of fructose to HMF.
Table 5 Dehydration of fructose into HMF in different solvents over LS-SO3H catalysta
| Entry |
Solvent |
Temperature, °C |
Time, h |
Conversion, % |
HMF yield, % |
| Reaction conditions: 1.08 g fructose, 0.2 g LS-SO3H, 30 mL solvent. The reaction was carried out without catalyst. |
| 1 |
DMF |
140 |
3 |
98.1 |
45.1 |
| 2 |
THF |
140 |
3 |
90.9 |
19.7 |
| 3 |
DMSO |
140 |
3 |
98.4 |
83.1 |
| 4b |
DMSO |
140 |
3 |
72.3 |
3.9 |
| 5 |
DMSO |
80 |
16 |
97.6 |
63.8 |
4. Conclusions
The LS-SO3H catalyst was efficiently synthesized by a foolproof one-step process of directly sulfonating sodium lignosulfonate. The catalyst preparation conditions (i.e., H2SO4 dosage, temperature, and time) markedly affected the preparation and properties of LS-SO3H. The as-prepared LS-SO3H catalyst was found to be highly active in the conversion of fructose into furan derivatives, giving a 57% yield of EMF in ethanol and a 83% yield of HMF in DMSO, respectively. Also, this catalyst was efficient for the one-pot transformation of fructose-based inulin, and a 47% yield of EMF could be achieved in ethanol medium. The used LS-SO3H catalyst was readily separated from the resulting reaction mixture by filtration, and reused with slight deactivation that is attributed to the adsorption of humin by-products on the catalyst surface. The activity of slightly-deactivated catalyst could be completely regained by a simple re-doping process in concentrated H2SO4 solution. In summary, this study not only develops an easy-prepared and low-cost recyclable solid acid catalyst for the efficient synthesis of promising furan derivatives from biorenewable feedstocks, but also provides a novel and complementary strategy for the resource utilization of pulping by-product lignosulfonate.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to the financial support from the National Natural Science Foundation of China (21566019).
Notes and references
-
(a) J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed;
(b) K. Srirangan, L. Akawi, M. Moo-Young and C. P. Chou, Appl. Energy, 2012, 100, 172–186 CrossRef.
-
(a) X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS;
(b) A. A. Rosatella, S. P. Simeonov, R. F. Frade and C. A. Afonso, Green Chem., 2011, 13, 754–793 RSC;
(c) B. Agarwal, K. Kailasam, R. S. Sangwan and S. Elumalai, Renewable Sustainable Energy Rev., 2018, 82, 2408–2425 CrossRef CAS.
- S. Alipour, H. Omidvarborna and D. S. Kim, Renewable Sustainable Energy Rev., 2017, 71, 908–926 CrossRef CAS.
-
(a) A. Liu, Z. Zhang, Z. Fang, B. Liu and K. Kuang, J. Ind. Eng. Chem., 2014, 20, 1977–1984 CrossRef CAS;
(b) G. Morales, M. Paniagua, J. A. Melero and J. Iglesias, Catal. Today, 2017, 279, 305–316 CrossRef CAS.
-
(a) C. M. Lew, N. Rajabbeigi, M. Tsapatsis and M., Ind. Eng. Chem. Res., 2012, 51, 5364–5366 CrossRef CAS;
(b) J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC.
- A. Osatiashtiani, A. F. Lee, D. R. Brown, J. A. Melero, G. Morales and K. Wilson, Catal. Sci. Technol., 2014, 4, 333–342 CAS.
-
(a) Y. Yang, M. M. Abu-Omar and C. Hu, Appl. Energy, 2012, 99, 80–84 CrossRef CAS;
(b) Y. Ren, B. Liu, Z. Zhang and J. Lin, J. Ind. Eng. Chem., 2015, 21, 1127–1131 CrossRef CAS.
-
(a) B. Liu, Z. Zhang and K. Deng, Ind. Eng. Chem. Res., 2012, 51, 15331–15336 CrossRef;
(b) A. Herbst and C. Janiak, New J. Chem., 2016, 40, 7958–7967 RSC.
-
(a) Z. Yuan, Z. Zhang, J. Zheng and J. Lin, Fuel, 2015, 150, 236–242 CrossRef CAS;
(b) J. Wang, Z. Zhang, S. Jin and X. Shen, Fuel, 2017, 192, 102–107 CrossRef CAS.
-
(a) B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24–38 RSC;
(b) B. Liu and Z. Zhang, ChemSusChem, 2016, 9, 2015–2036 CrossRef CAS PubMed.
-
(a) F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS;
(b) N. E. EI Mansouri and J. Salvadó, Ind. Crops Prod., 2006, 24, 8–16 CrossRef.
-
(a) P. J. M. Carrott and M. R. Carrott, Bioresour. Technol., 2007, 98, 2301–2312 CrossRef PubMed;
(b) S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS;
(c) B. M. Upton and A. M. Kasko, Chem. Rev., 2015, 116, 2275–2306 CrossRef PubMed.
-
(a) M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS;
(b) P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS.
-
(a) F. L. Pua, Z. Fang, S. Zakaria, F. Guo and C. H. Chia, Biotechnol. Biofuels, 2011, 4, 56 CrossRef PubMed;
(b) F. Guo, Z. L. Xiu and Z. X. Liang, Appl. Energy, 2012, 98, 47–52 CrossRef CAS;
(c) S. Kang, J. Ye, Y. Zhang and J. Chang, RSC Adv., 2013, 3, 7360–7366 RSC;
(d) F. Liang, Y. Song, C. Huang, J. Zhang and B. Chen, Catal. Commun., 2013, 40, 93–97 CrossRef CAS;
(e) L. Hu, X. Tang, Z. Wu, L. Lin, J. Xu, N. Xu and B. Dai, Chem. Eng. J., 2015, 263, 299–308 CrossRef CAS;
(f) S. Hu, F. Jiang and Y. L. Hsieh, ACS Sustainable Chem. Eng., 2015, 3, 2566–2574 CrossRef CAS;
(g) M. Huang, J. Luo, Z. Fang and H. Li, Appl. Catal., B, 2016, 190, 103–114 CrossRef CAS;
(h) J. Zhu, L. Gan, B. Li and X. Yang, Korean J. Chem. Eng., 2016, 34, 110–117 CrossRef.
- A. Duval, S. Molina-Boisseau and C. Chirat, Ind. Crops Prod., 2013, 49, 66–74 CrossRef CAS.
-
(a) D. Y. Kim, Y. Nishiyama, M. Wada and S. Kuga, Cellulose, 2001, 8, 29–33 CrossRef CAS;
(b) J. Laohapornchaiphan and C. B. Smith, Chem.–Asian J., 2017, 12, 3178–3186 CrossRef CAS PubMed;
(c) I. F. Nata, M. D. Putra, C. Irawan and C. K. Lee, J. Environ. Chem. Eng., 2017, 5, 2171–2175 CrossRef CAS;
(d) X. Liu, L. Zhang, W. Sun, M. Zhang and S. Yu, Res. Chem. Intermed., 2017, 43, 1–16 CrossRef.
- R. J. A. Gosselink, E. De Jong, B. Guran and A. Abächerli, Ind. Crops Prod., 2004, 20, 121–129 CrossRef CAS.
-
(a) A. Nadif, D. Hunkeler and P. Käuper, Bioresour. Technol., 2002, 84, 49–55 CrossRef CAS PubMed;
(b) T. Danner, H. Justnes, M. Geiker and R. A. Lauten, Cem. Concr. Res., 2016, 79, 333–343 CrossRef CAS.
- L. J. Konwar, J. Boro and D. Deka, Renewable Sustainable Energy Rev., 2014, 29, 546–564 CrossRef CAS.
- J. Wang, W. Xu, J. Ren, X. Liu, G. Lu and Y. Wang, Green Chem., 2011, 13, 2678–2681 RSC.
-
(a) B. Liu, Z. Zhang, K. Huang and Z. Fang, Fuel, 2013, 113, 625–631 CrossRef CAS;
(b) X. Yu, X. Gao, R. Tao and L. Peng, Catalysts, 2017, 7, 182 CrossRef.
- L. Wang, X. Dong, H. Jiang, G. Li and M. Zhang, Bioresour. Technol., 2014, 158, 392–395 CrossRef CAS PubMed.
- W. J. Liu, K. Tian, H. Jiang and H. Q. Yu, Sci. Rep., 2013, 3, 2419 CrossRef PubMed.
- N. Tsubouchi, C. Xu and Y. Ohtsuka, Energy Fuels, 2003, 17, 1119–1125 CrossRef CAS.
-
(a) H. Zhao, J. H. Kwak, Z. C. Zhang, H. M. Brown, B. W. Arey and J. E. Holladay, Carbohydr. Polym., 2007, 68, 235–241 CrossRef CAS;
(b) F. Kallel, F. Bettaieb, R. Khiari, A. García, J. Bras and S. E. Chaabouni, Ind. Crops Prod., 2016, 87, 287–296 CrossRef CAS.
- Y. Wang, L. Lin, B. S. Zhu, Y. X. Zhu and Y. C. Xie, Appl. Surf. Sci., 2008, 254, 6560–6567 CrossRef CAS.
- K. Nakajima and M. Hara, ACS Catal., 2012, 2, 1296–1304 CrossRef CAS.
- X. Li, Y. Jiang, L. Shuai, L. Wang, L. Meng and X. Mu, J. Mater. Chem., 2012, 22, 1283–1289 RSC.
- G. Chen and B. Fang, Bioresour. Technol., 2011, 102, 2635–2640 CrossRef CAS PubMed.
- T. Chen, L. Peng, X. Yu and L. He, Fuel, 2018, 219, 344–352 CrossRef CAS.
- H. Wikberg, T. Ohra-aho, F. Pileidis and M. M. Titirici, ACS Sustainable Chem. Eng., 2015, 3, 2737–2745 CrossRef CAS.
- X. Dong, Y. Jiang, W. Shan and M. Zhang, RSC Adv., 2016, 6, 17118–17124 RSC.
- M. Balakrishnan, E. R. Sacia and A. T. Bell, Green Chem., 2012, 14, 1626–1634 RSC.
- X. Qi, N. Liu and Y. Lian, RSC Adv., 2015, 5, 17526–17531 RSC.
- F. Guo, Z. Fang and T. J. Zhou, Bioresour. Technol., 2012, 112, 313–318 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Lincai Peng
,
Lincai Peng