
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfur-doped mesoporous carbon via thermal reduction of CS2 by Mg for high-performance supercapacitor electrodes and Li-ion battery anodes†
Lu Suna,
Jinzhang Liu *a,
Zehui Liua,
Teng Wangb,
Hongxia Wangb and
Yan Li*a
*a,
Zehui Liua,
Teng Wangb,
Hongxia Wangb and
Yan Li*a
aSchool of Materials Science and Engineering, Beihang University, Beijing 100191, China. E-mail: ljz78@buaa.edu.cn; liyan@buaa.edu.cn
bSchool of Chemistry, Physics, and Mechanical Engineering, Queensland University of Technology, Brisbane, 4001 QLD, Australia
First published on 30th May 2018
Abstract
This paper demonstrates a facile method based on vapor–solid reaction between magnesium powder and carbon disulfide vapor to produce S-doped porous carbon. The property of the as-prepared carbon is tunable by varying the synthesis temperature. The sample synthesized at 600 °C shows the highest specific surface area, suitable for supercapacitor electrodes. A high specific capacitance of 283 F g−1 in H2SO4 aqueous electrolyte is achieved. The best performance of porous carbon for a Li-ion battery anode is obtained at the optimal temperature of 680 °C. Owing to the well-balanced soft and hard carbon compositions in the material, this porous carbon exhibits a high reversible capacity of 1440 mA h g−1 and excellent rate performance.
1. Introduction
Carbon materials play an important role in electrochemical energy storage devices.1 The electrodes of commercial supercapacitors are based on porous activated carbon (AC), which possesses high specific surface areas for ensuring high capacitance because the energy storage depends on the physical adsorption of ions from the electrolyte onto the electrode surface when charged. Supercapacitors have advantages over conventional batteries such as fast charge/discharge rate, high power density, and long cycling life. However, due to the unsatisfactory specific capacitance of the AC-based electrode, commercial supercapacitors suffer from low energy density, which is about one twentieth of that of a Li-ion battery (LIB). The most common anode material of LIBs is graphite, in which Li ions are electrochemically intercalated between the graphite layer during the charging process. Although LIBs have been widely used in practice, today's battery technology still lags behind the requirement of energy storage for portable information devices. This obstacle mainly comes from the limit of energy storage capability of the graphite anode, which has a theoretical specific capacity of only 372 mA h g−1. Hence, new LIB anodes with higher specific capacity are in high demand.Carbon materials are diverse in terms of allotrope and their physical properties. Among different carbon materials, graphene has been considered as an ideal candidate to replace AC for making high-capacitance supercapacitor electrodes,2,3 as the theoretical specific capacitance of graphene is 550 F g−1. Nevertheless, though much efforts have been paid in this area, the specific capacitance of graphene-based electrode is only in the range of 200–300 F g−1 in most reports.4,5 One reason for the much lower performance of graphene compared to its theoretical value is the low quality of graphene material which is normally derived from graphene oxide prepared by the well-known Hummer's method. Other reasons such as restacking of graphene sheets due to strong π–π interaction are also responsible for the unsatisfactory electrochemical performance of graphene-based electrodes.6 For LIB anodes, though researchers have investigated alternative materials, such as Si,7 Ge,8 and Sn,9 to replace graphite to enhance the capacity, other carbon structures that enable higher capacity than that of graphite are more attractive because of the natural advantage of carbon in aspect of material abundance, resistance to oxidation, and low cost. Hard carbon, also called non-graphitizable carbon, consists of randomly oriented graphitic layers and can provide much higher Li+ storage capacity compared to graphite.10 However, the disordered microstructure of hard carbon leads to low conductivity and poor electrochemical stability. Soft carbon, on the other hand, has numerous intercalation channels for Li+ transport and has a low reaction voltage profile owing to its amorphous feature. It is a suitable choice to achieve good rate capability and cycle performance of LIBs, while its specific capacity is relatively low. Therefore, a method for synthesizing carbonaceous materials containing both hard and soft carbons is of great interest. In most previous reports, a top-down strategy is used to prepare carbonaceous materials for electrochemical energy storage application. For instance, natural graphite is converted into graphene sheets for supercapacitors, and hard carbon for LIB anodes is generally via the pyrolysis of resin.11 In this work, we report a bottom-up method to grow S-doped porous carbon composed of both soft and hard carbons. This method utilizes the chemical reaction between Mg powder and CS2 vapor, which is very effective in terms of producing carbon materials with controlled properties to meet requirements of either supercapacitors electrodes or LIB anodes. This method also has the advantage of doping sulfur in carbon structures due to the use of CS2 precursor. Carbon structures doped with other elements, such as N and S, could exhibit enhanced performance when used in supercapacitor electrodes or LIB anodes. For instance, porous N-doped carbon derived from silk was used to make both supercapacitor electrode and LIB anode, showing 242 F g−1 in ionic liquid electrolyte and a reversible capacity of 1865 mA h g−1, respectively.12 The effect of S-doping in amorphous carbon or graphene on enhancing the capacitance and Li-ion storage capacity was also demonstrated.13,14
Previously, researchers reported the preparation of few-layer graphene by using active metals, such as Li,15 Na,16 and Mg,17 to thermally reduce CO2. The chemical reaction between Mg and CO2 occurs at high temperatures above the melting point of Mg (650 °C). Chakrabarti et. al. reported the production of few-layer graphene by burning Mg ribbon inside a dry ice bowl.18 In their product a small amount of MgO nanoparticles embedded in the carbon product was difficult to remove. Xing et al. heated a Mg/Zn powder mixture in a CO2 flow at 680 °C to produce nanoporous graphene.19 Their product, though after an acid purification process, contained ZnO nanoparticles wrapped by carbon. In our work, the CS2 vapor can be thermally reduced by Mg at lower temperature compared to CO2, according to the reaction Gibbs energy. The formation of porous carbon by the Mg-CS2 system even can occur at temperature as low as 520 °C, and the byproduct of MgS can be thoroughly removed using acid. Hence, there is a wider temperature range for implementing this reaction and tuning the morphology and microstructure of resultant carbon products. In addition, the splitting of CS2 enables S-dopant in the resultant carbon material, introducing more active sites for enhanced electrochemical energy storage performance.
2. Experimental section
2.1. Materials synthesis
The experimental setup is illustrated in Fig. 1. In a typical experiment, an alumina boat containing 4 g Mg powder was firstly loaded into the central region of a tube furnace. The tube was then evacuated by a rotary pump to remove oxygen. After this, the tube was heated up with heating rate of ∼12 °C min−1 and the inner pressure was maintained to be minimum with the help of continuous pumping. When reaching to 600 °C, Ar flow together with CS2 vapor were supplied into the tube at a constant flow rate and the pressure was adjusted at 20 kPa. The bubbling CS2 liquid was maintained at 30 °C by using a water bath, and the reaction temperature was kept at 600 °C for 90 min. The hose connecting to the pump for gas outlet was placed inside a fume hood, and unreacted CS2 vapor was captured by porous activated carbon while flowing out. After the reaction, the tube was naturally cooled down and the black product was weighed to deduce the percentage of Mg that participated in the reaction 2Mg + CS2 → 2MgS + C. The calculated carbon weight from the mass gain of the powder in alumina boat is equivalent to the actual weight of purified carbon product. In general, ∼95% Mg can be consumed after one experimental process. For purification, the product was washed with water and HCl solution in sequence, and filtered. The black product was re-dispersed in diluted HNO3 and magnetically stirred for 30 min to completely remove residues of Mg compounds, then filtered and washed with de-ionized water thoroughly. Finally, the wet product was freeze-dried. This sample is named as S600. Other samples were prepared in a similar way, except for different reaction temperature employed.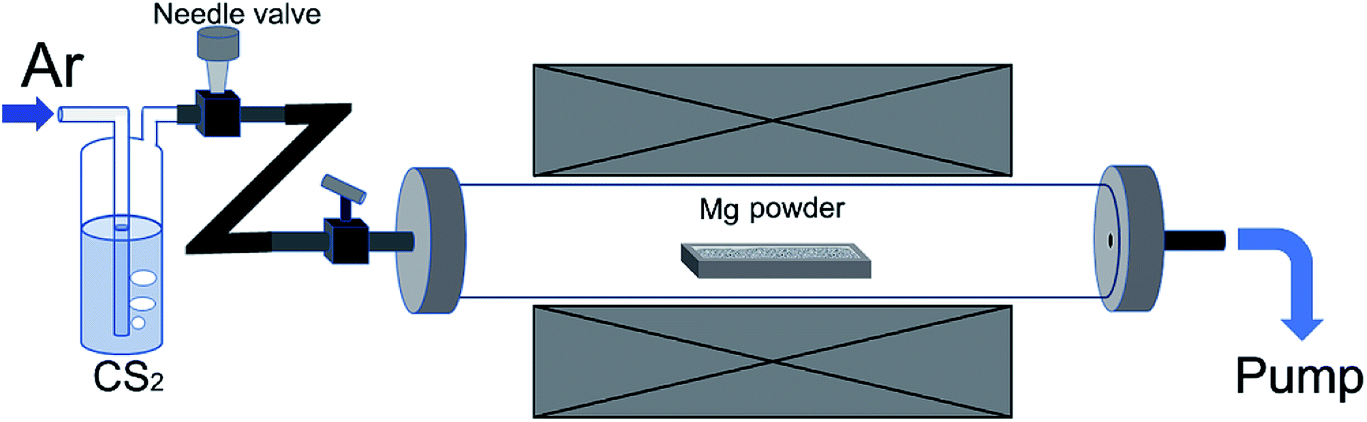 | ||
| Fig. 1 Illustration for the experimental setup. | ||
2.2. Materials characterization
Morphologies of the carbon products were studied by scanning electron microscopy (SEM, Zeiss Supra55) and transmission electron microscopy (TEM, FEI Tecnai F30). Raman spectroscopy (Horiba Jobin-Yvon HR800, laser wavelength 523 nm), X-ray photoelectron spectroscopy (XPS, Thermo Scientific Escalab 250Xi), and X-ray diffraction (XRD, D/MAX-2500) were also employed to determine chemical composition and chemical state as well as material structure of the samples.2.3. Electrode preparation and electrochemical test
The obtained carbon powder was blended with carbon black and polyvinylidene fluoride at a mass ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. N-Methyl-2-pyrrolidone solvent was added to the blend and mixed to make a slurry, which was subsequently blade-coated onto a Pt foil and dried to make the electrode. The areal density of active material was about 1.2 mg cm−2. In a three-electrode cell containing 1 M H2SO4 aqueous electrolyte, the carbon electrode was used as working electrode, and a Pt foil was the counter electrode. The reference electrode was Ag/AgCl.
:1:1. N-Methyl-2-pyrrolidone solvent was added to the blend and mixed to make a slurry, which was subsequently blade-coated onto a Pt foil and dried to make the electrode. The areal density of active material was about 1.2 mg cm−2. In a three-electrode cell containing 1 M H2SO4 aqueous electrolyte, the carbon electrode was used as working electrode, and a Pt foil was the counter electrode. The reference electrode was Ag/AgCl.
For LIB, half cells were made using 2032 coin-type cases and LiPF6-based organic electrolyte. The anode was composed of 80 wt% of as-synthesized S-doped porous carbon, 10 wt% carbon black and 10 wt% poly (vinyl difluoride) as binder. The mixed slurry was pasted onto a copper foil, following a dry process at 120 °C for 12 h in vacuum. Pure Li foil was used as the counter electrode, and the separator was a disk-shaped polypropylene film. The charge and discharge measurements were carried out on Land CT 2001A system at various current densities in the voltage range of 0.01–3.0 V versus Li/Li+.
3. Results and discussion
3.1. Morphological and structural studies
Two representative samples grown at 600 and 680 °C, named as S600 and S680, respectively, are compared in Fig. 2. The low- and high-magnification scanning electron microscopy (SEM) images of the S600 sample shown in Fig. 2a and b reveal a porous structure consisting of interconnected carbon sheets. Fig. 2c shows the high-resolution transmission electron microscopy (HRTEM) image of this sample. Disordered graphitic layers in a matrix of amorphous carbon are disclosed. The SEM images in Fig. 2d and e correspond to S680, which has a similar porous structure as S600. During the reaction process, carbon sheets and MgS were produced simultaneously. The porous carbon structure was left after the removal of MgS in the purification process. However, the HRTEM image in Fig. 2f shows more distinct textures of atomic layers that are bundled up and randomly oriented. The interspace between two layers is 0.33 nm, in good agreement with interspace of [002] planes of graphite. But the crystalline carbon layers are curved, forming a microstructure analogous to that of hard carbon.20 The evolution of a Mg grain in CS2 vapor at high temperature is illustrated in Fig. 2g. The Mg grain is firstly etched by CS2 vapor to form fluffy carbon structures mixed with solid MgS. Gaps between patches of carbon hulls allow CS2 vapor diffusion to further reduce the size of Mg core, and finally Mg is exhausted. By comparing Fig. 2c and f, it can be concluded that the reaction temperature is a key factor to adjust the portion of soft or hard carbon components.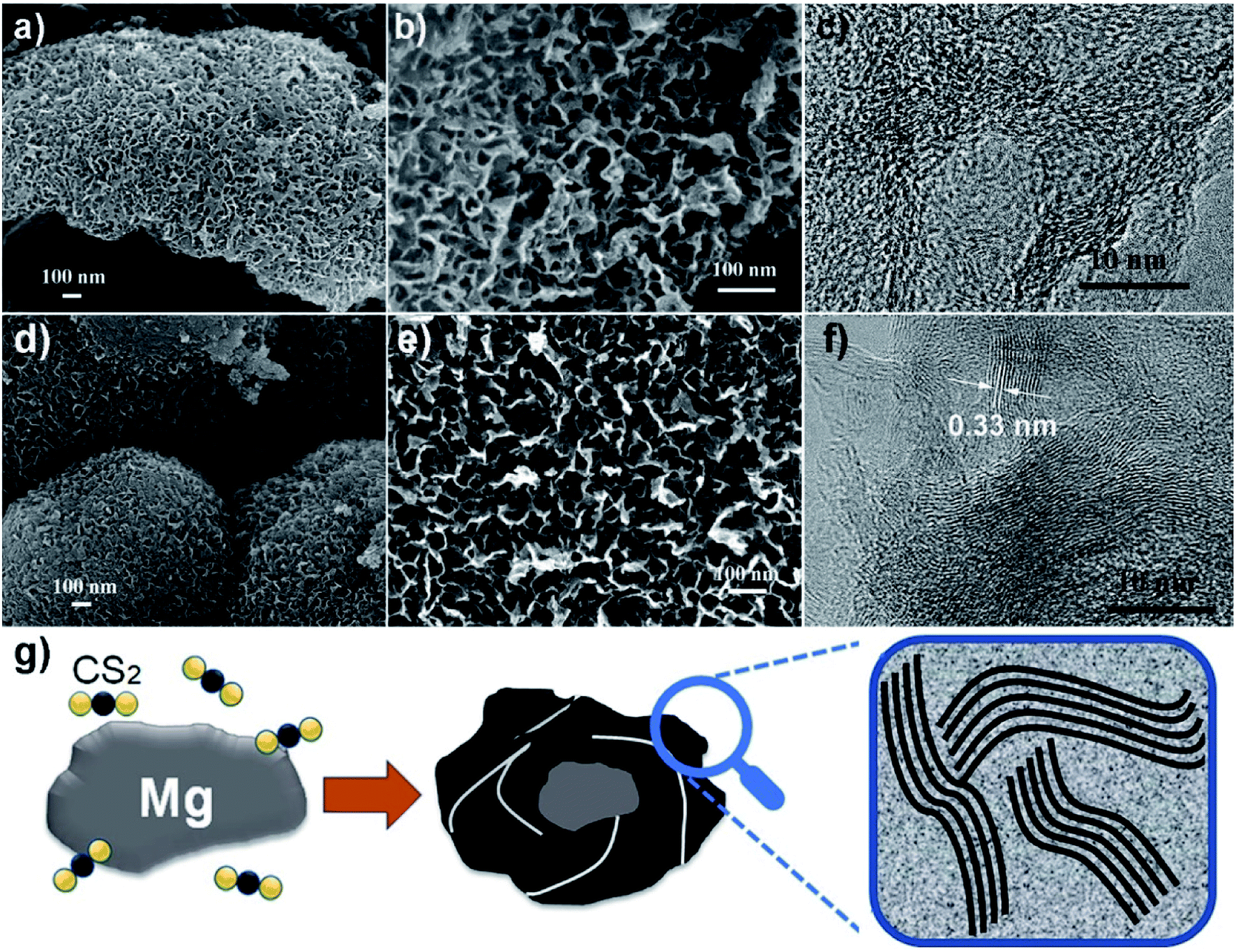 | ||
| Fig. 2 SEM images (a and b) and HRTEM image (c) of the S600 sample. SEM images (d and e) and HRTEM image (f) of the S680 sample. (g) Illustration for the formation of porous carbon from a solid–vapor reaction between Mg and CS2. | ||
The surface area and porosity of the two samples prepared at different temperatures were further investigated by Brunauer, Emmett, and Teller measurement. The nitrogen adsorption/desorption isotherms are compared in Fig. 3a. Apparently, specific surface areas of the two samples are very different. S600 possesses a surface area of 1721 m2 g−1 whereas a much lower surface area of 735 m2 g−1 was obtained with S680. The analysis of the nitrogen adsorption data reveals the mesoporosity of both as-prepared carbon products. Pore size characterizations of the two samples are obtained (Fig. 3b) by using nonlocal density functional theory method. It is found that both samples show pore diameters below 10 nm. However, the S600 has more small pores (less than 2 nm) compared to S680. Fig. 3c and d show XRD and Raman spectra of the two samples, respectively. The two XRD patterns are almost identical except the intensity difference. The broad peak at 22° can be attributed to distorted [002] planes of graphite, and the width of this peak indicates the nanoscale thickness of stacked graphitic layers. For few-layer graphene the (002) diffraction peak is normally obtained at 26° and is sharper.21 From the XRD and TEM analysis, we can conclude that the as-prepared carbon is a blend of soft and hard carbons. First, our XRD patterns resemble those of hard carbon microtubes derived from cotton;22 second, HRTEM images reveal disordered soft carbon around layered carbon atoms. Fig. 3d shows Raman spectra of the two samples. The amorphous structure is confirmed by two separate characteristic bands of D-band (disorder-induced) and G-band (sp2-related) observed with both samples. XPS analysis was employed to investigate the surface chemistry of the as-synthesized carbon materials. Two survey XPS spectra shown in Fig. 3e reveal oxygen and sulfur contents in both samples. The observed oxygen was introduced in the samples during the HNO3-purification process. The S atomic percentages in S600 and S680 samples are 6.0% and 6.6%, respectively. The high-resolution S 2p XPS peaks of S600 and S680 shown in Fig. 3f and g reveal different chemical states of S in the form of S and –SOn–. The signal from –SOn– bonds can be attributed to acid activation of S dopants. However, XPS is responsible for signals from atoms in shallow depth of 0–10 nm beneath the material surface. It is believed that oxygenated groups only reside in the surface and have a minor influence on Li+ intercalation. However, S-doping inside the carbon material can help enhance the capacity when used as LIB anode, which has been demonstrated by S-doped graphene.14,23
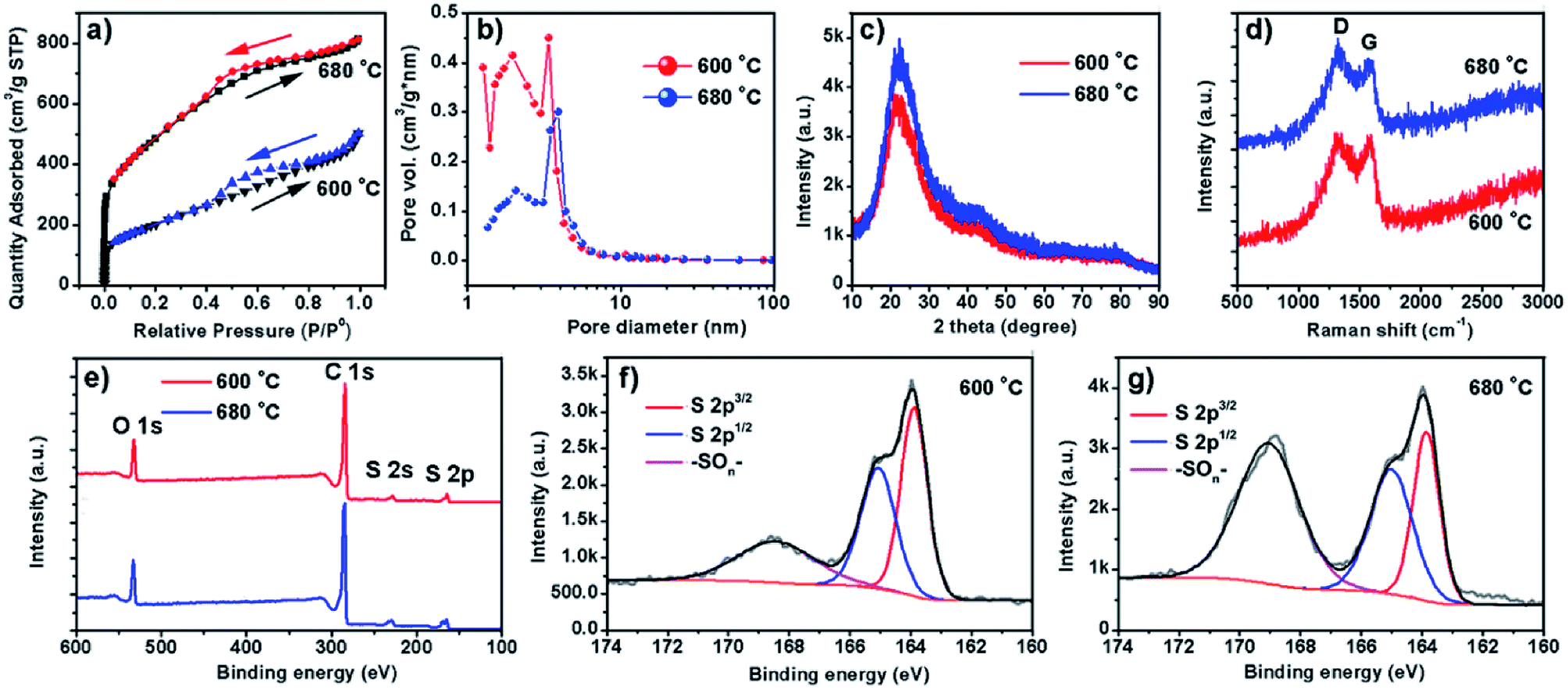 | ||
| Fig. 3 (a) Nitrogen adsorption/desorption plots at room temperature for S600 and S680. (b) BJH pore-size distributions of the two samples in mesopore regime. (c) XRD patterns. (d) Raman analysis. (e) XPS survey spectra. (f and g) High-resolution S 2p XPS spectra of S600 and S680, respectively. | ||
3.2. Electrochemical performance of supercapacitor electrode
Since the carbon powder of S600 has high specific surface area, this sample was used to make supercapacitor electrode and tested in a three-electrode cell. Fig. 4a and b show cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) curves. The CV loops in the voltage range of −0.2 to 0.8 V show a typical quasi-rectangular shape of carbon material, indicating good capacitive behavior. The symmetrical linear shape of the GCD curves is in good agreement with the electrical double layer capacitance of carbon based electrode for supercapacitors. The specific capacitance can be determined by the slope of discharge section. Fig. 4c shows the specific capacitance over a range of different current densities. The electrode exhibits a high specific capacitance of 283 F g−1 at 1 A g−1, which outperforms most graphene-based electrodes reported previously. In 2008, Ruoff et al. for the first time reported supercapacitors based on reduced graphene oxide (RGO), of which the specific capacitance of the RGO electrode was around 100 F g−1 when using KOH aqueous electrolyte.24 In 2013, Duan et al. reported supercapacitor electrodes based on 3D graphene hydrogel films, which showed 196 F g−1 at 1 A g−1 in 1 M H2SO4 aqueous solution.25 In 2014, the same group reported the use of holey graphene to make supercapacitor electrodes, which showed specific capacitance of 310 F g−1 at 1 A g−1 in 6 M KOH aqueous electrolyte.26 In our case, the electrode made of S600 sample also shows excellent cycling performance. After continuously running GCD at 5 A g−1 for 10000 cycles, the retention rate of the material remained 98.5% (Fig. S1†). The excellent electrochemical performance of our carbon material is related with its specific surface area and porous characteristics, which is dependent on the reaction temperature between Mg and CS2. Fig. 4d shows the maximum specific capacitance and the specific surface area of the carbon samples prepared at different temperature. Apparently, 600 °C is the optimal temperature for the formation of porous carbon with the highest specific surface area and specific capacitance. For the few-layer graphene product obtained by exploiting the chemical reacting between Mg and CO2, it showed a specific surface area of 829 m2 g−1. After procced into supercapacitor electrode, the material showed 170 F g−1 in 6 M KOH electrolyte.19 Chang et al. used Na and CO2 as precursors to produce 3D surface-microporous graphene that shows a specific surface area of 890 m2 g−1, and the electrode based on this material showed 175 F g−1 at 1 A g−1 in 2 M KOH aqueous electrolyte.27 Therefore, our method has the advantage of producing mesoporous carbon with much higher specific surface area for enhancing the specific capacitance. On the other hand, sulfur doping to the carbon lattice also plays a key role in enhancing the capacitance. A sulfur atom doped in carbon changes the charge on the neighboring carbon to be positive, which is proposed to enhance the capacitive performances.28,29
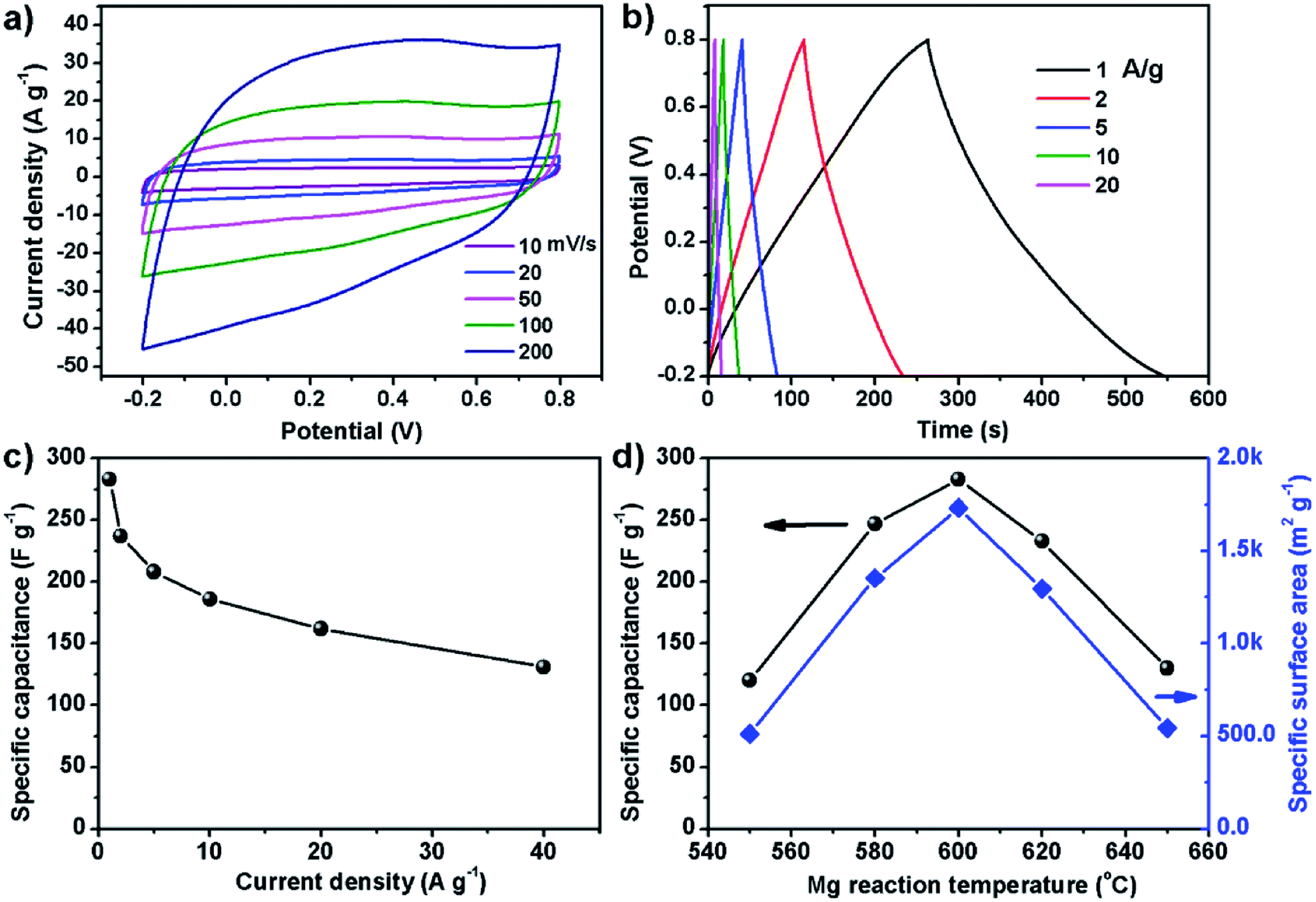 | ||
| Fig. 4 Electrochemical performance of the supercapacitor electrode made of S600 sample. (a) CV curves. (b) Galvanostatic CD curves at different current densities. (c) Specific capacitance versus current density. (d) Carbon preparation temperature dependent specific capacitance of the electrode and specific surface area of the carbon powder. | ||
3.3. Electrochemical performance of LIB anodes
Though the S600 sample has the highest specific capacitance, it is inferior when used as LIB anodes. Instead, a better performance is obtained with the carbon material formed at higher temperature of 680 °C. Fig. 5 shows the electrochemical performance of LIB anode based on the S680 sample. The initial discharge and charge profile measured at 0.05 A g−1 within a voltage range of 0–3 V is displayed in Fig. 5a. This electrode exhibits a reversible specific capacity of 1440 mA h g−1, which is almost four times as much as the theoretical capacity of graphite (372 mA h g−1). This remarkable value indicates a more complicated lithium storage mechanism rather than the graphite intercalation mechanism. Fey et al. prepared LIB anode materials by pyrolysis of rice husk, which naturally contain Si.30 Their hard carbon material showed a high reversible capacity of 1055 mA h g−1. In our work, the S-doping may play an important role in enhancing the capacity of the porous carbon material. Ning et al. prepared S-doped (∼10 at%) porous carbon by carbonization of pitch using MgSO4 whiskers as both templates and S source.14 They found that the Li storage capacity of S-doped carbon is more than double compared to that of the undoped carbon. Apart from the high capacity, the initial coulombic efficiency of our carbon material is 57%, which is much higher than that of recently reported 3D porous carbon (∼29%).31 Moreover, after 20 cycles, the reversible capacity was retained at 1045 mA h g−1 with a 73% capacity retention, whereas the capacity values of S620 and S750 samples were decreased to 892 and 943 mA h g−1, corresponding to capacity retentions of 63% and 70%, respectively (Fig. 5b). The XPS analysis reveals that the sulfur content of S750 sample is equivalent to that of the sample prepared at much lower temperature, like S600. Since all samples have mesoporous feature (Fig. S2†), their different anode performances might be related to the ratio of soft to hard carbons that is dependent on the growth temperature. In addition, the fluffy carbon sheets containing nanoholes (Fig. S3†) provide pathways for efficient ions diffusion inside the anode.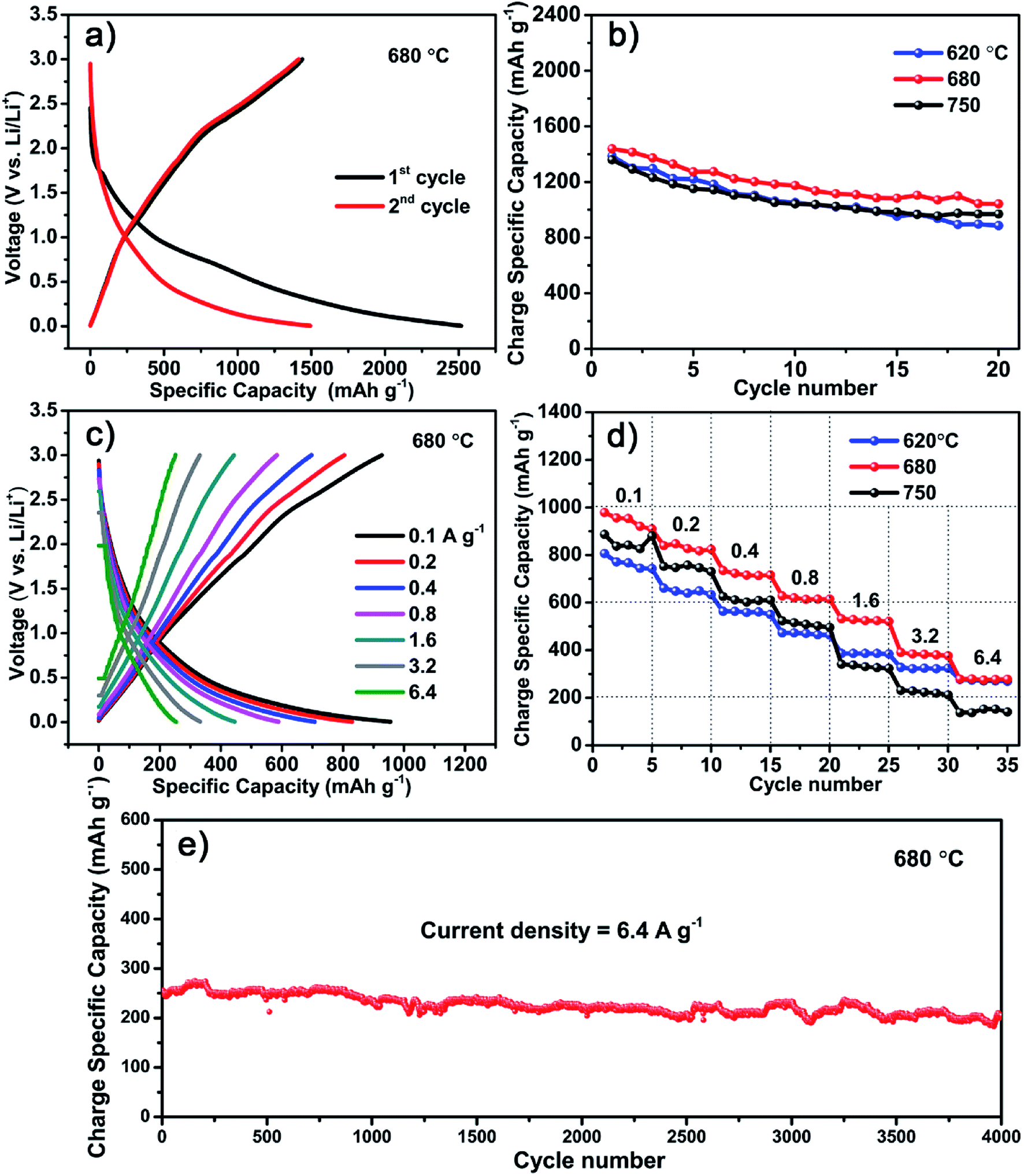 | ||
| Fig. 5 (a) Galvanostatic discharge/charge curves at 50 mA g−1 after the first and second cycles. (b) Cycling performances at 50 mA g−1 for S620, S680, and S750 samples. (c) Galvanostatic discharge/charge profiles of the S680 anode at various current densities gradually increasing from 0.1 to 6.4 A g−1. (d) Rate performances of three different anodes at various rates in A g−1. (e) Superlong cycle performance of the S680 electrode. | ||
Fig. 5c shows the rate performances of the S680 sample tested by charging and discharging the device at different current densities ranging from 0.1 to 6.4 A g−1. For comparison, the specific capacities of the three samples, S620, S680, and S750 at different current densities and after various cycle numbers are shown in Fig. 5d. With increasing the current density, the S750 sample undergoes the largest capacity drop, while the capacity loss of S620 is the smallest. The carbon growth temperature is a key factor influencing the ratio of soft to hard carbons. Higher temperature favors the production of hard carbon component,22 which decreases the rate performance. In S680, the soft and hard carbon components might be well balanced, which results in a high Li storage capacity. The long term electrochemical stability of this sample is remarkable even at a high current density of 6.4 A g−1, as shown in Fig. 5e. After ultralong 4000 cycles, a large reversible capacity of 216 mA h g−1 is obtained with a nearly 100% capacity retention.
4. Conclusions
In summary, S-doped porous carbon was synthesized using a thermal reduction approach involving Mg powder and CS2 based precursors. The merit of this approach is embodied by the controlled properties of resultant carbon materials, of which the specific surface area and microstructure are strongly affected by the growth temperature. The carbon material contains both soft and hard carbons and material structure is in the form of inter-connected nanosheets. It was found that the porous carbon formed at 600 °C has the highest specific surface area of 1730 m2 g−1, and the supercapacitor electrode based on this material shows a high specific capacitance of 283 F g−1 at 1 A g−1 in H2SO4 aqueous electrolyte. The carbon material synthesized at 680 °C exhibits the best electrochemical performance when used as LIB anode despite its much lower specific capacitance. A high reversible capacity of 1440 mA h g−1 was achieved. Furthermore, this anode material also showed good rate performance. We believe that the synthesis approach developed in this work is promising for producing S-doped mesoporous carbon on a large scale for practical applications.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Key R&D Program of China (Grant No. 2016YFE0204200), and National Natural Science Foundation of China (NSFC, Grant No. 51702009 and 21771017).References
- F. Yao, D. T. Pham and Y. H. Lee, ChemSusChem, 2015, 8, 2284 CrossRef PubMed.
- W. K. Chee, H. N. Lim, Z. Zainal, N. M. Huang, I. Harrison and Y. Andou, J. Phys. Chem. C, 2016, 126, 4153 Search PubMed.
- Y. Huang, J. Liang and Y. Chen, Small, 2012, 8, 1805 CrossRef PubMed.
- H. Yang, S. Kannappan, A. S. Oandian, J. H. Jang, Y. S. Lee and W. Lu, J. Mater. Chem. A, 2017, 5, 23720 Search PubMed.
- B. Wang, J. Liu, Y. Zhao, Y. Li, W. Xian, M. Amjadipour, J. MacLeod and N. Motta, ACS Appl. Mater. Interfaces, 2016, 8, 22316 Search PubMed.
- J. Li and M. Östling, Crystals, 2013, 3, 163 CrossRef.
- X. Su, Q. Wu, J. Li, X. Xiao, A. Lott, W. Lu, B. W. Sheldon and J. Wu, Adv. Energy Mater., 2014, 4, 1300882 CrossRef.
- L. Y. Lim, N. Liu, Y. Cui and M. F. Toney, Chem. Mater., 2014, 26, 3739 CrossRef.
- J. Lu and D. Xue, Electrochim. Acta, 2010, 56, 243 CrossRef.
- H. Fujimoto, K. Tokumitsu, A. Mabuchi, N. Chinnasamy and T. Kasuh, J. Power Sources, 2010, 195, 7452 CrossRef.
- A. Piotrowska, K. Kierzek, P. Rutkowski and J. Machnikowski, J. Anal. Appl. Pyrolysis, 2013, 102, 1 CrossRef.
- J. Hou, C. Cao, F. Idrees and X. Ma, ACS Nano, 2015, 9, 2556 CrossRef PubMed.
- Y. P. Wu, S. Fang, Y. Jiang and R. Holze, J. Power Sources, 2002, 108, 245 CrossRef.
- G. Ning, X. Ma, X. Zhu, Y. Cao, Y. Sun, C. Qi, Z. Fan, Y. Li, X. Zhang, X. Lan and J. Gao, ACS Appl. Mater. Interfaces, 2014, 6, 15950 Search PubMed.
- W. Wei, K. Sun and Y. H. Hu, J. Mater. Chem. A, 2016, 4, 12054 Search PubMed.
- W. Wei, K. Sun and Y. H. Hu, J. Mater. Chem. A, 2014, 2, 16842 Search PubMed.
- C. Li, X. Zhang, K. Wang, X. Sun, G. Liu, J. Li, H. Tian, J. Li and Y. Ma, Adv. Mater., 2017, 29, 1604690 CrossRef PubMed.
- A. Chakrabarti, J. Lu, J. C. Skrabutenas, T. Xu, Z. Xiao, J. A. Maguire and N. S. Hosmane, J. Mater. Chem., 2011, 21, 9491 RSC.
- Z. Xing, B. Wang, W. Gao, C. Pan, J. K. Halsted, E. S. Chong, J. Lu, X. Wang, W. Luo, C. H. Chang, Y. Wen, S. Ma, K. Amine and X. Ji, Nano Energy, 2015, 11, 600 CrossRef.
- Z. L. Yu, S. Xin, Y. You, L. Yu, Y. Lin, D. W. Xu, C. Qiao, Z. H. Huang, N. Yang, S. H. Yu and J. B. Goodenough, J. Am. Chem. Soc., 2016, 138, 14915 CrossRef PubMed.
- A. Malesevic, R. Vitchev, K. Schouteden, A. Volodin, L. Zhang, G. V. Tendeloo, A. Vanhulsel and C. V. Haesendonck, Nanotechnology, 2008, 19, 305604 CrossRef PubMed.
- Y. Li, Y. S. Hu, M. M. Titirici, L. Chen and X. Huang, Adv. Energy Mater., 2016, 6, 1600659 CrossRef.
- S. Zhang, F. Yao, L. Yang, F. Zhang and S. Xu, Carbon, 2015, 93, 143 CrossRef.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef PubMed.
- Y. Xu, Z. Lin, X. Huang, Y. Liu, Y. Huang and X. Duan, ACS Nano, 2013, 7, 4042 CrossRef PubMed.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CrossRef PubMed.
- L. Chang, D. J. Stacchiola and Y. H. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 24655 Search PubMed.
- W. Kiciński, M. Szala and M. Bystrzejewski, Carbon, 2014, 68, 1 CrossRef.
- X. Ma, G. Ning, Y. Kan, Y. Ma, C. Qi, B. Chen, Y. Li, X. Lan and J. Gao, Electrochim. Acta, 2014, 150, 108 CrossRef.
- G. T. K. Fey and C. L. Chen, J. Power Sources, 2001, 97–98, 47–51 CrossRef.
- V. Etacheri, C. Wang, M. J. O’conell, C. K. Chan and V. G. Pol, J. Mater. Chem. A, 2015, 3, 9861 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01729h |
| This journal is © The Royal Society of Chemistry 2018 |