
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of oxidation degree of graphite oxide and chemical structure of corresponding reduced graphite oxide by selecting different-sized original graphite†
Lu Shen‡
 ,
Lihua Zhang‡,
Kui Wang,
Lijing Miao,
Qiaofeng Lan,
Kemin Jiang,
Huanming Lu,
Ming Li,
Yong Li*,
Bin Shen* and
Wenge Zheng*
,
Lihua Zhang‡,
Kui Wang,
Lijing Miao,
Qiaofeng Lan,
Kemin Jiang,
Huanming Lu,
Ming Li,
Yong Li*,
Bin Shen* and
Wenge Zheng*
Ningbo Institute of Material Technology and Engineering, Chinese Academy of Sciences (CAS), Ningbo, Zhejiang province 315201, China. E-mail: liyong@nimte.ac.cn; shenbin@nimte.ac.cn; wgzheng@nimte.ac.cn
First published on 10th May 2018
Abstract
The thermal exfoliation and reduction of graphite oxide (GO) is the most commonly used strategy for large-scale preparation of graphene, and the oxidation degree of GO would influence the chemical structure of prepared graphene, thereby affecting its final physical and chemical properties. In addition to serving as the precursor for synthesizing graphene, GO also possesses great potential for various important applications owing to its abundant oxygen-containing groups and hybrid electronic structure. Therefore, systematically studying the influencing factors on the oxidation degree of GO and clarifying the effect of oxidation degree on the corresponding graphene is particularly important. Herein, we have studied the effect of the lateral size of the original graphite on the oxidation degree of GO in order to control the oxidation degree of GO. GOs with different degrees of oxidation were synthesized using a modified Hummers method. The results of X-ray diffraction (XRD), X-ray photoelectron spectra (XPS) and Raman spectroscopy revealed that decreased lateral size of the original graphite would lead to increased oxidation degree of GO. Furthermore, the interlayer spacing of the GO samples achieved 0.9–1.0 nm, which indicated that the modified Hummers method could make well oxidized graphite. The corresponding reduced graphite oxide (rGO) was also prepared by low-temperature exfoliation of GO at 140 °C under ambient atmosphere. It was found that a larger lateral size of GO resulted in rGO with fewer oxygen-containing functional groups, but a smaller lateral size of graphite possessed a higher exfoliation degree with a larger specific surface area. More importantly, the relationship between binding energy (EB) of photoelectron of C atom in oxygen-containing groups and the number of oxygen-containing groups in GO and rGO samples was analyzed theoretically.
Introduction
Graphene has attracted much attention owing to its unique properties and potential applications. Until now, a variety of approaches have been utilized to prepare graphene, including micromechanical or chemical exfoliation of graphite,1 chemical vapor deposition (CVD) growth,2 epitaxial growth, as well as thermal exfoliation and reduction of graphite oxide (GO).3,4 Among them, the thermal exfoliation-reduction of GO is considered to be the most promising approach for preparing graphene on a large scale.5 Since GO is always obtained through strong oxidation of graphite and possesses abundant oxygen-containing functional groups, such as hydroxyl, epoxy, carbonyl and carboxyl groups,6,7 the graphene prepared by thermal exfoliation-reduction of GO usually contains residual oxygen-containing groups.8 Generally, the oxidation degree of GO would inevitably influence the residual functional groups as well as defects on the basal plane of prepared graphene,9 thereby affecting its final physical and chemical properties.10 It is essential to study about the influence factors on the oxidation degree of GO systematically and clarify the effect of oxidation degree and structure of GO on the chemical structure of the corresponding graphene for preparing high-quality graphene with desired characteristic.In addition to acting as the precursor for preparation of graphene, GO itself exhibits plenty of distinguished properties and possesses huge potentials for extensive technological applications such as optical materials, supercapacitors, composite materials, lithium-ion batteries, biosensors and as a drug delivery agent.11–14 For example, the oxygen-containing groups render GO with hydrophilicity and well dispersed in many solvents, especially in aqueous medium without using surfactants.15 The covalent and non-covalent interactions between GO and other organic or inorganic materials enable them to effectively form hybrid or nanocomposites with enhanced performance.16 More importantly, some of the distinguished properties of GO result from its hybrid electronic structure since it features both the conducting π state from the sp2 carbon domains and the σ state from sp3 carbon domains.17 The controllable synthesis of GO with different oxidation degree could endow GO with different ratios of sp2/sp3 domains, which may bring new properties that can be useful for promoting graphene-based research.
Furthermore, there are three major methods for solution-based synthesis of GO, involving Staudenmaier method (including fuming nitric acid, concentrated sulfuric acid and potassium chlorate),18 Hofmann method (which uses concentrated nitric acid, concentrated sulfuric acid and potassium chlorate),19 and Hummers method (including concentrated sulfuric acid, sodium nitrate and potassium permanganate).20 Among them, the former two methods involve toxic reactions due to the generation of toxic gases and highly reactive species.21 In contrast, Hummers method is widely used for preparing GO owing to its nontoxicity involved.
Gaoquan Shi et al. studied the influence of graphite particle size (3–20 μm and 10–100 μm) on the oxidation degree and yield of GO. They found that both of oxidation degree and yield of GO obtained from small-sized graphite particle is higher than that obtained from large-sized graphite particle.22 Herein, we have systematically studied the effect of the lateral size of the original graphite on the oxidation degree of GO to control the oxidation degree of GO. Original graphite was oxidized thoroughly by a modified Hummers method. The results revealed that the lateral size of original graphite played an important role in the oxidation degree of GO. The decreased lateral size of original graphite would lead to the increased oxidation degree of GO. The corresponding rGO was also prepared by low-temperature exfoliation of GO at 140 °C under ambient atmosphere. It is worth mentioning that the oxidation degree of GO and structure of graphite have a significant impact on the chemical structure of rGO. It was found that the larger lateral size of graphite resulted in rGO with less oxygen-containing functional groups, but smaller lateral size of graphite resulted in rGO with larger specific surface area. Additionally, the variation of binding energy (EB) of photoelectron of C atom in oxygen-containing groups of GO and rGO samples was analyzed theoretically.
Results and discussion
Fig. 1 depicts the typical SEM images of the original graphite with different lateral size including 100 mesh, 325 mesh and 2000 mesh (Fig. 1a–c) and the resultant GO (GO-100, GO-325, GO-2000) (Fig. 1d–f). It is reported that GO actually consists of two distinct components noncovalently complexed together: the majority by mass being large functionalized graphene-like sheets together with small, more highly oxidized, fragments or debris. The debris is strongly adhered to the graphene-like sheets of GO by a combination of π–π stacking and hydrogen bonds in acidic or neutral conditions.23 In this work, GO consists of functionalized graphene-like sheets and debris. It can be found that all of the graphite shows rough surfaces after oxidation. The existence of oxygen-containing groups makes GO sheets rough. The structure of GO is amorphous due to distortions from the fraction of sp3 C–O bonds.24 As the lateral size decreases, GO sheets becomes more rough. Especially, GO-2000 presents obvious layered structure. The different levels of roughness of GO-100, GO-325 and GO-2000 may suggest the graphite with different lateral size might be oxidized to different degrees under the same conditions. The representative SEM images of rGO-100, rGO-325 and rGO-2000 which are exfoliated at 140 °C (Fig. 1g–i), compared to GO-100, GO-325 and GO-2000 (Fig. 1d–f), showed that GO were turned to rGO sheets with only a few parts connected with each other. It is reported that multilayer GO is a metastable material undergoing spontaneous chemical modifications and reduction with a relaxation time of ∼35 days at room temperature. GO exhibits a structure deprived of epoxide groups and enriched in hydroxyl groups, with a C/O ratio of 2.63 (ref. 25). Lv et al. demonstrated that few-layered graphene can be produced at a low temperature of 200 °C in about 5 h under high vacuum.26 The time required for exfoliation is a major drawback. The synthesis of few-layered graphene via hydrogen induced reduction-exfoliation of graphite oxide at 200 °C is reported.27 But it is unsafe for the existence of hydrogen. | ||
| Fig. 1 (a–c) SEM images of graphite of 100 mesh, 325 mesh and 2000 mesh; (d–f) SEM images of GO-100, GO-325 and GO-2000; (g–i) SEM images of rGO-100, rGO-325 and rGO-2000; (j–l) high-magnification SEM images of rGO-100, rGO-325 and rGO-2000. | ||
Fig. 2 presents TEM micrographs of rGO-100, rGO-325 and rGO-2000, respectively. All of the three materials show wrinkled sheets structure, confirming an effective reduction of GO. Selected area electron diffraction (SAED) patterns of the rGO-100, rGO-325 and rGO-2000 show a clear hexagonal spots pattern (inset of Fig. 2a–c), indicating that they are well-crystallized rGO. The high-resolution TEM images of rGO depict that they contain a corrugated surface morphology (Fig. 2d–f). Moreover, all of rGO-100, rGO-325 and rGO-2000 present single-layer structure according to high-resolution TEM images. This result reveals that the low-temperature exfoliation of GO can yield rGO with high quality.
 | ||
| Fig. 2 (a–c) TEM images of rGO-100, rGO-325 and rGO-2000 and their corresponding SAED; (d–f) high-resolution TEM images of rGO-100, rGO-325 and rGO-2000. | ||
Fig. 3 reveals the XRD patterns of original graphite (100 mesh), three kinds of GO and rGO-100. It can be observed that the diffraction peak of pure graphite is located at around 26°, corresponding to a typical interlayer spacing of 0.34 nm.28 After successful oxidation, GO-100, GO-325 and GO-2000 show diffraction peaks centered at 2θ = 9.51°, 9.41°, and 9.36°, respectively. The GO prepared using graphite with different lateral size exhibit different interlayer spacing. GO-2000 prepared using the 2000 mesh graphite presents the largest spacing (0.944 nm) followed by GO-325 at 0.939 nm. The smallest spacing is determined to be GO-100 with spacing of 0.929 nm. The interlayer spacing of GO synthesized in the modified method is bigger than some other GO synthesized in literatures,22,26,29,30 which confirmed our modified method could oxidize graphite thoroughly. The various interlayer spacing suggests that different amounts of oxygen-containing functional groups are inserted into the sheets of graphite for different original graphite by harsh oxidation treatment of graphite. It can be seen in Fig. 3, the (002) diffraction line of pure graphite totally disappeared after oxidation. This reveals that a complete oxidation has occurred for all of the original graphite. After low-temperature exfoliation, the characteristic peak of GO disappeared, indicating that the periodic layered structure of GO was destroyed. The data from XRD are consistent with the data of Raman spectroscopy below that was performed on GO-2000, GO-325 and GO-100.
 | ||
| Fig. 3 XRD patterns of pristine graphite, GO and rGO-100. | ||
Raman spectroscopy is usually employed to investigate the structure of carbon materials. Raman spectrum of original graphite was also measured and is shown in Fig. S1 (ESI document†). Generally, a D-band at 1300–1400 cm−1 in a spectrum corresponds to defect and disorder, while a G-band at around 1500–1600 cm−1 represents the crystallinity.31 It shows a strong G band at 1581 cm−1 because of first order scattering of the E2g mode. It also contains a small band at 1352 cm−1 named the D band, which confirms the presence of defects in the graphite such as bond-length disorder, bond-angle disorder, vacancies, edge defects and so on. It is known that harsh chemical oxidation process can result in prominent structural changes in graphite lattice owing to the formation of different types of oxygen-containing groups at both the basal plane and at the edges. Raman spectra of GO and rGO are depicted in Fig. 4. In GO-100, GO-325 and GO-2000, the G bands shifted towards higher wavenumber of 1602 cm−1, 1604 cm−1, and 1605 cm−1 due to the oxidation of graphite. It indicates that G band shift towards a higher wavenumber with the increase in oxidation level. The shift in G band is associated with the formation of new sp3 carbon atoms in the graphite lattice.32 The I(D)/I(G) ratio is often used as an indication of disorder degree in a carbon structure. As shown in Fig. 4, I(D)/I(G) ratios were calculated to be 0.87, 0.91 and 0.92 for GO-100, GO-325 and GO-2000, respectively. This result indicates that the disorder degree in GO increased owing to oxidation degree increase with the decrease of lateral size of graphite. The corresponding I(D)/I(G) ratios of obtained rGO are measured as 0.89, 0.93 and 0.93, respectively. It can be seen that all of the I(D)/I(G) ratios of GO increased after reduction, which implies that the average size of sp2 carbon domains decreased. It is possibly because the reduction process brings more smaller sp2 carbon domains and some unrepaired defects still exist.33,34
 | ||
| Fig. 4 Raman spectra of GO (a) and resultant rGO (b). | ||
On the basis of the above results, we hold the opinion that the differences of oxidation degree are inherently attributed to the different lateral size of the original graphite. It is known to all that GO synthesis involves the oxidation of graphite with strong oxidants.35,36 The exposed carbon atoms, especially those on the edges of the graphite, are more easily to contact with oxidants in oxidation process.37 The smaller the lateral size is, the shorter the diffusion route of the oxidant is. In consequence, the oxidation and intercalation developed more easily for graphite with smaller lateral size, which could form well-oxidized GO easily compared to the graphite with larger lateral size. Accordingly, we suggest that the synthesis of GO with different oxidation degree is substantially attributed to the selected original graphite with different lateral size in this work. In the process of oxidation, phenolic groups formed firstly both on the basal plane of graphite and at the edges of graphite. With further development of oxidation, part of phenolic groups converted to C–O–C or quinone groups. Then quinone groups were converted to ketone groups and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.38,39 As the oxidation degree became deeper, more O–CO formed. According to the data in Table 1, the content of O–CO in GO increases with the decrease of lateral size of original graphite. The results could also manifest that as the lateral size of graphite decreases, the oxidation degree increases.
O.38,39 As the oxidation degree became deeper, more O–CO formed. According to the data in Table 1, the content of O–CO in GO increases with the decrease of lateral size of original graphite. The results could also manifest that as the lateral size of graphite decreases, the oxidation degree increases.
| CC |
C–C | C–O/C–O–C | CO/O–CO |
π–π* | A/B | C/O | |
|---|---|---|---|---|---|---|---|
| rGO-100 | 55.14 | 17.56 | 17.08 | 8.34 | 1.88 | 2.86 | 3.30 |
| rGO-325 | 54.62 | 16.27 | 19.46 | 7.81 | 1.84 | 2.60 | 3.18 |
| rGO-2000 | 53.98 | 11.86 | 22.91 | 8.78 | 2.47 | 2.08 | 3.05 |
| GO-100 | 1.33 | 49.35 | 45.26 | 4.07 | — | 1.03 | 2.70 |
| GO-325 | 1.52 | 48.13 | 43.76 | 6.59 | — | 0.99 | 2.35 |
| GO-2000 | 0.98 | 48.16 | 44.16 | 6.71 | — | 0.97 | 1.67 |
XPS spectrum was employed to further analyze the chemical structure of GO and rGO (Fig. 5). XPS spectra of GO-100 exfoliated in 300, 500, 700, 900, 1000 °C were shown in Fig. S2 (ESI document†). As shown in Fig. 5, the C1s XPS spectrum of GO with peak deconvolution shows four carbon components. The peak intensities ratios of intact carbon (C–C/CC) and oxygenated carbon atoms in the XPS spectra were calculated to be 1.03, 0.99 and 0.97 for GO-100, GO-325 and GO-2000, respectively. This tendency is consistent with the results of XRD and Raman data. And we found that a small amount of S and N existed in these GO. The traces of S and N that we observed may originate from the covalently bonded sulfates and nitrates or the adsorbed sulfuric acid and nitric acid.28,38,39 Careful curve fitting was performed on each C1s spectrum to differentiate four different carbon stages of GO-100, GO-325 and GO-2000. The EB at ∼284.50 eV, ∼285.20 eV, ∼287.24 eV and ∼288.60 eV are assigned to sp2-hybridized carbon atoms, sp3-hybridized carbon atoms, C–O/C–O–C of hydroxyl or epoxy group, CO/O–CO of carbonyl groups, carboxy acid or ester groups.22,30,37,40 It should be noted that EB of C–O/C–O–C of rGO-140 is 286.22 eV. It shows the difference of 1.02 eV compared with GO. Furthermore, the EB of C–O/C–O–C, CO and O–CO of rGO-140 and rGO-h presents obvious differences. Table 2 shows the differences of 0.12 eV, 0.68 eV and 0.74 eV for C–O/C–O–C, CO and O–CO, respectively. It is observed that the EB of C–O/C–O–C, CO and O–CO revealed a trend of increase with the increase of oxygen-containing groups in GO and rGO structure (in order to reduce the error, CO and O–CO were not separated for GO. We just compared rGO-140 and rGO-h for the content of CO and O–CO). The results in Tables 1 and 2 are illustrated in Fig. 6. The XPS spectra were measured at an interval of 0.1 eV thus the difference of 0.1 eV is an allowable error, but that of more than 0.2 eV is not.
 | ||
| Fig. 5 C1s spectra of GO and resultant rGO (rGO-140). | ||
| Sample | CC |
C–C | C–O/C–O–C | CO |
O–CO |
π–π* |
|---|---|---|---|---|---|---|
| a GO represents GO-100, GO-325 and GO-2000. rGO-140 represents rGO-100, rGO-325 and rGO-2000. rGO-h represents GO-100 was expanded at 300, 500, 700, 900 and 1000 °C (rGO-300, rGO-500, rGO-700, rGO-900, rGO-1000). | ||||||
| GO | 284.50 | 285.20 | 287.24 | 288.60 | — | |
| rGO-140 | 284.50 | 285.20 | 286.22 | 288.33 | 289.57 | 290.98 |
| rGO-h | 284.50 | 285.20 | 286.10 | 287.65 | 288.83 | 290.99 |
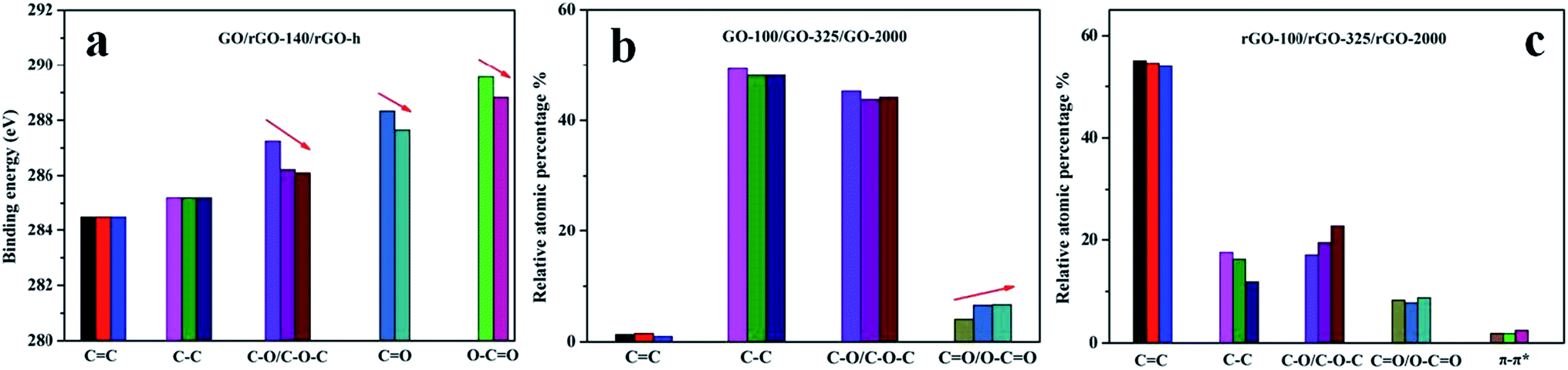 | ||
| Fig. 6 (a). Fitting of the C1s peak binding energy of GO, rGO-140 and rGO-h; (b). Quantitative comparison of XPS of C1s core level for GO-100, GO-325 and GO-2000; c. Quantitative comparison of XPS of C1s core level for rGO-100, rGO-325, and rGO-2000. | ||
In general, EB is calculated by eqn (1) where hν is the energy of the X-ray irradiated onto samples (in this case 1486.6 eV), EK is the kinetic energy of the photoelectron emitted outside by X-ray irradiation, ϕ is a work function of the instrument.
| EB = hν − EK − ϕ | (1) |
O, O–CO) increases. As a result, EB of the photoelectron in C atom of C–O increases with the increase of oxygen-containing groups in GO or rGO. Similarly, the trend of EB of the photoelectron in C atom of CO or O–CO can be stated clearly in the same way.
A great many of oxygen-containing groups were eliminated during the thermal exfoliation process for GO. A quantitative comparison of various oxygen-containing groups is listed in Table 1. It could be clearly observed that the total content of oxygen-containing groups decreases with the increasing of lateral size of the original graphite for the rGO prepared. The exfoliation of GO occurred at low temperature (140 °C) under ambient atmosphere. During the synthesis process, hydrochloric acid was introduced to the GO (with 3.6% hydrochloric acid solution). The residual hydrochloric acid in GO could vaporize and expand because of the external heating as well as the internal heating during the GO decomposition, supplying additional pressure in the interlayer of GO sheets, which results in an effective reduction of GO at low temperature.8 As is well known, the oxygen-containing groups of GO decompose and produce gases like H2O and CO2 during the thermal reduction process. Only when the pressure from these gases exceed the van der Waals forces, GO could be exfoliated successfully. The gases formed in larger GO sheets are hard to escape, compared with those in smaller GO sheets. And this could accelerate heat accumulation, temperature raising and decomposition of oxygen-containing groups. Meanwhile, decomposition of oxygen-containing groups produced a large amount of heat. And finally, the heat promoted the decomposition of oxygen-containing groups again. This may be the reason for the total content of oxygen-containing groups decreased with the increase of lateral size of the original graphite for rGO.
In Table 1, it can be seen that the content of C–O–C and hydroxyls decreases significantly in the reduction process. However, the number of edge-located O–CO increases marginally. This trend is similar with the work of Maxim K. Rabchinskii and his co-workers'.41 The effect might be related to the formation of nanoscale holes in the reduction process in the air atmosphere. The newly formed vacancies with the formation of nanoholes make the edge of rGO with O–CO.41
rGO layers have a tendency to interact with each other to form a structure of “layer upon layer” (as can be observed in SEM images in Fig. 1), which must be taken seriously when considering some potential applications of graphene-based materials (like energy storage materials). Accordingly, N2 adsorption–desorption was carried out for rGO-100, rGO-325 and rGO-2000 samples to probe the “layer upon layer” structure. As shown in Fig. 7, all of the rGO samples are characterized by type II isotherms and possess hysteretic loops of type H3. Type II isotherm suggests that no micropores or small mesopores exist in the rGO samples. The type H3 hysteresis loop indicates that there exist asymmetrically slit-shaped pores of a large pore size (Fig. 7). As a matter of fact, many slits shaped resulting from the overlap of numerous rGO layers. This result is consistent with the pore shape of H3 type hysteretic loop in Fig. 7. The specific surface area of rGO-100, rGO-325 and rGO-2000 was calculated to be 200 m2 g−1, 276 m2 g−1 and 305 m2 g−1 using Brunauer–Emmett–Teller (BET) model. The results clearly show that with the decreasing of the lateral size of graphite, the specific surface area of rGO increases. Generally speaking, specific surface area of rGO depends on both the lateral size of graphite and the number of oxygen-containing groups of GO. According to the results, lateral size of graphite played a leading role in this work.
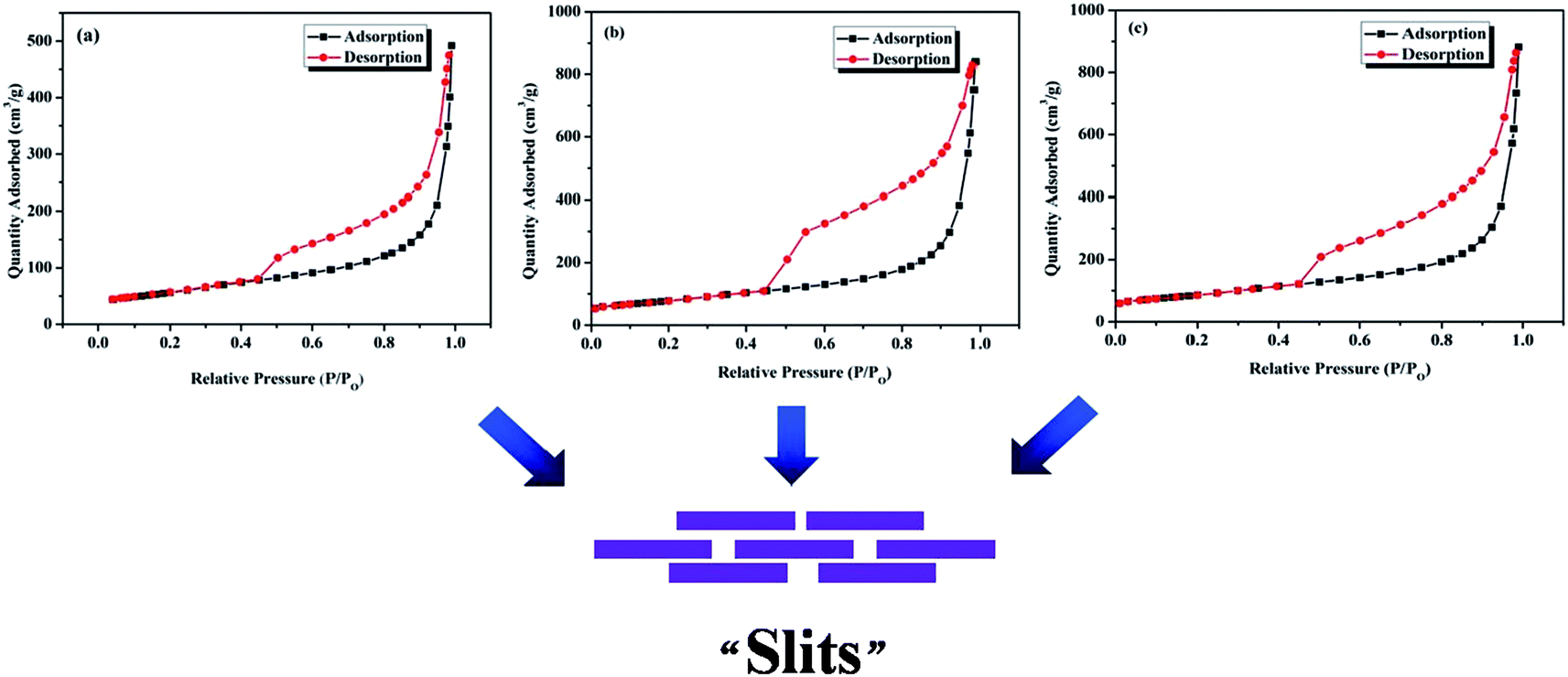 | ||
| Fig. 7 N2 adsorption–desorption isotherms of (a) rGO-100, (b) rGO-325 and (c) rGO-2000. | ||
GO-100 was choze to be thermally treated at 300, 500, 700, 900 and 1000 °C (rGO-300, rGO-500, rGO-700, rGO-900, rGO-1000) under ambient atmosphere. The obvious reduction of GO-100 at 300, 500, 700, 900 and 1000 °C was observed in SEM images (Fig. 8). The SEM images of rGO in Fig. 8, compared to that of GO-100 (Fig. 1d), show that GO sheets are exfoliated to a very large extent during the reduction process. Compared with rGO-300, rGO-500, rGO-700, rGO-900 and rGO-1000 show no significant differences from morphological observations. This morphology is very similar to graphene prepared at low-temperature exfoliation under high-vacuum.26 C1s XPS spectra of rGO are shown in Fig. S2.† The C/O atomic ratios of the prepared rGO increased from 3.45 to 7.00 by increasing treated temperature from 300 °C to 1000 °C. Accordingly, parts of oxygen-containing functional groups still existed in rGO even at 1000 °C. The reason might be that GO were oxidized during thermally treated process under ambient atmosphere. Moreover, as the reduction temperature increased, reduction time reduced from 10 min to 30 s (Fig. 9). The specific surface area of these five kinds rGO was calculated to be 250, 274, 292, 367, 372 m2 g−1 using BET model (Fig. 10). The result manifests that raising the temperature can promote reduction of GO under ambient atmosphere.
 | ||
| Fig. 8 SEM images of (a) rGO-300, (b) rGO-500, (c) rGO-700, (d) rGO-900 and (e) rGO-1000. | ||
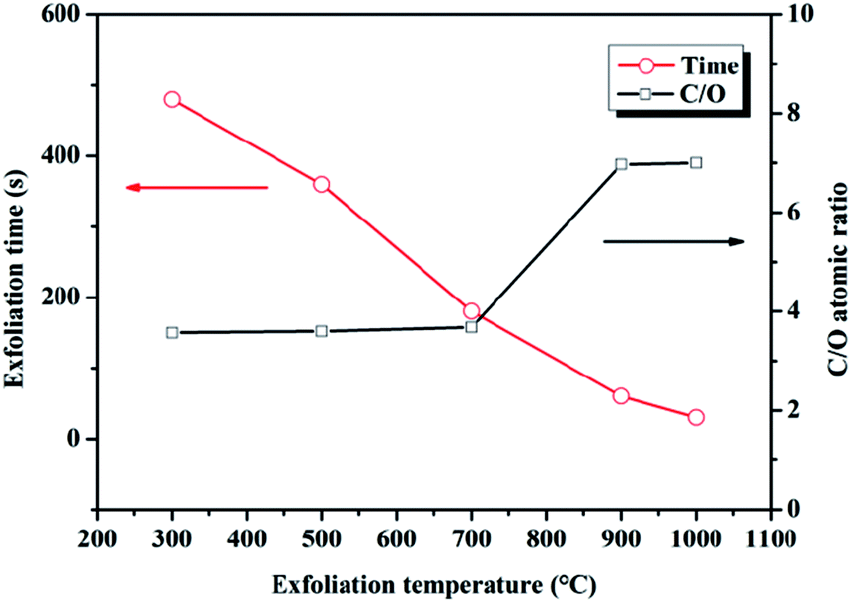 | ||
| Fig. 9 The reduction time and C/O atomic ratios of rGO exfoliated at different temperature. | ||
 | ||
| Fig. 10 BET Surface plots of rGO-300, rGO-500, rGO-700, rGO-900 and rGO-1000. | ||
Experimental
Materials
Three kinds of natural flake graphite materials with different lateral size (100 mesh, 325 mesh, 2000 mesh) were kindly supplied by Qingdao Huatai Lubricant Sealing S&T. Sulfuric acid (98%), potassium permanganate (98%), hydrogen peroxide (30%), and hydrochloric acid (36–38%) were purchased from Sinopharm Chemical Reagent Co., Ltd.Preparation of GO
GO was synthesized by oxidation of natural flake graphite using the modified Hummer's method. Briefly, 2.3 g graphite and 1.5 g sodium nitrate were stirred with 69 mL sulfuric acid (98%) in an ice bath for 10 min. Then, 9 g potassium permanganate was added slowly over a period of 35 min. In the subsequent 1 hour, the reaction mixture was stirred in an ice bath. After that, the ice bath was removed and the temperature of water bath was stayed at 35 °C. The reaction mixture continued to react in the water bath for 4 h. Then, it was poured into a beaker with 700 mL distilled water slowly instead of adding distilled water to the product in the literatures.22,42,43 Compared with other methods, this method can effectively control the temperature of product, which can avoids destructing the structure of GO by high temperature caused by the heat of exothermic reaction. The product was stirred for several minutes. Then, it was treated with 30% hydrogen peroxide (20 mL) to remove the unreacted potassium permanganate and manganese dioxide. Finally, the resulting suspension was filtered, washed with hydrochloric acid (3.6%) for twice, and dried in a vacuum oven at 50 °C for 48 h.Exfoliation and reduction of GO
The as-prepared GO was expanded at 140 °C under ambient atmosphere to obtain rGO. A slice of GO was placed in a beaker covered with lid. Then the beaker was quickly placed in an oven at 140 °C for 10 min. Dramatic volume expansion of GO accompanied by the voice of the pop was observed. In addition, GO was also thermally treated at high temperature including 300 °C, 500 °C, 700 °C, 900 °C and 1000 °C using a muffle oven.Characterizations
Scanning electron microscopy (SEM) observation was performed with a FEI QUANTA 250 FEG field emission SEM at an accelerating voltage of 5 kV. A Tecnai G2 F20 transmission electron microscopy (TEM) operating at 100 kV was used to acquire TEM images. The X-ray diffraction (XRD) was measured with a Bruker AXS X-ray diffractometer with CuKα radiation at a generator voltage of 40 kV and a generator current of 40 mA. Raman spectra were excited with a laser of 532 nm and record with Labram spectrometer (Super LabRam II system). X-ray photoelectron spectroscopy (XPS) was performed with a Kratos AXIS ULTRA Multifunctional XPS using Al (mono) Kα radiation (1486.6 eV) under 1.2 × 10−9 Torron. Specific surface area analysis was carried out with a Micromeritics ASAP 2020 Analyzer by using Brunauer, Emmett and Teller (BET) model.Conclusions
In conclusion, GO with big interlayer spacing (0.9–1.0 nm) was successfully synthesized by a modified Hummers method. A systematic investigation for the effect of lateral size of original graphite on the oxidation degree of GO was carried out. The results indicated that the lateral size of original graphite played an important part in controlling oxidation degree of GO. As the lateral size decreased, the oxidation degree of GO increased. This result was confirmed by XRD data, Raman spectra and XPS data. Furthermore, rGO was successfully obtained through reduction at 140 °C under ambient atmosphere, and the exfoliated rGO exhibited single-layer structure from TEM images without any surfactant. Moreover, we found that the chemical structure of rGO is strongly correlated to the oxidation degree of GO and lateral size of graphite. The results indicated that the larger lateral size of graphite led to rGO with less oxygen-containing groups. The specific surface area of rGO increased with the decreasing of lateral size of original graphite. Additionally, the relationship between binding energy of photoelectron of C atom in oxygen-containing groups and the number of oxygen-containing groups in GO and rGO samples was analyzed theoretically according to the C1s spectra. Besides, compared with the situation of low-temperature exfoliation of GO-100, it was also thermally treated at 300, 500, 700, 900 and 1000 °C under ambient atmosphere. It was confirmed that raising the temperature could promote reduction of GO under ambient atmosphere on the basis of XPS data and specific surface area data. These findings figured out the effect of lateral size of original graphite on the oxidation degree of GO and analyzed the chemical structure of corresponding rGO by selecting different-sized original graphite.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the financial supports from National Natural Science Foundation of China (51603218 and 51473181), Science and Technology Major Project of Ningbo (2015S1001), and Ningbo Key Lab of Polymer Materials (2010A22001).Notes and references
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed.
- Z. Z. Sun, Z. Yan, J. Yao, E. Beitler, Y. Zhu and J. M. Tour, Nature, 2010, 468, 549–552 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- H.-B. Zhang, W.-G. Zheng, Q. Yan, Y. Yang, J.-W. Wang, Z.-H. Lu, G.-Y. Ji and Z.-Z. Yu, Polymer, 2010, 51, 1191–1196 CrossRef CAS.
- G. Wang, X. Sun, C. Liu and J. Lian, Appl. Phys. Lett., 2011, 99, 1–3 Search PubMed.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- S. Wang, P. K. Ang, Z. Wang, A. L. L. Tang, J. T. L. Thong and K. P. Loh, Nano Lett., 2010, 10, 92–98 CrossRef CAS PubMed.
- Y. W. Zhu, S. Murali, W. W. Cai, X. S. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 5226 CrossRef CAS.
- C. Gomez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144–1148 CrossRef CAS PubMed.
- K. Krishnamoorthy, R. Mohan and S. J. Kim, Appl. Phys. Lett., 2011, 98, 244101 CrossRef.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, Carbon, 2011, 49, 2917–2925 CrossRef CAS.
- G.-H. Yang, D.-D. Bao, H. Liu, D.-Q. Zhang, N. Wang and H.-t. Li, J. Inorg. Organomet. Polym. Mater., 2017, 27, 1129–1141 CrossRef CAS.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- M. Veerapandian, M.-H. Lee, K. Krishnamoorthy and K. Yun, Carbon, 2012, 50, 4228–4238 CrossRef CAS.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481 CrossRef CAS.
- E. K. U. Hofmann, Z. Anorg. Allg. Chem., 1937, 234, 311 CrossRef.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- J. P. Rourke, P. A. Pandey, J. J. Moore, M. Bates, I. A. Kinloch, R. J. Young and N. R. Wilson, Angew. Chem., Int. Ed., 2011, 50, 3173–3177 CrossRef CAS PubMed.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9, 1058–1063 CrossRef CAS PubMed.
- S. Kim, S. Zhou, Y. Hu, M. Acik, Y. J. Chabal, C. Berger, W. de Heer, A. Bongiorno and E. Riedo, Nat. Mater., 2012, 11, 544–549 CrossRef CAS PubMed.
- W. Lv, D.-M. Tang, Y.-B. He, C.-H. You, Z.-Q. Shi, X.-C. Chen, C.-M. Chen, P.-X. Hou, C. Liu and Q.-H. Yang, ACS Nano, 2009, 3, 3730–3736 CrossRef CAS PubMed.
- A. Kaniyoor, T. T. Baby and S. Ramaprabhu, J. Mater. Chem., 2010, 20, 8467–8469 RSC.
- L. Zhang, X. Li, Y. Huang, Y. Ma, X. Wan and Y. Chen, Carbon, 2010, 48, 2367–2371 CrossRef CAS.
- H. L. Poh, F. Sanek, A. Ambrosi, G. Zhao, Z. Sofer and M. Pumera, Nanoscale, 2012, 4, 3515–3522 RSC.
- B. Shen, D. Lu, W. Zhai and W. Zheng, J. Mater. Chem. C, 2013, 1, 50–53 RSC.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B, 2000, 61, 14095–14107 CrossRef CAS.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 1–6 CrossRef PubMed.
- Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950–2956 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS PubMed.
- J. L. Li, K. N. Kudin, M. J. McAllister, R. K. Prud'homme, I. A. Aksay and R. Car, Phys. Rev. Lett., 2006, 96, 176101 CrossRef PubMed.
- G. Shao, Y. Lu, F. Wu, C. Yang, F. Zeng and Q. Wu, J. Mater. Sci., 2012, 47, 4400–4409 CrossRef CAS.
- A. Dimiev, D. V. Kosynkin, L. B. Alemany, P. Chaguine and J. M. Tour, J. Am. Chem. Soc., 2012, 134, 2815–2822 CrossRef CAS PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Photoelectron Spectroscopy, 1992 Search PubMed.
- M. K. Rabchinskii, V. V. Shnitov, A. T. Dideikin, A. E. Aleksenskii, S. P. Vul, M. V. Baidakova, I. I. Pronin, D. A. Kirilenko, P. N. Brunkov, J. Weise and S. L. Molodtsov, J. Phys. Chem. C, 2016, 120, 28261–28269 CAS.
- Z.-S. Wu, W. Ren, L. Gao, B. Liu, C. Jiang and H.-M. Cheng, Carbon, 2009, 47, 493–499 CrossRef CAS.
- J. Zhao, S. Pei, W. Ren, L. Gao and H.-M. Cheng, ACS Nano, 2010, 4, 5245–5252 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01486h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |