
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent synthesis of dual 1,4-dihydropyridines with different substituted patterns from enaminones and aldehydes through domino reactions†
Fu-Jun Liua,
Tian-Tian Suna,
Yun-Gang Yanga,
Chao Huang*b and
Xue-Bing Chen *a
*a
aKey Laboratory of Natural Pharmaceutical and Chemical Biology of Yunnan Province, School of Science, Honghe University, Mengzi, Yunnan 661100, China. E-mail: orangekaka@126.com
bSchool of Chemistry and Environment, Engineering Research Center of Biopolymer Functional Materials of Yunnan, Yunnan Minzu University, Kunming, Yunnan 650503, China
First published on 3rd April 2018
Abstract
A concise and efficient protocol for the regioselective synthesis of dual 1,4-dihydropyridines with several substituted patterns has been developed from a cascade cyclization of enaminones and aldehydes in different media (EtOH/CH3CN). The one-pot cascade reaction involves at least five reactive sites and generates multiple C–C and C–N bonds. The established protocol explores the chemistry of enaminones by employing their three reactive sites. The method has several advantages including mild conditions, operational simplicity, and high bond-forming efficiency. It may offer promise in a variety of biochemical applications.
Introduction
Molecularly diverse and complex heterocycles play an important role in both organic synthesis and discovery of new pharmaceutical reagents.1 1,4-Dihydropyridines (1,4-DHPs) derivatives represent one of the most important classes of heterocycles due to their application in biological activities and molecular functions. For example, 1,4-DHPs are contained in marketed drugs such as the calcium channel blockers nifedipine, felodipine, nicardipine, and nimodipine. During the last several decades, it has been demonstrated that 1,4-DHPs are involved in various biological applications, i.e. antibacterial,2 anticonvulsant,3 anti-HIV,4 anti-tumour,5 radioprotective,6 and neuroprotection7 activities; they could also act as adenosine-A3 receptor antagonism8 and sirtuin inhibitor.9 In view of their high significance, numerous methodologies for 1,4-DHPs have been introduced.10 However, few strategies have been successfully developed for efficient and concise access to highly functionalized and molecularly diverse target products. Therefore, it is of great importance to build libraries of target compounds in a parallel manner.Multicomponent domino reactions (MCDRs) are powerful tools in modern organic synthesis because they enable more than two building blocks to combine in a single-step operation to generate complex structures through the simultaneous formation of multiple bonds.11 With their inherent characteristics, i.e., step efficiency, atom economy, target molecules diversity, operational simplicity. MCDRs have become an essential tool for generating complex molecular libraries in the screening of biologically and pharmacologically active candidates.
As a group of versatile and powerful synthons, enaminones have been used for construction of diverse heterocyclic systems.12 These compounds include acridines,13 indoles,14 naphthyridines,15 quinolones,16 pyrroles,17 thiazines,18 pyridines,19 and thiazoles.20 Some of these heterocycles which are synthesized from enaminones also demonstrate promising biological activity.21 So far, a number of natural product-like heterocycles have been successfully synthesized based on enaminones. Their synthetic methodology and associated medicinal activity should be engaged in the chemistry community.
In pursuit of our research interests regarding the development of MCRs based on enaminones, we report a convenient and efficient protocol for the regioselective synthesis of two libraries of 1,4-DHPs 3 and 4 via a tandem annulation of enaminones 1 and aldehydes 2 in EtOH and CH3CN, respectively (Scheme 1). At least five distinct reactive sites participate in this process.
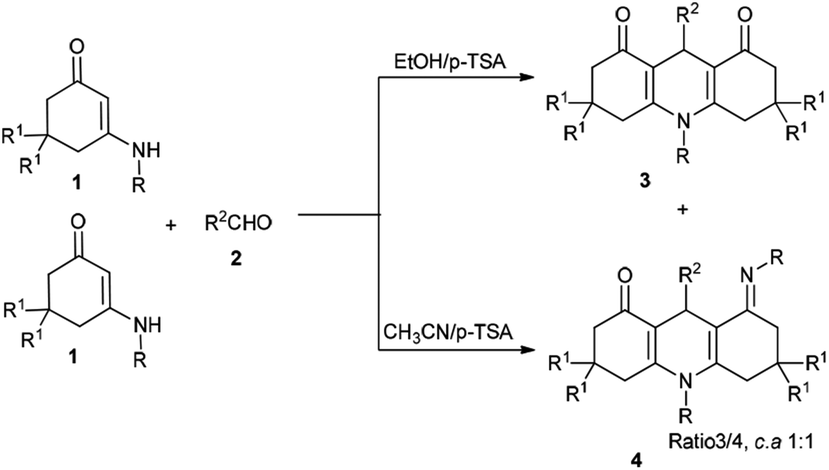 | ||
| Scheme 1 Proposed mechanism for the construction of 1,4-DHPs 3 and 4. | ||
Results and discussion
On the basis of our recent work, we examined the domino reaction which employed N-p-F-Ph substituted enaminone 1a and 4-chlorobenzaldehyde 2a as the substrates (Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalystb | Time (h) | Yieldc (%) | |
| 3a | 4a | ||||
| a The reaction was performed with 1a (1.0 mmol), 2a (0.6 mmol), and the solvent (15 mL) was under reflux temperature.b Catalyst (0.3 mmol) was added to the reaction.c Isolated yields were based on enaminone 1a. | |||||
| 1 | EtOH | — | 4 | n.r | n.r |
| 2 | EtOH | Piperidine | 3 | n.r | n.r |
| 3 | EtOH | Et3N | 3 | n.r | n.r |
| 4 | EtOH | DBU | 3 | n.r | n.r |
| 5 | EtOH | DABCO | 3 | n.r | n.r |
| 6 | EtOH | p-TSA | 3 | 66 | n.r |
| 7 | EtOH | TFA | 2 | 64 | n.r |
| 8 | EtOH | HOAc | 2 | n.r | n.r |
| 9 | EtOH | L-proline | 2 | n.r | n.r |
| 10 | CH3CN | p-TSA | 2 | 43 | 36 |
| 11 | CH3CN | TFA | 2 | 50 | 34 |
| 12 | CH3CN | TfOH | 2 | n.r | Trace |
| 13 | 1,4-Dioxane | p-TSA | 2 | 30 | 25 |
| 14 | DMF | p-TSA | 2 | 45 | 28 |
| 15 | CH3OH | p-TSA | 2 | n.r | n.r |
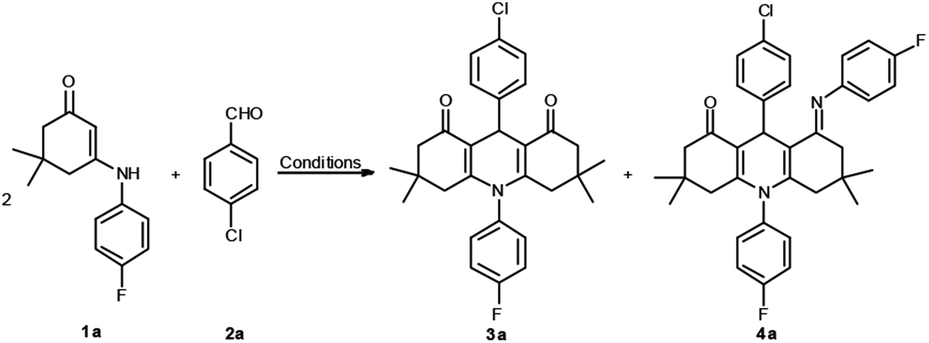
First, the reaction was executed without any catalyst in anhydrous ethanol at reflux; no reaction occurred even within 3 hours (Table 1, entry 1). Next, different types of organic bases, such as piperidine, Et3N, DBU, and DABCO, were employed as catalysts, but no transformation was observed (Table 1, entries 2–5). Nevertheless, when p-TSA (0.2 equiv.) was added, compound 3a was obtained in a yield of 66% (Table 1, entry 6). Other acids, such as TFA, HOAc and L-proline were also investigated, but could not generate the reaction efficiently (Table 1, entries 7–9). The reaction conditions were then optimized by screening several solvents. When p-TSA was added to a solution of 1a and 2a in CH3CN (Table 1, entry 10), a new compound 4a was generated in 36% yield. In order to efficiently obtain a single final product, additional attempts were made. For example, three types of solvents, i.e., 1,4-dioxane, DMF, and CH3OH, and acidic catalysts TFA, and TfOH were used in the reaction. However, the yields of 3a and 4a were decreased, and likewise, the vying preference for the formation of 4a was unsatisfied (Table 1, entries 11–15). Thus, ethanol was the best medium for selectively obtaining product 3a. It was discovered that the optimum reaction conditions for the synthesis of 3a include p-TSA as the catalyst and EtOH as the medium at reflux for 3 h. Conversely, the best conditions for the preparation of 4a are conducted in CH3CN.
With the optimal reaction conditions, the synthesis of 1,4-DHPs 4 was tested by using a range of readily available enaminones and different substituted aldehydes. As shown in Table 2, for precursors 1 and 2 bearing either electron-withdrawing or electron-donating substituents on the aromatic ring, or the aromatic groups (for 2) with diverse substitution patterns (para or meta), all reactions proceeded smoothly to provide the corresponding 1,4-DHPs derivatives. The reaction was also investigated by using alkyl enaminone 1h, which yielded the corresponding target molecule 3m.
|
||||
|---|---|---|---|---|
| Entry | 1 (R/R1) | 2 (R2) | 3 | Yieldb (%) |
| a The reaction was performed with 1 (1.0 mmol), 2 (0.6 mmol), and p-TSA (0.3 mmol) in EtOH (15 mL) at reflux for 3 hours.b Isolated yields based on enaminones 1. | ||||
| 1 | 1a (4-FC6H5/CH3) | 2a (4-ClC6H5) | 3a | 66 |
| 2 | 1a (4-FC6H5/CH3) | 2b (C6H5) | 3b | 58 |
| 3 | 1b (3-FC6H5/CH3) | 2b (C6H5) | 3c | 62 |
| 4 | 1c (3-ClC6H5/CH3) | 2a (4-ClC6H5) | 3d | 63 |
| 5 | 1c (3-ClC6H5/CH3) | 2b (C6H5) | 3e | 68 |
| 6 | 1d (4-BrC6H5/CH3) | 2a (4-ClC6H5) | 3f | 65 |
| 7 | 1d (4-BrC6H5/CH3) | 2b (C6H5) | 3g | 62 |
| 8 | 1e (3-BrC6H5/CH3) | 2b (C6H5) | 3h | 60 |
| 9 | 1f (C6H5/CH3) | 2a (4-ClC6H5) | 3i | 65 |
| 10 | 1f (C6H5/CH3) | 2b (C6H5) | 3j | 63 |
| 11 | 1f (C6H5/CH3) | 2c (4-OCH3C6H5) | 3k | 64 |
| 12 | 1g (3-CH3C6H5/CH3) | 2b (C6H5) | 3l | 62 |
| 13 | 1h (n-butyl/CH3) | 2a (4-ClC6H5) | 3m | 70 |
| 14 | 1i (4-ClC6H5/H) | 2a (4-ClC6H5) | 3n | 71 |
| 15 | 1j (3-ClC6H5/H) | 2a (4-ClC6H5) | 3o | 72 |
| 16 | 1k (3-BrC6H5/H) | 2c (4-OCH3C6H5) | 3p | 68 |
| 17 | 1l (4-CH3C6H5/H) | 2a (4-ClC6H5) | 3q | 60 |
| 18 | 1a (4-FC6H5/CH3) | 2d (n-propyl) | 3r | 62 |
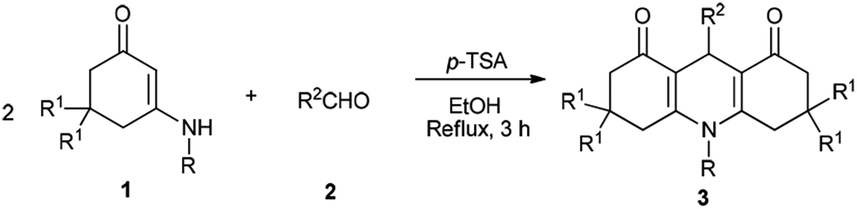
After completing the synthesis of compounds 3, the domino reaction was studied for preparing other target molecules 4 (Scheme 2). First, the scope of substrates 1 was tested by using 3-amino-5,5-dimethylcyclohex-2-enones. The reaction results of various 3-aminocyclohex-2-enonephenyls with aldehydes demonstrate that the process tolerates both 3-amino-5,5-dimethylcyclohex-2-enones 1 and aromatic aldehydes 2 bearing either electron-donating or electron-withdrawing substituents on the aryl group. However, as both compounds 3 and 4 are generated from this reaction, the yields of 4 would be relatively low (yields 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, c.a 1:1).
:4, c.a 1:1).
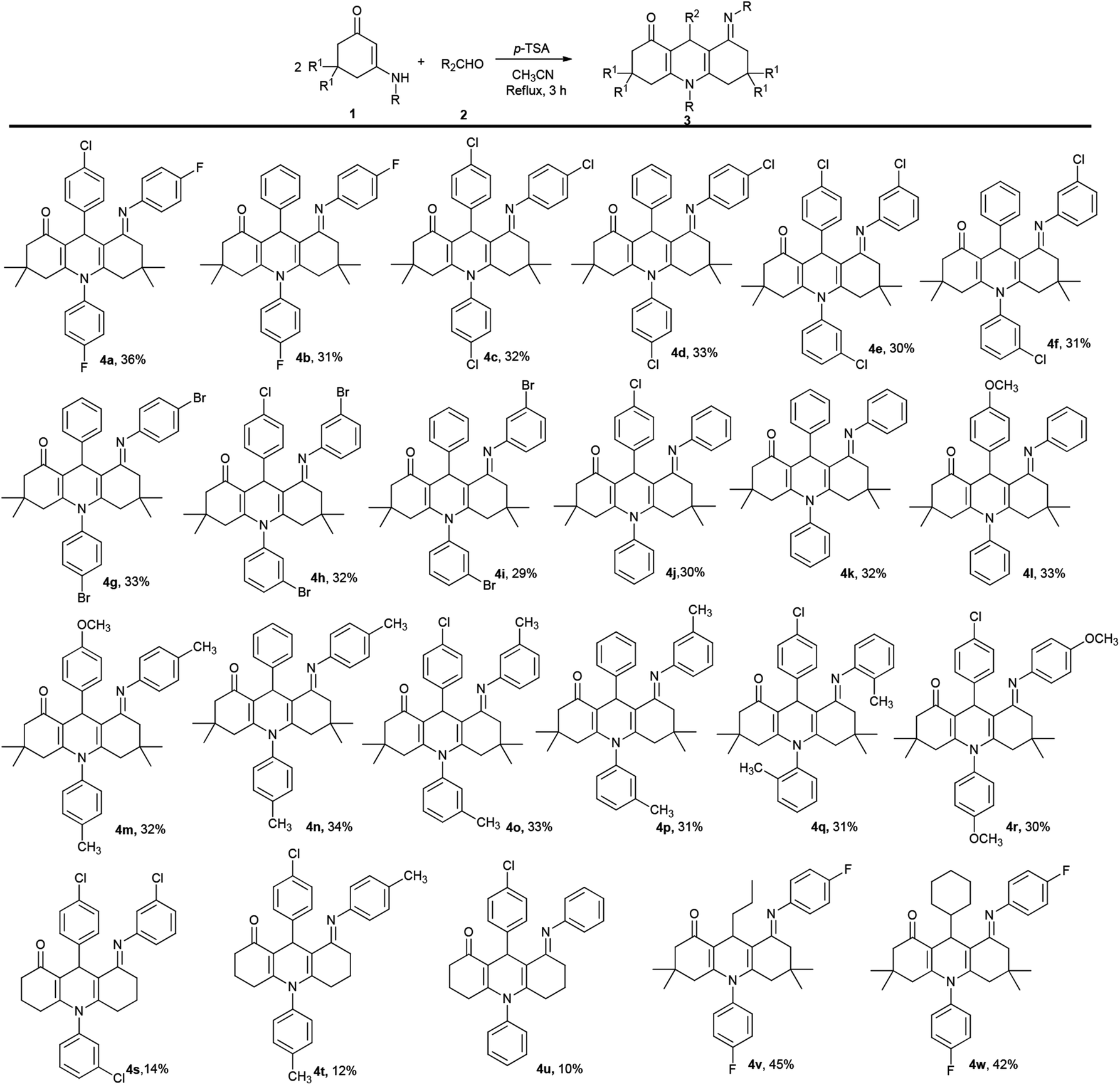 | ||
| Scheme 2 Substrate scope of the reaction for 1,4-DHPs 4. The reaction was performed with 1 (1.0 mmol), 2 (0.6 mmol), and p-TSA (0.3 mmol) in CH3CN (10 mL) at reflux for 2 h. The yields were isolated based on enaminones 1. | ||
Next, the reaction was examined using N-substituted 3-aminocyclohex-2-enonephenyls. Unfortunately, the results did not yield much success. Most of the N-substituted 3-aminocyclohex-2-enonephenyls generated a low yield of 4 but a high yield of 3 (Scheme 2, 4s–4u). It is worthwhile noting that compounds 4 are difficult to separate by conventional methods, like column chromatography and recrystallization. Hitherto, only three compounds were readily obtained by using N-substituted 3-aminocyclohex-2-enonephenyls.
Finally, aliphatic aldehydes butyraldehyde and cyclohexanecarboxaldehyde were also employed in this process, and both reactions proceeded well giving the corresponding 1,4-DHPs 4v and 4w. And it was observed that the aliphatic aldehydes gave the major compounds 4 while products 3 were scarcely generated.
The chemical structures of 1,4-DHPs 3 and 4 were characterized by IR, 1H NMR, 13C NMR, and HRMS, and unequivocally confirmed by X-ray diffraction analysis of a monocrystal of 3n (Fig. 1, CCDC 1819954).
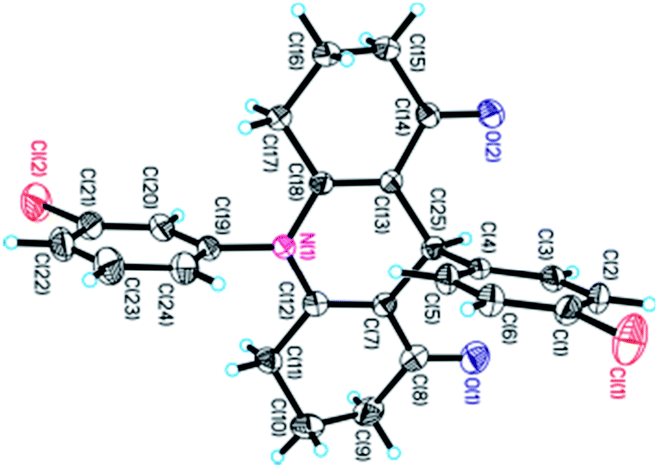 | ||
| Fig. 1 X-ray crystal structures of 3n; ellipsoids are drawn at 30% probability level. | ||
The possible mechanism for the formation of products 3/4 is shown in Scheme 3. Take aromatic aldehyde as the example. First, the benzaldehyde 2 accepts a proton to form 5. Enaminone 1 serves as a heteroene component to react with 5 accompanying the loss of one molecule of H2O to generate intermediate 6 via an aza-ene reaction. Following this, compound 6 undergoes imine-enamine tautomerization to produce 7. Intermediate 7 accepts a proton to form 8, and the elimination of water from intermediate 7 gives iminium ion 9. Enaminone 1 then participates in a Michael addition with compound 9 to form intermediate 10. Two potential reactions then occur in intermediate 10. In pathway 1, the –NH group undergoes an intramolecular attack of the carbonyl group, resulting in a cyclization reaction that forces it to lose a molecule of water, affording 4. In pathway 2, intermediate 10 undergoes imine–enamine tautomerization to give intermediate 12 and elimination of aniline gives 1,4-DHPs 3.
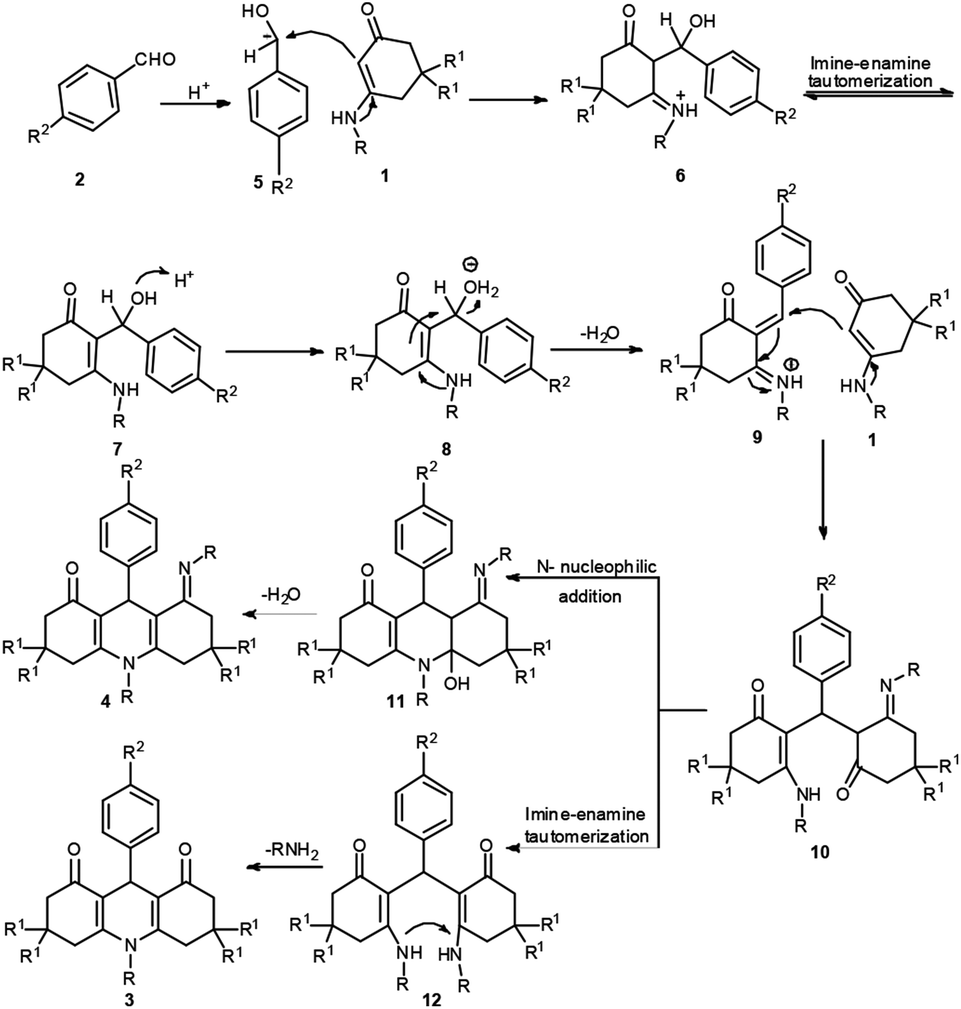 | ||
| Scheme 3 Proposed mechanism for the construction of 1,4-DHPs 3 and 4. | ||
Conclusions
In conclusion, we have successfully developed an operationally simple, one-pot three component reaction for the selective synthesis of two libraries of highly functionalized 1,4-DHPs derivatives by using of EtOH or CH3CN as media via a tandem cyclization of enaminones 1 with aldehydes 2. The reaction displays many attractive features, including excellent regioselectivity, mild and environmentally friendly conditions, and molecular diversities. Moreover, these series of 1,4-DHPs may provide potential biological activities for medical treatment. Further studies will be conducted in our laboratories in due course.Experimental
General
All compounds were fully characterized by spectroscopic data. The NMR spectra were recorded on Bruker AVIII-400 (1H: 400 MHz, 13C: 100 MHz) or Bruker AVIII-500 (1H: 500 MHz, 13C: 125 MHz) and chemical shifts (δ) are expressed in ppm, and J values are given in Hz, CDCl3 was used as solvent. IR spectra were recorded on a FT-IR Thermo Nicolet Avatar 360 using a KBr pellet. The reactions were monitored by thin-layer chromatography (TLC) using silica gel GF254. The melting points were determined on XT-4A melting point apparatus and are uncorrected. HRMS were performed on a Agilent LC/Msd TOF and monoisotopic mass instrument.General procedure for the synthesis of compounds 3
A mixture of enaminones 1 (1.0 mmol), aldehydes 2 (0.6 mmol), p-TSA (0.3 mmol), and EtOH (15 mL) was stirred at reflux for 3 h. After the desired product was formed as indicated by TLC, the reaction mixture was quenched with saturated NH4Cl solution (2 mL) and extracted with ethyl acetate (40 mL). The organic phase was dried over Mg2SO4, and concentrated under vacuum. The residue was purified by flash chromatography (petroleum ether/ethyl acetate = 2:3) giving a white solid 3.
General procedure for the synthesis of compounds 4
A mixture of enaminones 1 (1.0 mmol), aldehydes 2 (0.6 mmol), p-TSA (0.3 mmol), and CH3CN (10 mL) was stirred at reflux for 2 h. After the desired product was formed as indicated by TLC, the reaction mixture was quenched with saturated NH4Cl solution (2 mL) and extracted with ethyl acetate (40 mL). The organic phase were dried over Mg2SO4, and concentrated under vacuum. The residue was purified by flash chromatography (petroleum ether/ethyl acetate = 1:1) giving a yellow solid 4.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 61361002, 21662046), Applied Basic Research Project of Yunnan (2017FD156), and Yunnan Province Department of Education Fund (2016ZZX217).Notes and references
- (a) J. D. Sunderhaus, C. Dockendorff and S. F. Martin, Org. Lett., 2007, 9, 4223 CrossRef CAS PubMed; (b) F. Liéby-Muller, T. Constantieux and J. Rodriguez, J. Am. Chem. Soc., 2005, 127, 17176 CrossRef PubMed; (c) C. Lamberth and J. Dinges, in Bioactive Heterocyclic Compound Classes: Pharmaceuticals, ed. C. Lamberth and J. Dinges, Wiley-VCH, Weinheim, 2012, ch. 1, pp. 3–5 Search PubMed; (d) C. Hulme and V. Gore, Curr. Med. Chem., 2003, 10, 51 CrossRef CAS PubMed.
- H. M. Refat and A. A. Fadda, Eur. J. Med. Chem., 2013, 70, 419 CrossRef CAS PubMed.
- (a) R. Surendra Kumar, A. Idhayadhulla, A. Jamal Abdul Nasser, S. Kavimani and S. Indumathy, Indian J. Pharm. Sci., 2010, 72, 719 CrossRef PubMed; (b) B. B. Subudhi, P. K. Panda, S. P. Swain and P. Sarangi, Acta Pol. Pharm., 2009, 66, 147 CAS.
- (a) A. Hilgeroth, Mini-Rev. Med. Chem., 2002, 2, 235 CrossRef CAS PubMed; (b) C. K. Chu, V. S. Bhadti, K. J. Doshi, J. T. Etse, J. M. Gallo, F. D. Boudinot and R. F. Schinazi, J. Med. Chem., 1990, 33, 2188 CrossRef CAS PubMed; (c) A. Hilgeroth and H. Lilie, Eur. J. Med. Chem., 2003, 38, 495 CrossRef CAS PubMed.
- (a) H. A. Abbas, W. A. El Sayed and N. M. Fathy, Eur. J. Med. Chem., 2010, 45, 973 CrossRef CAS PubMed; (b) S. R. Morshed, K. Hashimoto, Y. Murotani, M. Kawase, A. Shah, K. Satoh, H. Kikuchi, H. Nishikawa, J. Maki and H. Sakagami, Anticancer Res., 2005, 25, 2033 CAS; (c) J. Robert and C. Jarry, J. Med. Chem., 2003, 46, 4805 CrossRef CAS PubMed.
- I. O. Donkor, X. Zhou, J. Schmidt, K. C. Agrawal and V. Kishore, Bioorg. Med. Chem., 1998, 6, 563 CrossRef CAS PubMed.
- V. Klusa, Drugs Future, 1995, 20, 135 CrossRef.
- A. H. Li, S. Moro, N. Forsyth, N. Melman, X.-D. Ji and K. A. Jacobson, J. Med. Chem., 1999, 42, 706 CrossRef CAS PubMed.
- A. Mai, S. Valente, S. Meade, V. Carafa, M. Tardugno, A. Nebbioso, A. Galmozzi, N. Mitro, E. D. Fabiani, L. Altucci and A. Kazantsev, J. Med. Chem., 2009, 52, 5496 CrossRef CAS PubMed.
- (a) J.-P. Wan and Y.-Y. Liu, RSC Adv., 2012, 2, 9763 RSC; (b) G. Li and J. C. Antilla, Org. Lett., 2009, 11, 1075 CrossRef CAS PubMed; (c) D. Zhang, L.-Z. Wu, L. Zhou, X. Han, Q.-Z. Yang, L.-P. Zhang and C.-H. Tung, J. Am. Chem. Soc., 2004, 126, 3440 CrossRef CAS PubMed; (d) P. R. Girling, A. S. Batsanov, H. C. Shen and A. Whiting, Chem. Commun., 2012, 4893 RSC; (e) Y. Nakaike, N. Nishiwaki, M. Ariga and Y. Tobe, J. Org. Chem., 2014, 79, 2163 CrossRef CAS PubMed; (f) J.-P. , Wan, R.-H. Zhou, Y.-Y. Liu and M.-Z. Cai, RSC Adv., 2013, 3, 2477 RSC.
- (a) V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2010, 39, 4402 RSC; (b) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234 CrossRef CAS PubMed; (c) N. Isambert, M. d. M. S. Duque, J. C. Plaquevent, Y. Genisson, J. Rodriguez and T. Constantieux, Chem. Soc. Rev., 2011, 40, 1347 RSC; (d) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156 CrossRef CAS PubMed; (e) B. Ganem, Acc. Chem. Res., 2009, 42, 463 CrossRef CAS PubMed; (f) A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; (g) J.-P. Wan, L. Gan and Y.-Y. Liu, Org. Biomol. Chem., 2017, 15, 9031 RSC.
- (a) B. Stanovnik and J. Svete, Chem. Rev., 2004, 104, 2433 CrossRef CAS PubMed; (b) J.-P. Wan and Y. Gao, Chem. Rec., 2016, 16, 1164 CrossRef CAS PubMed.
- (a) B. Jiang, X. Wang, H.-W. Xu, M.-S. Tu, S.-J. Tu and G.-G. Li, Org. Lett., 2013, 15, 1540 CrossRef CAS PubMed; (b) W.-J. Hao, J.-Q. Wang, X.-P. Xu, S.-L. Zhang, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2013, 78, 12362 CrossRef CAS PubMed; (c) X.-B. Chen, X.-Q. Wang, Y. Hong and W. Liu, Asian J. Org. Chem., 2016, 5, 907 CrossRef CAS; (d) H.-Y. Wang, L.-L. Li, W. Lin, P. Xu, Z.-B. Huang and D.-Q. Shi, Org. Lett., 2012, 14, 4598 CrossRef CAS PubMed.
- (a) H. Xu, B. Zhou, P. Zhou, J. Zhou, Y.-H. Shen, F.-C. Yu and L.-L. Li, Chem. Commun., 2016, 52, 8002 RSC; (b) X.-B. Chen, T.-B. Luo, G.-Z. Gou, J. Wang and W. Liu, Asian J. Org. Chem., 2015, 4, 921 CrossRef CAS; (c) B. Jiang, M.-S. Yi, F. Shi, S.-J. Tu, S. Pindi, P. McDowell and G.-G. Li, Chem. Commun., 2012, 48, 808 RSC; (d) J.-D. Hu, C.-P. Cao, W. Lin, M.-H. Hu, Z.-B. Huang and D.-Q. Shi, J. Org. Chem., 2014, 79, 7935 CrossRef CAS PubMed; (e) L. Wang, L.-X. Shi, L. Liu, Z.-X. Li, T. Xu, W.-J. Hao, G.-G. Li, S.-J. Tu and B. Jiang, J. Org. Chem., 2017, 82, 3605 CrossRef CAS PubMed.
- (a) J.-Y. Li, Q.-Y. Li, B. Jiang and S.-J. Tu, RSC Adv., 2013, 3, 5056 RSC; (b) B. Jiang, Y. Li, M.-S. Tu, S.-L. Wang, S.-J. Tu and G.-G. Li, J. Org. Chem., 2012, 77, 7497 CrossRef CAS PubMed.
- (a) M.-Y. Li, H.-W. Xu, W. Fan, Q. Ye, X. Wang, B. Jiang, S.-L. Wang and S.-J. Tu, Tetrahedron, 2014, 70, 1004 CrossRef CAS; (b) F.-C. Yu, B. Zhou, H. Xu, Y.-M. Li, J. Lin, S.-J. Yan and Y.-H. Shen, Tetrahedron, 2015, 71, 1036 CrossRef CAS; (c) X.-B. Chen, H.-R. Bai and C. Huang, Chin. J. Org. Chem., 2017, 31, 881 CrossRef; (d) X.-B. Chen, J.-W. Gong, X.-D. Zhang, X.-L. Liu, W. Liu and Y.-C. Wang, Tetrahedron, 2018, 74, 401 CrossRef CAS.
- (a) B. Jiang, X. Wang, M.-Y. Li, Q. Wu, Q. Ye, H.-W. Xu and S.-J. Tu, Org. Biomol. Chem., 2012, 10, 8533 RSC; (b) C.-P. Cao, W. Lin, M.-H. Hu, Z.-B. Huang and D.-Q. Shi, Chem. Commun., 2013, 49, 6983 RSC.
- Z.-Y. Gu, J.-J. Gao, Y.-S. Wang and S.-J. Ji, Chem. Sci., 2016, 7, 4067 RSC.
- (a) N. Ma, B. Jiang, G. Zhang, S.-J. Tu, W. Waver and G.-G. Li, Green Chem., 2010, 12, 1357 RSC; (b) S. Rana, M. Brownb and C. Mukhopadhyay, RSC Adv., 2013, 3, 3291 RSC; (c) F.-C. Yu, B. Zhou, H. Xu, K.-J. Chang and Y.-H. Shen, Tetrahedron Lett., 2015, 56, 837 CrossRef CAS; (d) P. Zhou, B. Hu, J. Yang, L.-D. Li, K.-R. Rao, D.-F. Zhu and F.-C. Yu, Eur. J. Org. Chem., 2017, 29, 7256 CrossRef; (e) X.-J. Tu, W.-J. Hao, Q. Ye, S.-S. Wang, B. Jiang, G.-G. Li and S.-J. Tu, J. Org. Chem., 2014, 79, 11110 CrossRef CAS PubMed; (f) J.-P. Wan, Y.-F. Lin, Y.-F. Jin, M.-L. Xu and Y.-Y. Liu, Tetrahedron, 2014, 70, 7874 CrossRef CAS; (g) S. Cao, S.-S. Zhong, C.-F. Hu, J.-P. Wan and C.-P. Wen, Chin. J. Chem., 2015, 33, 568 CrossRef CAS.
- X.-B. Chen, X.-Q. Wang, J.-N. Song, Q.-L. Yang, W. Liu and C. Huang, Org. Biomol. Chem., 2017, 15, 3611 CAS.
- (a) J. A. Makawana, M. P. Patel and R. G. Patel, Chin. Chem. Lett., 2012, 23, 427 CrossRef CAS; (b) C. Hundsdörfer, H. J. Hemmerling, C. Götz, F. Totzke, P. Bednarski, M. L. Borgne and J. Jose, Bioorg. Med. Chem., 2012, 20, 2282 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures. CCDC 1819954. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra01236a |
| This journal is © The Royal Society of Chemistry 2018 |