
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToward simultaneous toughening and reinforcing of trifunctional epoxies by low loading flexible reactive triblock copolymers†
Bing Tanga,
Miqiu Kongb,
Qi Yanga,
Yajiang Huang *a and
Guangxian Li*a
*a and
Guangxian Li*a
aCollege of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering of China, Sichuan University, Chengdu 610065, P. R. China. E-mail: hyj@scu.edu.cn; guangxianli@scu.edu.cn
bSchool of Aeronautics and Astronautics, Sichuan University, Chengdu 610065, P. R. China
First published on 14th May 2018
Abstract
Flexible reactive poly(glycidyl methacrylate)-b-poly(propylene glycol)-b-poly(glycidyl methacrylate) (GPG) and nonreactive poly(ethylene glycol)-b-poly(propylene glycol)-b-poly(ethylene glycol) (EPE80) were utilized to toughen a trifunctional epoxy (diglycidyl 4, 5-epoxycyclohexane-1, 2-dicarboxylate, TDE-85). In comparison with the nonreactive EPE80 and reactive GPG92 with long reactive blocks (Lreactive), the incorporation of reactive GPG83 with short Lreactive improved the comprehensive mechanical properties of the epoxy. Upon an optimal GPG83 loading of 2.5 wt%, the tensile strength, elongation at break and critical strain energy release rate (G1c) increased by ca. 31%, 45.9% and 130.8%, respectively, without sacrificing the modulus and thermal stability. Morphology characterization evidenced that micro-scale domains and nanosized vesical micelles coexisted in the nonreactive EPE80 toughened systems. However, homogeneous morphologies were formed in reactive GPG83 and GPG92 toughened systems. Fracture morphology analysis suggested that GPG can toughen epoxy thermosets by incorporating flexible PPG blocks into the epoxy network, thereby enabling an energy dissipation mechanism. The good balance between the mobility of flexible PPG and degree of cross-link density leads to the simultaneous toughening and reinforcing effect of GPG83 toward the trifunctional epoxy.
Introduction
Although epoxy resins have been widely used in coatings, adhesives and electronic encapsulates due to their superior mechanical properties and excellent processability, their employment in high performance areas such as aerospace and defense is still greatly limited. A major reason associated with this limitation is their highly cross-linked structure which endows epoxies with intrinsic brittleness. Therefore, improving the fracture toughness of epoxies has been recognized as a key task required to expanding their applications.During the past few decades, considerable efforts have been devoted to the toughening of epoxies. The most common toughening way is incorporating a second phase, such as elastomers,1–3 thermoplastic polymers,4–6 nanoparticles7–9 and block copolymers (BCPs).10–16 For elastomers or thermoplastic polymers, it usually requires an optimizing loading of ca. 10–20 wt% to obtain a desirable toughening effect. However it inevitably results in a noticeable deterioration in the thermomechanical properties or a series of processing problems due to the increase in viscosity. In comparison, BCPs which consist of at least one epoxy-philic block and epoxy-phobic block, such as poly(ethylene glycol)-b-poly(propylene glycol)-b-poly(ethylene glycol) (EPE),17 have attracted special attention because of their superior toughening efficiencies at relatively low loadings (usually ≤5 wt%) without significantly reducing the modulus or glass transition temperature (Tg).11,13,18 The superior toughening effect of BCPs usually stems from the formation of nanostructures with spherical, vesical and wormlike morphologies within the epoxy matrix through self-assembly or reaction induced phase separation (RIPS) mechanism.13,15,18–21
According to the interaction between epoxy-philic block and epoxy resin, the BCPs could be classified into two types: the first one is nonreactive (e.g., poly(ethylene oxide),17 polycaprolactone22 and poly(methyl methacrylate)23) which can improve the miscibility of BCPs with the epoxy precursors by forming noncovalent interaction (e.g., intermolecular hydrogen bonding). While the other one can react with epoxy matrix, preventing a macrophase separation of BCPs (e.g., epoxidized polybutadiene,24–26 poly(glycidyl methacrylate),19,27 and poly(methacrylic acid)15,28). Due to the covalent bonding between nanostructures and epoxy matrix after curing, reactive BCPs would be a more promising strategy in improving the thermomechanical properties of epoxy resins. Rebizant et al.27 and Guo et al.19 have reported that upon a low loading of glycidyl methacrylate-based copolymers in epoxy, various nanosized structures were formed and noticeable improvement in the toughness and tensile properties were observed without a compromise in Tg possibly due to the improved cross-link density of the epoxy matrix. Recently, Zhang et al.15 have systematically studied the mechanical propertied of diglycidyl ether of bisphenol (DGEBA)/diaminodiphenyl methane (DDM) system modified by poly[styrene-alt-(maleic anhydride)]-b-polystyrene-b-poly(n-butyl acrylate)-b-polystyrene with different reactivities. They found that the inclusion size of BCPs decreased from micrometer to nanometer when simply adjusting the fraction of reactive blocks and the toughening effect was improved with the decrease in inclusion size.
However, there still exists an interesting question for reactive BCP toughened epoxies that whether a longer reactive block length or a higher reactivity of BCPs will result in better mechanical properties and toughening effect. Moreover, although toughening by reactive BCPs has made a great success in common epoxies like DGEBA, its effectiveness for multifunctional epoxies with higher cross-link degrees, such as aerospace-grade high performance epoxies, has not been explored. Bates et al.18 has demonstrated that the efficacy of block copolymers in promoting fracture toughness declined noticeably when the epoxy network possessed higher cross-link densities. Based on the above analysis, poly(glycidyl methacrylate)-b-poly(propylene glycol)-b-poly(glycidyl methacrylate) (GPG) with different reactive block lengths were synthesized. The mechanical properties of trifunctional TDE-85 epoxies modified with low loadings (1–5 wt%) of reactive GPG triblock copolymers were systematically analysed by tensile tests, fracture toughness and dynamic mechanical analysis (DMA). And the results were compared with commercial nonreactive EPE modified epoxy systems. Finally, the morphology features and toughening mechanism of epoxies modified with different BCPs were analysed by transmission electron microscopy (TEM), scanning electron microscopy (SEM) and differential scanning calorimetry (DSC).
Experimental
Materials
Poly(ethylene glycol)-b-poly(propylene glycol)-b-poly(ethylene glycol) (PEG-PPG-PEG). EPE80 for short, Mn = 8400 g mol−1 by supplier, Mn = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 was characterized by BreezeTM 2 gel permeation chromatography (GPC) with 80 wt% PEG were purchased from Sigma Corp. Poly(glycidyl methacrylate)-b-poly(propylene glycol)-b-poly(glycidyl methacrylate) (PGMA-PPG-PGMA) was synthesized in our laboratory (see ESI†) and the parameters of these polymers are listed in Table 1. The PGMA34-PPG34-PGMA34 with short reactive blocks (short Lreactive) possessed a similar weight fraction of epoxy-philic blocks (GPG83, WPGMA = 83 wt%) with PEG76-PPG29-PEG76 (nonreactive EPE80, WPEG = 80 wt%). And, the polymerization degree of the PGMA81-PPG34-PGMA81 (GPG92, WPGMA = 92 wt%) with a long reactive blocks (long Lreactive) was similar with that of EPE80. The trifunctional epoxy resin, TDE-85, with an epoxide equivalent weight of ∼118 g per equivalent was obtained from Tianjin Institute of Synthetic Materials Industry, China. 4,4-Diaminodiphenyl sulfone (DDS, 98%), supplied by Shanghai Huayi resins Co., Ltd., China, was used as the curing agent. This amino hardener was dried in vacuum oven at 60 °C overnight prior to use. All materials were used as received without further purification unless stated otherwise.
700 was characterized by BreezeTM 2 gel permeation chromatography (GPC) with 80 wt% PEG were purchased from Sigma Corp. Poly(glycidyl methacrylate)-b-poly(propylene glycol)-b-poly(glycidyl methacrylate) (PGMA-PPG-PGMA) was synthesized in our laboratory (see ESI†) and the parameters of these polymers are listed in Table 1. The PGMA34-PPG34-PGMA34 with short reactive blocks (short Lreactive) possessed a similar weight fraction of epoxy-philic blocks (GPG83, WPGMA = 83 wt%) with PEG76-PPG29-PEG76 (nonreactive EPE80, WPEG = 80 wt%). And, the polymerization degree of the PGMA81-PPG34-PGMA81 (GPG92, WPGMA = 92 wt%) with a long reactive blocks (long Lreactive) was similar with that of EPE80. The trifunctional epoxy resin, TDE-85, with an epoxide equivalent weight of ∼118 g per equivalent was obtained from Tianjin Institute of Synthetic Materials Industry, China. 4,4-Diaminodiphenyl sulfone (DDS, 98%), supplied by Shanghai Huayi resins Co., Ltd., China, was used as the curing agent. This amino hardener was dried in vacuum oven at 60 °C overnight prior to use. All materials were used as received without further purification unless stated otherwise.
| Sample abbreviationa | Mn,GPC (g mol−1) | PDIGPC | Mn,NMR (g mol−1) | WPEG or PGMA in BCPb (wt%) |
|---|---|---|---|---|
| a Polymerization degree of each block (the subscript number in the abbreviation).b Weight ratio of PEG or PGMA blocks were calculated from GPC data. | ||||
| PEG76-PPG29-PEG76 (EPE80) | 12700 (8400) |
1.02 | — | 80 |
| PGMA34-PPG34-PGMA34 (GPG83) | 11900 |
1.30 | 12600 |
83 |
| PGMA81-PPG34-PGMA81 (GPG92) | 25000 |
1.37 | 31500 |
92 |
The chemical structure of epoxy, hardener and block copolymers are shown in Scheme 1.
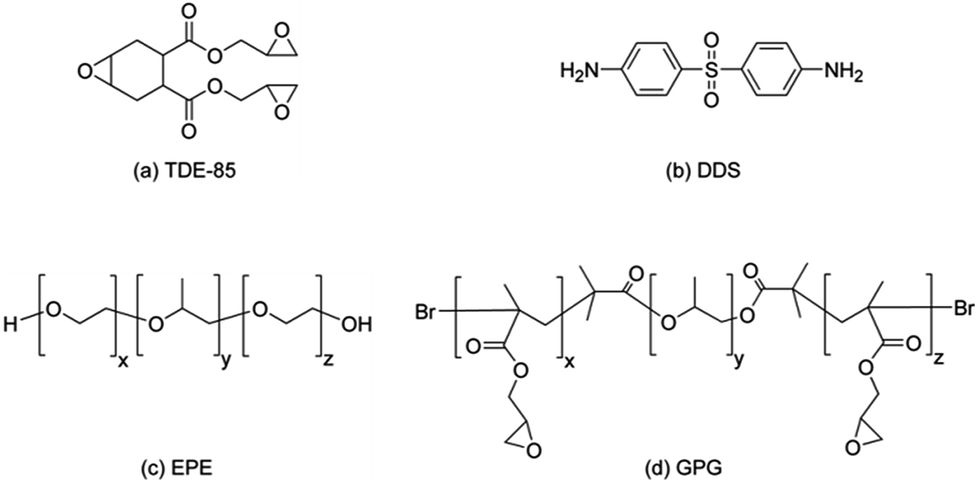 | ||
| Scheme 1 Chemical structures of (a) TDE-85; (b) DDS; (c) EPE80; (d) GPG. | ||
Preparation of BCP-modified thermosets
To prepare BCP-modified epoxy samples, the BCPs were first blended with the epoxy resin at 130 °C for 2 h until full dissolution. Then stoichiometric amounts of the curing agent DDS was added to make the mole ratio of the epoxide group to the reactive hydrogen on the amine group to be 1:1. The blends were stirred vigorously at 130 °C and degassed under vacuum for 10 min. Once a clear solution was obtained, the mixtures were poured into preheated polytetrafluoroethylene molds and cured in a multistage process at 120 °C for 3 h, 150 °C for 2 h, 180 °C for 1 h and post-cured at 200 °C for 4 h and then slowly cooled down to room temperature.
Characterization
Calorimetric measurements were made on a Q20 differential scanning calorimeter (TA instruments) under a nitrogen flow of 50 mL min−1 as purge gas. Samples of approximately 5–10 mg were placed in aluminum pans. The samples were heated from 40 to 250 °C at 5 °C min−1.FTIR were characterized by a Nicolet 6700 spectrometer (Thermo Fisher Scientific) in the transmission or reflection mode in a wavelength range from 4000 to 650 cm−1.
SEM images were obtained by a FEI Inspect F50 scanning electron microscope operated at an accelerating voltage of 20 kV. Samples were coated with a thin layer of gold prior to observation. TEM images were obtained on a FEI Tecnai G2 F20 transmission electron microscope using an accelerating voltage of 200 kV. Samples were microtomed at room temperature with a Leica EM UC6/FC6 Ultramicrotome S equipped with a glass knife. The resulting ultrathin sections (∼100 nm thick) were floated on water and were then picked up on Cu grids and stained in the vapor of a 0.5 wt% aqueous solution of RuO4 for 10 min.
Dynamic mechanical properties were carried out for cured samples on a DMA200 dynamic mechanical analyzer (TA instruments). The sample dimensions were 44 × 10 × 3 mm3. The testing was performed in dual cantilever mode at frequency of 1 Hz with a heating rate of 3 °C min−1 between 0 °C and 300 °C.
Tensile tests were performed on an Instron 5567 universal testing machine equipped with a 30 kN load cell at a loading rate of 1 mm min−1. The sample size used for the test was according to the ASTM D638-2003. For each sample, at least 5 tests were performed. Fracture toughness behaviors were measured with crack-opening tests on single-edge notch-bend (SENB) specimens which were molded according to ASTM D5045-99. The dimensions of specimens were 44 × 10 × 5 mm3. A precrack was initiated by tapping a fresh razor blade into the notch. Samples were loaded to failure at 10 mm min−1 using the universal testing machine equipped with a 1 kN load cell. For each sample, at least 8 samples were tested. The plain strain condition was verified in ESI.†
Results and discussion
Tensile properties
Fig. 1 shows the effect of triblock copolymers on the static mechanical properties of TDE-85 thermosets. For the neat thermoset, the tensile strength, Young's modulus and elongation at break are 105.4 MPa, 3.46 GPa and 5.27%, respectively. Depending on the BCP loading, the incorporation of nonreactive EPE80 increases the tensile strength and elongation at break by 6.7–17.3% and 26.4–43.3%, respectively, as compared to the neat thermoset. However, the Young's modulus decreases by 4–9.5% probably owning to the plasticization or dilute effect of soft EPE80 molecules.11,19,24 For GPG92 with a long Lreactive, moderate improvements in mechanical properties with uncompromised Young's moduli are observed. The tensile strength and elongation at break are barely increased by 9.8% and 14.8% at best, respectively. The most noticeable mechanical improvements are found in thermosets modified by GPG83 with a short Lreactive. For example, at an optimal loading of 2.5 wt%, the tensile strength and elongation at break increase by 31% and 45.9% compared with the neat thermoset, respectively. More importantly, the Young's modulus is not decreased upon the addition of GPG83. | ||
| Fig. 1 Tensile properties of epoxy thermosets modified with triblock copolymers: (a) tensile strength; (b) Young's modulus; (c) elongation at break; (d) tensile toughness determined from stress–strain curves. | ||
The toughening effect of BCPs on the thermosets is also evaluated from engineering stress–strain (σ − ε) curves. The tensile toughness was obtained by integrating the area under the stress–strain curves. Fig. 1d shows that the tensile toughness of the neat epoxy is 3.07 MJ m−3. All BCP-modified thermosets are toughened in various degrees compared with the neat epoxy. Particularly, with the incorporation of 2.5 wt% EPE80, GPG83 and GPG92, the toughness of thermosets increased by 55%, 120.8% and 24.4%, respectively. Overall, GPG83 is more efficient in toughening the trifunctional thermoset than the EPE80 and GPG92.
Fracture toughness
The stress intensity factor (K1c) and critical energy release rate (G1c) of BCPs modified thermosets determined from SENB test are shown in Fig. 2 as a function of BCP loading. The modified thermosets show various degrees of improvement in fracture toughness depending on the specie or content of BCPs. In the range of BCP loading (1–5 wt%) investigated, the toughness of the nonreactive EPE80-modified epoxy thermosets increases with increasing EPE80 loading and reaches a maximum up to 5 wt%. The K1c and G1c increase by 48.6% and 140%, respectively, as compared with the neat thermoset. The addition of 2.5 wt% GPG83 with a short Lreactive leads to 54.2% increase in K1c, from 1.05 MPa m1/2 for the neat thermoset to 1.62 MPa m1/2 for the modified thermoset. The G1c increases from 286.3 J m−2 for the neat thermoset by 130.8% to 660.7 J m−2. However, incorporation of 2.5 wt% of GPG92 with a longer Lreactive leads to a much less profound increase in K1c (25.7%) and G1c (43.6%). Especially for G1c, the amplitude of increment is only 1/3 of that of GPG83. Otherwise, it seems that the improvement in fracture toughness is independent of the GPG92 loading within the range investigated. Above results confirm again that 2.5 wt% GPG83 could significantly toughen the trifunctional thermoset while exhibiting a reinforcement effect simultaneously. | ||
| Fig. 2 Fracture toughness of different epoxies as a function of BCP loading: (a) critical stress intensity factor K1c; (b) critical strain energy release rate G1c. | ||
Dynamical mechanical properties
Representative DMA spectra shown in Fig. 3 reveal the influence of BCPs on the thermomechanical properties of epoxy thermosets. The storage modulus (E′) and glass transition temperature (Tg) estimated from the temperature at maximum tanδ value are summarized in Table 2. The E′ of the neat epoxy at room temperature is 2.85 GPa, while it decreases dramatically by ca. 24% upon adding 2.5 wt% nonreactive EPE80. This is consistent with the results reported in rubber modified epoxy thermosets, which can be ascribed to the rubber particles in the epoxy matrix and the weak interfacial interaction.1,29 However, the E′ values of epoxies modified with reactive GPG83 or GPG92 are comparable to that of pristine epoxy. These results are in good agreement with the trend of Young's modulus (Fig. 1). The neat thermoset exhibited a high Tg of 222 °C. Upon the addition of nonreactive EPE80, the Tg shifts to a lower temperature, for example, the Tg is 207 °C at 5 wt% EPE80 loading. The reduction of Tg in EPE80 systems might be attributed to two factors: (1) plasticization of partially miscible EPE80 molecules in the epoxy thermosets;21 (2) the cross-link density may decline as a result of delaying of curing process by the incorporation of PEG blocks.30,31 For GPG-modified thermosets where the nonreactive PEG blocks are substituted by reactive PGMA blocks, Tg (also the tensile properties) are similar with that of neat thermosets possibly because their cross-link density did not decrease because of the formation of covalent bonding between PGMA blocks of GPG and epoxy matrix.19
 | ||
| Fig. 3 Representative dynamical mechanical analysis results of epoxy thermosets: (a) storage modulus; (b) tanδ. | ||
| Sample | BCP loading (wt%) | E′30 °C (GPa) | E′rubbery plateau (MPa) | Tg,tanδ (°C) |
|---|---|---|---|---|
| Neat EP | 0 | 2.85 ± 0.04 | 24.77 ± 1.20 | 222 ± 4 |
| EPE80 | 1 | 2.46 ± 0.26 | 22.78 ± 0.49 | 217 ± 3 |
| 2.5 | 2.16 ± 0.27 | 21.13 ± 1.42 | 211 ± 2 | |
| 5 | 2.31 ± 0.24 | 17.97 ± 0.91 | 207 ± 2 | |
| GPG83 | 1 | 2.71 ± 0.06 | 24.78 ± 0.74 | 222 ± 3 |
| 2.5 | 2.78 ± 0.20 | 23.05 ± 1.17 | 222 ± 1 | |
| 5 | 2.68 ± 0.12 | 26.02 ± 1.00 | 225 ± 4 | |
| GPG92 | 1 | 2.64 ± 0.15 | 23.16 ± 1.34 | 222 ± 3 |
| 2.5 | 2.80 ± 0.11 | 26.11 ± 1.36 | 217 ± 2 | |
| 5 | 2.74 ± 0.05 | 27.60 ± 2.30 | 219 ± 3 |
To verify these speculations, the cross-link density is semi-quantificationally assessed by an estimated molecular weight between cross-links (Mc) which is in inverse proportion to the cross-link density.31 The Mc of the epoxy networks is calculated by using the rubber elasticity equation Mc = ρRT/Gr, where ρ is the corresponding density of epoxy thermosets at rubbery plateau (list in Table S3†), T and Gr are the Kelvin temperature and the shear equilibrium storage modulus in the rubbery plateau region (Tg + 30 K), respectively. R is the gas constant (8.314 J K−1 mol−1). The Gr value can be obtained from the following equation Gr = E′/2(1 + v), where E′ were obtained from DMA curves in the rubber plateau region (Tg + 30 K). The values of E′ increase slightly around 250 °C, as the curing process of high glass transition temperature epoxy resins do not always complete.32 So the cross-link density calculated is only valid for qualitative comparison. v is the Poisson's ratio of rubbery material which is taken to be 0.5.31 The calculated Mc values are shown in Fig. 4.
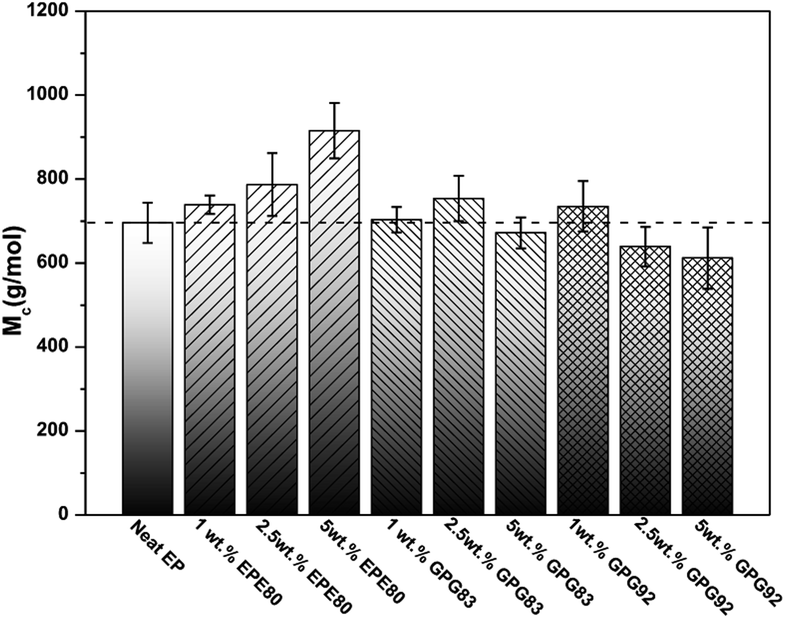 | ||
| Fig. 4 Molecular weight between the cross-links (Mc) of neat and BCPs modified epoxy thermosets are calculated from DMA data. | ||
The cross-link density of nonreactive EPE80 systems decrease gradually with the increasing of EPE loading (means a higher Mc), which can be attributed to the dilute effect of EPE toward the curing process.30,31 However, the values of Mc of both GPG-modified thermosets do not increase as a result of introduction of reactive PGMA blocks. Thus, the negative effect of lower cross-link density caused by rubbery BCP molecules can be diminished, as the reactive PGMA blocks can react with epoxy matrix simultaneously during the curing process. Moreover, with the addition of GPG92, the cross-link densities even slightly improve due to the longer reactive blocks of GPG92. In addition, the cross-link density of GPG-modified thermosets increase with increasing concentration of PGMA blocks, which is agreement with the observation by Guo et al.19
Phase behaviour
To understand the reason why the reactive BCPs can effectively toughen and reinforce the high performance TDE-85/DDS thermosets and affect their Tg, the miscibility and reactivity of the modified epoxy blends were investigated with representative BCPs loadings. For BCPs in this work, the center PPG block is epoxy immiscible while the ending PEG and PGMA blocks are epoxy miscible. Before curing, all the BCP-modified epoxy blends are homogeneous and transparent at least on the wavelength of visible light. And no scattering peaks of these blends were detected by small-angle X-ray scattering (SAXS). These results indicated that good miscibility exists between epoxy blends and BCPs through specific interactions (viz. hydrogen bonding), which have been reported by others.12,20 For BCP/TDE-85/DDS blends, oxygen atoms of epoxy-philic blocks (e.g., PEG) can form hydrogen bonding with the amino groups of DDS (or with hydroxyl groups inevitably formed from TDE-85 via ring-opening reaction during melt blending), which can result in miscibility. After curing, the nonreactive EPE80-modified thermoset becomes obscure (Fig. 5b), indicating the occurrence of reaction induced macroscopic phase separation. However, both thermosets containing reactive GPG copolymers are transparent (Fig. 5c and d), indicating the lack of macroscopic phase separation at a visual wavelength scale. To further investigate the morphologies of modified thermosets, the SEM images of cryogenic fractured surfaces of representative samples are shown in Fig. 6. After the incorporation of EPE80, lots of nanoscale voids (ca. 80 nm) coexist with few microscale voids (ca. 850 nm) on the fracture surface (Fig. 6a), which are generated during crack propagation. This can be indicative of the existence of micro and nanoscale phase separation in EPE80-modified systems, which lead to the opacity of this sample (Fig. 5b). However, the fracture surface of GPG-modified thermosets (Fig. 6b and c) does not display any apparent voids as revealed in EPE80-modified sample, implying that at least macrophase separation was hindered with the incorporation of reactive PGMA blocks. | ||
| Fig. 5 Optical images of cured epoxy thermosets: (a) neat thermoset; 2.5 wt% (b) EPE80; (c) GPG83 and (d) GPG92 modified thermosets. | ||
 | ||
| Fig. 6 Representative SEM images of cryogenic fractured surfaces of thermosets containing 2.5 wt% (a) EPE80, (b) GPG83 and (c) GPG92. | ||
The finer phase structures of the modified epoxy thermosets are further revealed by TEM (Fig. 7) considering its higher resolution and contrast in electron density. In this study, the preferentially stained order is PEG, PGMA > PPG, epoxy.10 In this case, the dark parts in the TEM images are assignable to PEG or PGMA whereas the grey regions to PPG or epoxy matrix. Fig. 7a show that a mass of blurred and irregular nanosized vesicle structures disperse uniformly in the EPE80 modified epoxy matrixes, which consist of epoxy as the core and EPE80 as the shell. Moreover, the shell is composited by inner layer of PPG and outer layer of PEG. At higher loading (5 wt%), more distinct boundary are observed as shown in Fig. S3.† In addition, a few of microscale voids are presented as a result of pulled-out of macrophase separated rubber domains during preparation of ultrathin slice, implying the interfacial interaction between particles and epoxy matrix is weak. When the nonreactive PEG blocks are replaced by the reactive PGMA blocks, a homogeneous morphology are formed in both GPG-modified epoxy thermosets (Fig. 7b and c). It should be pointed out that the differences of TEM images between those two systems are ascribed to different contrast and artifact during the slice preparation. Additional TEM images were acquired from different parts of TEM sight as shown in Fig. S4.† According to SAXS analysis (Fig. S5†), a broad scattering peak is detected in the EPE-modified system, suggesting that heterogeneous morphology was established during curing due to the reaction induced phase separation of EPE molecules. Moreover, this result indicates that the EPE particles are dispersed disorderly in the EPE-modified thermosets, which is consistent with the observation of SEM and TEM. However, no scattering signal is detected in reactive GPG systems, confirming that homogeneous morphology are formed in GPG-modified thermosets. It seems that microphase separation is hindered in these systems. Therefore, the changes in the morphology imply that the miscibility or interaction between reactive BCPs molecules and epoxy may be improved by forming covalent bonds which is much stronger than noncovalent hydrogen bonds (nonreactive EPE80 system). To further confirm this speculation, model DSC tests of BCPs/DDS, TDE-85/DDS and 50 wt% BCPs modified TDE-85/DDS blends are performed (50 wt% BCPs are used to increase the intensity of thermal effect in DSC tests). As shown in Fig. 8, EPE80/DDS blend shows two endothermic peaks with the intensive one around 51 °C corresponding to the melting of PEG blocks and the other one is associated with the melt of DDS. No apparent exothermic peak is detected in such system, confirming the nonreactivity of EPE80. As for both GPG/DDS blends, single exothermic peaks and similar exothermic range are observed compared with TDE-85/DDS, indicating that the epoxy groups of GPG and TDE-85 have similar reactivity toward the curing agent DDS. Moreover, it should be mentioned that Tp of GPG/DDS blends are slightly higher than that of TDE-85/DDS blends (Table 3) as a result of insufficient contact between GPG and DDS molecules in solid state. With regard to TDE-85/DDS, the temperature of exothermic peak (Tp) is 191.4 °C. While, with the incorporation of nonreactive EPE in TDE-85/DDS blends, Tp shifts significantly to higher temperatures (212.2 °C). This indicates that the cure kinetics is delayed due to the dilute effect of nonreactive EPE or the decline in autocatalytic process of the curing reaction as a result of the formation of hydrogen bonding between EPE80 and the epoxy mixtures.33 For TDE-85/DDS blends modified with GPG, Tp does not change significantly and a single exothermic peak is presented in DSC curve, suggesting the reactivity of PGMA blocks in GPG. The cure enthalpies are also shown in Table 3. It shifts to lower values with the addition of nonreactive EPE80 as compared with that of neat blends, due to the dilute effect of nonreactive PEG blocks. Furthermore, for such blends, a secondary peak located in the major peak (indicated by an arrow), which is associated with the macrophase separation.33 As to reactive GPG, this value is not altered apparently by the blending of GPG83 with a short Lreactive. But on the contrary, while adding of GPG92 with a long Lreactive, it is increased slightly possibly as a result of higher concentration of epoxy groups in the corresponding blend.
 | ||
| Fig. 7 Representative TEM images of TDE-85 thermosets containing 2.5 wt% of (a) EPE80, (b) GPG83 and (c) GPG92. The inset shows many nanoscale and few macroscale voids of 2.5 wt% EPE system at a lower magnification. | ||
 | ||
| Fig. 8 DSC thermograms of BCPs/DDS, TDE-85/DDS and 50 wt% BCPs modified TDE-85/DDS blends. | ||
Otherwise, the reactivity of GPG with DDS or epoxy network can be investigated by FTIR (Fig. 9). As can be seen, characteristic band of epoxy group at 906 cm−1 are found in all blends. Furthermore, the same wavelength of epoxy group were found in TDE-85 blends (Fig. S6†) might because of the similar adjacent chemical structure of epoxy group between GMA and TDE-85 (as shown in Scheme 1). The strong peaks for the bending stretching of NH2 groups of curing agent DDS also can be observed at 1630 and 3234 cm−1 in DDS and the blends containing DDS. After a completely curing process for GPG83/DDS blend, the intensity of epoxy band decreased apparently. But it is not consumed completely and normalized intensity of epoxy groups of GPG are listed in Table S4.† Moreover, the peaks of NH2 of DDS completely disappeared as well. These results confirm again that PGMA blocks of GPG can react with curing agent DDS, which have been demonstrated in DSC analysis. Whereas, the residual epoxy group of cured GPG might be the result of insufficient contact between GPG and DDS molecules in solid state. In order to prove this speculation (Fig. S6†), 50 wt% GPG83 was dissolved in epoxy blend (50 wt% GPG83 was used to increase the intensity of GPG83 in FTIR test). The characteristic band vanished in cured 50 wt% GPG83 modified epoxy thermoset confirming that epoxy group of PGMA can react completely with DDS in a homogenous state.
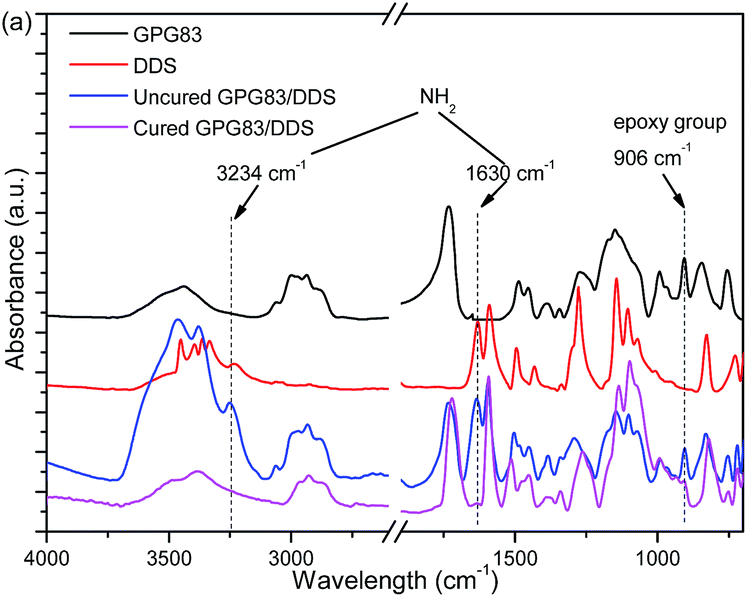 | ||
| Fig. 9 Represent FTIR spectra of uncured and cured of neat GPG83. | ||
In most types of epoxy resins, three kinds of functional reaction are wildly acceptable by many researchers.34–36 Thus possible chemical linkages of GPG with DDS or epoxy network were presented in Scheme 2 due to the similar chemical structure of epoxy groups between PGMA and TDE-85 epoxy resin.
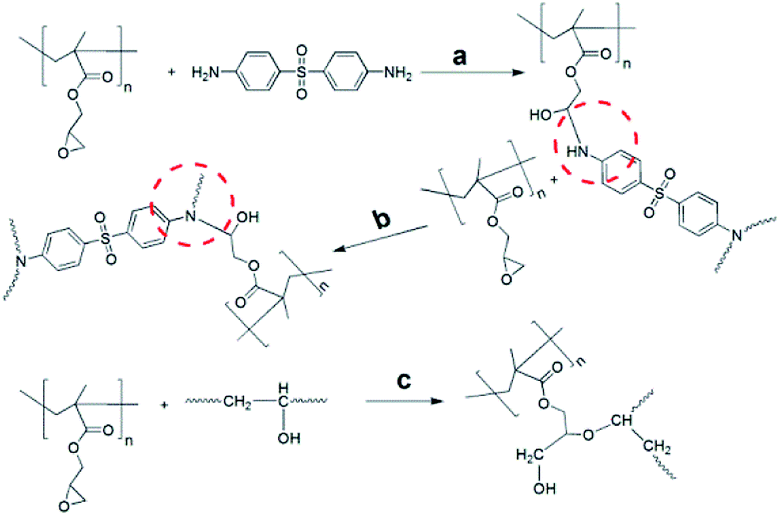 | ||
| Scheme 2 Possible chemical linkage of GPG with DDS or epoxy network. | ||
Firstly, the addition reaction between primary amine and epoxy groups (Scheme 2a); secondly, the secondary amine reacts with epoxy groups (Scheme 2b); finally, it is the etherification reaction between epoxy groups of PGMA and hydroxyl of epoxy network (Scheme 2c). Additionally, Qipeng Guo et al.19 reported that there were hydrogen bonding interactions between hydroxyl groups of epoxy and carbonyl of PGMA in a DGEBA/PDMS-PGMA/MDA system. So this interaction should not be ignored in our system either. Fig. S7† shows the FTIR of uncured and cured of neat GPG83, TDE-85, 50 wt% GPG83 modified TDE-85 blends at 2500–4000 cm−1. It is found that the bands of free hydroxyl groups and self-associated hydroxyl groups are presented at 3380 and 3490 cm−1, respectively, in the cured samples. Unfortunately, due to the similar reactive parts of PGMA and TDE-85, hydrogen bonding interactions between hydroxyl groups and carbonyl of PGMA cannot be distinguished from intramolecular hydrogen bonding of epoxy network.
Based on above analysis, the possible morphology evolution in BCP-modified systems with a change in the block structure of BCPs is illustrated in Scheme 3. For nonreactive EPE80 modified thermosets (Scheme 3a), epoxy-phobic PPG blocks will phase separate from the epoxy matrix during curing process, and the epoxy-philic PEG blocks would still stay mixed or partially mixed with the epoxy matrix due to their hydrogen bonding interaction even after the completion of curing.37 However, in this study, the hydrogen bonding interaction between EP and PEG blocks may be suppressed by intramolecular interaction between DDS and epoxy resin.38 Then, an inevitable aggregation of the nanosized particles occurred and sporadic macro domains are generated in epoxy matrix. For the GPG-modified epoxy (Scheme 3b and c), in which the nonreactive PEG epoxy-philic blocks of EPE are substituted by the reactive PGMA blocks with a proper length, homogeneous morphologies are present because the strong covalent bonding interaction prohibits the proceeding of phase separation. Furthermore, with the addition of reactive PGMA blocks, the cross-link densities of epoxy matrix are improved. And longer reactive PGMA blocks of GPG92 caused an even higher improvement of cross-link density compared with GPG83.
 | ||
| Scheme 3 Possible morphology evolution of thermosets modified with triblock copolymers with different reactive block lengths. | ||
Strengthen and toughening mechanism
The improvement of tensile strength in EPE and GPG modified thermosets at a proper BCPs loading could be related to the introduction of flexible polyether blocks into the epoxy network, which can significantly reduce the internal stress due to the shrinkage of epoxy thermosets during cooling process.39 Thus the defects (e.g., micro-cracks and voids) within epoxy thermosets caused by internal stress will be suppressed by the flexible segments, then the tensile strength is increased in our study. One the other hand, however, an unexpected reduction of modulus will occur with the incorporation of rubbery polyether blocks as it will result in a decrease of cross-link density.40 Thus, nonreactive EPE80 samples present an increase of tensile strength and a reduction of modulus. Unlike EPE80, the reactive PGMA blocks of GPG can react with the epoxy network. Then the reduction of cross-link densities caused by introducing PPG blocks can be weakened. As a result, the tensile strength is improved without a reduction in the modulus for the GPG83 samples. However, the improvement in the tensile strength is marginal for GPG92 samples, possibly due to the lager restriction of their higher cross-link density on the ability of flexible PPG block in reducing internal stress.To further investigate the toughening mechanism, representative SEM images of SENB test fracture surface of neat and modified epoxy thermosets are shown in Fig. 10. The fracture surface of the neat epoxy thermoset (Fig. 10a) is relatively smooth and mirror like due to the brittleness of the matrix. The fracture surfaces of BCPs modified epoxy are rougher than that of neat one. Nano and macroscale voids coexisted in EPE modified epoxy (Fig. 10b). During the fracture toughness test, these structures will trigger different toughening mechanisms. It has been reported that these voids are associated with the cavitation of the rubbery core of the nanostructures during sample fracture, which can dissipate fracture energy and contribute to an improved toughness41 such as voiding or debonding on the surface, and will contribute to an improvement in the toughness. Of course, a localized network damage mechanism proposed by Declet-Perez and Redline13,42 may also be responsible for the improvement of fracture toughness in such system. According to this mechanism, the presence of PEG corona will effectively disrupt the formation of epoxy network around the micelle core and could initiate the shear yielding of matrix. Furthermore, energy can be dissipated around the crack tip by plastic deformation of the epoxy network.43 In view of the work of Bates et al.,18,31 more plastic deformation can be generated by increasing the ductility of the epoxy networks due to the reduction of cross-link densities. Thus these deformation should be increase in this work as cross-link densities gradually decreased with the increasing EPE 80 loading (as shown in Fig. 4). However, the presence of these nonreactive and soft structures and the residual free EPE80 chains in the epoxy matrix will inevitably cause a noticeable decline in the modulus and Tg.
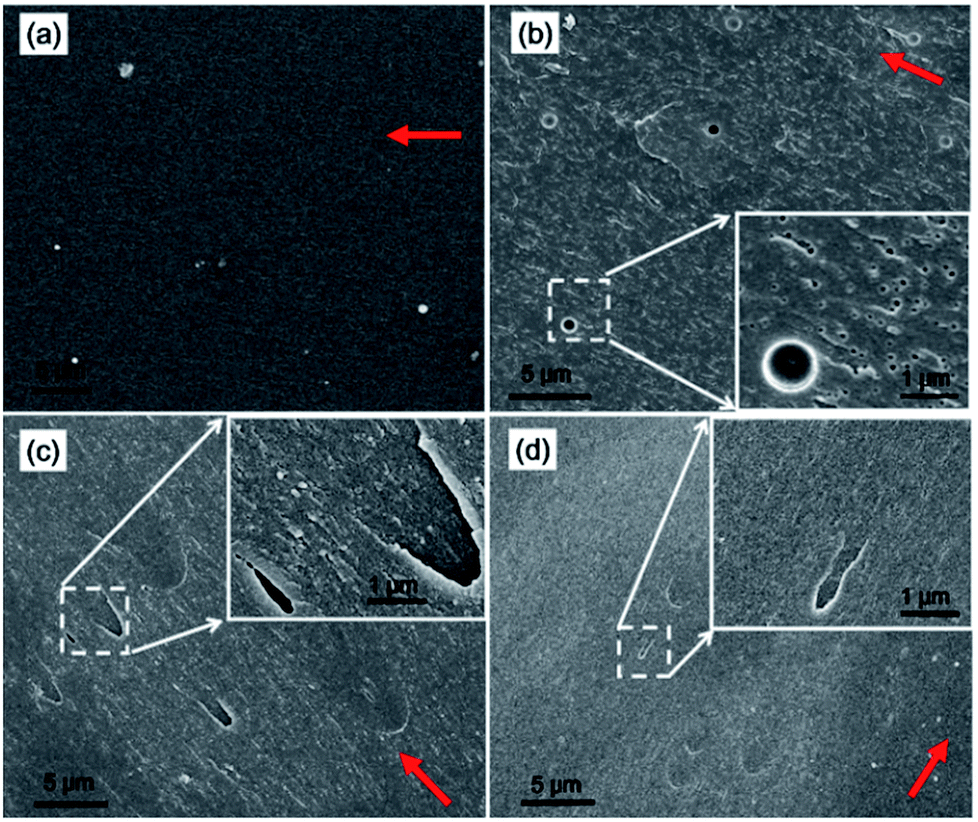 | ||
| Fig. 10 Representative SEM images of SENB test fracture surface of (a) neat epoxy and thermosets containing 2.5 wt% (b) EPE80, (c) GPG83 and (d) GPG92. The inset in (b)–(d) show the markings at an even higher magnification. Red arrows indicate the crack propagation direction. | ||
As to GPG-modified thermosets (Fig. 10c and d), the fracture surface are heterogeneous with the presence of some parabolic and elliptical markings as secondary cracks are initiated by some defects or flaws within the thermosets during crack propagation, which is related to the toughness improvement.13 Furthermore, the voids caused by the cavitation of second particles are not observed since GPG molecules are chemically bonded into the epoxy network and homogeneous morphologies are formed after curing. And the fracture surface of GPG83-modified thermosets is relatively rougher than that of GPG92-modified thermosets, demonstrating that more plastic deformation occurred in this thermosets. Thus the toughness will increase since more plastic deformation will dissipate more fracture energy during the crack propagation. This is consistent with the result presented in Fig. 2 that the GPG83-modified sample possesses the higher K1c and G1c values than the GPG92-modified thermoset.
Unlike EPE systems which toughen epoxy thermosets by forming second phase, GPG might toughen the epoxy thermosets by modification of the epoxy network. Therefore an energy dissipation mechanism will be involved due to the mobility of flexible PPG, which have been reported in reactive and homogeneous flexible polymers modified epoxy systems.40,44 However, there is also a disadvantage that the mobility of PPG blocks will be restricted by covalent bonding between PGMA and epoxy network. And this restriction will be more significant with the addition of higher GPG83 loading (5 wt%) as the cross-link density increase. Due to the higher reactivity, GPG92 shows even higher cross-link density compared with GPG83 at corresponding loading. And the cross-link density increased continuously with the increasing GPG92. As a result, the counterbalance between the mobility of PPG blocks and the contribution of PGMA blocks to cross-link density results in an optimal toughening effect in GPG83 and GPG92 at 2.5 wt% and 1 wt%, as show in Fig. 2c and d.
Conclusions
In this work, two kinds of GPG with different reactive block lengths (Lreactive) were synthesized and were used to improve the mechanical properties of trifunctional epoxy TDE-85. The incorporation of GPG83 with short Lreactive simultaneously toughened and reinforced the epoxy without compromising the Young's modulus and Tg. For thermoset modified with 2.5 wt% GPG83, the tensile strength, elongation at break and critical strain release rate increased by 31%, 45.9% and 130.8%, respectively. SEM, TEM and DSC results revealed that homogenous morphologies are formed and an energy dissipation mechanism via modification of epoxy network is involved in GPG toughened systems. However, a too high loading or too long reactive blocks of GPG may lead to deteriorated comprehensive thermomechanical properties due to the competitive effect between the mobility of flexible PPG and degree of cross-link densities. Therefore, a proper loading or length of reactive blocks of BCPs is highly desired to achieve well-balanced thermomechanical properties in high performance trifunctional epoxies like TDE-85. Overall, our results suggest that in reactive BCPs-modified thermosets, not only heterogeneous morphologies but also homogeneous morphologies can be used to toughen epoxy thermosets.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (51373109, 51721091), the Sichuan Youth Science and Technology Foundation (2015JQ0012), and the Scientific Research Foundation of International Cooperation and Exchanges of Sichuan Province (No. 2017HH0003).References
- A. J. Kinloch, S. J. Shaw, D. A. Tod and D. L. Hunston, Polymer, 1983, 24, 1341–1354 CrossRef.
- A. F. Yee and R. A. Pearson, J. Mater. Sci., 1986, 21, 2462–2474 CrossRef.
- A. J. Kinloch, MRS Bull., 2003, 28, 445–448 CrossRef.
- R. A. Pearson and A. F. Yee, Polymer, 1993, 34, 3658–3670 CrossRef.
- A. J. Kinloch, M. L. Yuen and S. D. Jenkins, J. Mater. Sci., 1994, 29, 3781–3790 CrossRef.
- R. D. Brooker, A. J. Kinloch and A. C. Taylor, J. Adhes., 2010, 86, 726–741 CrossRef.
- H. Gu, C. Ma, J. Gu, J. Guo, X. Yan, J. Huang, Q. Zhang and Z. Guo, J. Mater. Chem. C, 2016, 4, 5890–5906 RSC.
- Y. T. Park, Y. Qian, C. Chan, T. Suh, M. G. Nejhad, C. W. Macosko and A. Stein, Adv. Funct. Mater., 2015, 25, 575–585 CrossRef.
- T. H. Hsieh, A. J. Kinloch, K. Masania, A. C. Taylor and S. Sprenger, Polymer, 2010, 51, 6284–6294 CrossRef.
- Q. Guo, R. Thomann, W. Gronski and T. Thurn-Albrecht, Macromolecules, 2002, 35, 3133–3144 CrossRef.
- J. Liu, Z. J. Thompson, H.-J. Sue, F. S. Bates, M. A. Hillmyer, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Macromolecules, 2010, 43, 7238–7243 CrossRef.
- S. Wu, Q. Guo, M. Kraska, B. Stühn and Y.-W. Mai, Macromolecules, 2013, 46, 8190–8202 CrossRef.
- E. M. Redline, C. Declet-Perez, F. S. Bates and L. F. Francis, Polymer, 2014, 55, 4172–4181 CrossRef.
- L. Cano, J. Gutierrez and A. Tercjak, RSC Adv., 2015, 5, 102085–102095 RSC.
- R. He, X. Zhan, Q. Zhang, G. Zhang and F. Chen, Polymer, 2016, 92, 222–230 CrossRef.
- Y. Guo, W. Liu and Z. Wang, Acta Polym. Sin., 2016, 7, 919–924 Search PubMed.
- M. Larrañaga, E. Serrano, M. D. Martin, A. Tercjak, G. Kortaberria, K. de la Caba, C. C. Riccardi and I. Mondragon, Polym. Int., 2007, 56, 1392–1403 CrossRef.
- Z. J. Thompson, M. A. Hillmyer, J. Liu, H.-J. Sue, M. Dettloff and F. S. Bates, Macromolecules, 2009, 42, 2333–2335 CrossRef.
- N. Hameed, Q. Guo, Z. Xu, T. L. Hanley and Y.-W. Mai, Soft Matter, 2010, 6, 6119 RSC.
- M. A. Hillmyer, P. M. Lipic, D. A. Hajduk, K. Almdal and F. S. Bates, J. Am. Chem. Soc., 1997, 119, 2749–2750 CrossRef.
- H. Cong, L. Li and S. Zheng, Polymer, 2014, 55, 1190–1201 CrossRef.
- R. Yu, S. Zheng, X. Li and J. Wang, Macromolecules, 2012, 45, 9155–9168 CrossRef.
- H. Kishi, Y. Kunimitsu, J. Imade, S. Oshita, Y. Morishita and M. Asada, Polymer, 2011, 52, 760–768 CrossRef.
- H. Garate, I. Mondragon, N. B. D'Accorso and S. Goyanes, Macromolecules, 2013, 46, 2182–2187 CrossRef.
- E. Serrano, A. Tercjak, G. Kortaberria, J. A. Pomposo, D. Mecerreyes, N. E. Zafeiropoulos, M. Stamm and I. Mondragon, Macromolecules, 2006, 39, 2254–2261 CrossRef.
- R. B. Grubbs, J. M. Dean, M. E. Broz and F. S. Bates, Macromolecules, 2000, 33, 9522–9534 CrossRef.
- V. Rebizant, V. Abetz, F. Tournilhac, F. Court and L. Leibler, Macromolecules, 2003, 36, 9889–9896 CrossRef.
- V. Rebizant, A.-S. Venet, F. Tournilhac, E. Girard-Reydet, C. Navarro, J.-P. Pascault and L. Leibler, Macromolecules, 2004, 37, 8017–8027 CrossRef.
- C. Ocando, A. Tercjak, E. Serrano, J. A. Ramos, S. Corona-Galván, M. D. Parellada, M. J. Fernández-Berridi and I. Mondragon, Polym. Int., 2008, 57, 1333–1342 CrossRef.
- M. Larrañaga, M. D. Martín, N. Gabilondo, G. Kortaberria, M. A. Corcuera, C. C. Riccardi and I. Mondragon, Polym. Int., 2004, 53, 1495–1502 CrossRef.
- J. Liu, H.-J. Sue, Z. J. Thompson, F. S. Bates, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Polymer, 2009, 50, 4683–4689 CrossRef.
- O. Becker, R. J. Varley and G. P. Simon, Eur. Polym. J., 2004, 40, 187–195 CrossRef.
- M. Larrañaga, M. D. Martin, N. Gabilondo, G. Kortaberria, A. Eceiza, C. C. Riccardi and I. Mondragon, Colloid Polym. Sci., 2006, 284, 1403–1410 Search PubMed.
- N. A. St John and G. A. George, Polymer, 1992, 33, 2679–2688 CrossRef.
- V. Strehmel and T. Scherzer, Eur. Polym. J., 1994, 30, 361–368 CrossRef.
- J. Mijovic and S. Andjelic, Macromolecules, 1995, 28, 2787–2796 CrossRef.
- L. Cano, D. H. Builes and A. Tercjak, Polymer, 2014, 55, 738–745 CrossRef.
- R. Yu and S. Zheng, Macromolecules, 2011, 44, 8546–8557 CrossRef.
- G. Yang, S.-Y. Fu and J.-P. Yang, Polymer, 2007, 48, 302–310 CrossRef.
- X. Ren, S. Peng, W. Zhang, C. Yi, Y. Fang and D. Hui, Composites, Part B, 2017, 119, 32–40 CrossRef.
- C. Declet-Perez, L. F. Francis and F. S. Bates, ACS Macro Lett., 2013, 2, 939–943 CrossRef.
- C. Declet-Perez, E. M. Redline, L. F. Francis and F. S. Bates, ACS Macro Lett., 2012, 1, 338–342 CrossRef.
- H. J. Sue and A. F. Yee, Polym. Eng. Sci., 1996, 36, 2320–2326 Search PubMed.
- H. Abdollahi, A. Salimi and M. Barikani, J. Appl. Polym. Sci., 2016, 133, 44061–44074 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01017j |
| This journal is © The Royal Society of Chemistry 2018 |