
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonascus pigment rubropunctatin derivative FZU-H reduces Aβ(1-42)-induced neurotoxicity in Neuro-2A cells
Yunquan Zheng *ac,
Qisheng Pana,
Liuda Moa,
Wenyi Zhanga,
Yunjian Duana,
Chengqun Chenb,
Haijun Chen*a,
Yanghao Guoc,
Xianai Shic and
Jianmin Yangc
*ac,
Qisheng Pana,
Liuda Moa,
Wenyi Zhanga,
Yunjian Duana,
Chengqun Chenb,
Haijun Chen*a,
Yanghao Guoc,
Xianai Shic and
Jianmin Yangc
aCollege of Chemistry, Fuzhou University, 2 Xueyuan Road, Fuzhou 350116, Fujian 350108, China. E-mail: yunquanzheng@fzu.edu.cn; chenhaij@gmail.com; Fax: +86-591-22866237; Tel: +86-591-22866234
bDepartment of Chemical Engineering, Fuzhou University, Zhicheng College, 523 Gongye Road, Fuzhou 350002, China
cFujian Key Laboratory of Medical Instrument and Pharmaceutical Technology, Fuzhou University, 2 Xueyuan Road, Fuzhou 350116, China
First published on 14th May 2018
Abstract
Alzheimer's disease (AD) is an extremely complex disease, characterized by several pathological features including oxidative stress and amyloid-β (Aβ) aggregation. Blockage of Aβ-induced injury has emerged as a potential therapeutic approach for AD. Our previous efforts resulted in the discovery of Monascus pigment rubropunctatin derivative FZU-H with potential neuroprotective effects. This novel lead compound significantly diminishes toxicity induced by Aβ(1-42) in Neuro-2A cells. Our further mechanism investigation revealed that FZU-H inhibited Aβ(1-42)-induced caspase-3 protein activation and the loss of mitochondrial membrane potential. In addition, treatment of FZU-H was proven to attenuate Aβ(1-42)-induced cell redox imbalance and Tau hyperphosphorylation which caused by okadaic acid in Neuro-2A cells. These results indicated that FZU-H shows promising neuroprotective effects for AD.
1. Introduction
Alzheimer's disease (AD), characterized by abundant senile plaques (SP) comprised of the amyloid-beta (Aβ) peptide and neurofibrillary tangles (NFT) formed from the highly phosphorylated Tau protein, is the leading cause of dementia in the elderly.1–3 In vitro and in vivo studies demonstrated that Aβ peptide or Aβ peptide fragments induced the disruption of the mitochondrial membrane,4,5 resulting in the production of reactive oxygen species (ROS)6–8 and apoptosis of neurons directly or indirectly.9–12 Moreover, oxidative stress has an important function in the early stages of AD.13,14 Especially, overexpression of superoxide dismutase (SOD) in animals showed neuroprotective effects in Aβ-induced AD and neurotoxicity.15 On the other hand, several studies suggested that hyperphosphorylated Tau protein also caused mitochondrial dysfunction and induced oxidative stress.16,17 In addition, hyperphosphorylated Tau protein also exerted a neurotoxic effect when expressed in neuronal cells, via induction of chromatin condensation, DNA fragmentation, and caspase-3 activation, and finally resulted in a region-specific neurodegeneration.18–20 Therefore, inhibition of Aβ deposition, Tau phosphorylation, and oxidative stress is an important goal to alleviate the occurrence of AD.Red fermented rice (also called red yeast rice) has been widely used in East Asia for centuries as a source of folk medicine to improve food digestion and blood circulation.21,22 Several significant biological activities of red fermented rice were reported, including regulation of plasma cholesterol levels,23–25 reduction of blood glucose levels in diabetic rats26,27 and improvements of the memory and learning ability in Aβ-infused rats.28,29 Various metabolites were identified from the Monascus species, including azaphilones,30–32 furanoisophthalides, amino acids,33,34 polyketides,22 and fatty acids.35 However, the clear active ingredient in red fermented rice and the mechanisms of the function to repress Aβ peptide-induced neurotoxicity was largely undefined.
In recent years, it has been reported that azaphilones exhibited a wide range of biological activities including antimicrobial, antiviral, anticancer, antioxidant and anti-inflammatory activities.36,37 In particular, lovastatin and simvastatin (Fig. 1), one series of azaphilones, can reduce the production of Aβ fragments.38,39 In addition, azaphilones has been reported to exhibit anti-inflammatory and antioxidative effects which were regarded as the key factors against Aβ-induced neurotoxicity. These results indicated that it was reasonable to explore the neuroprotective effects of azaphilones against Aβ-induced apoptotic death and to investigate the associated potential mechanisms. In our continuing effort to develop potential therapeutic agents for AD, a series of rubropunctatin derivatives were designed, synthesized, and biological evaluated. Among them, compound FZU-H (Fig. 1) displayed significant neuroprotective effect. In this work, we further investigated the potential mechanisms of how FZU-H protected the neuronal cells against Aβ(1-42)-induced neurotoxicity and related signaling pathways.
 | ||
| Fig. 1 Chemical structures of lovastatin, simvastatin, rubropunctatin and rubropunctatin derivative FZU-H. | ||
2. Experimental section
2.1 General
Neuro-2A cell line was purchased from the Cell Resource Center (Beijing, China). Minimum Essential Medium Eagles with Earle's Balanced Salts (MEM-EBSS) and fetal bovine serum (FBS) were obtained from HyClone (Logan, UT, USA). Mouse Aβ(1-42) was purchased from GL Biochem (Shanghai, China). Hexafluoroisopropanol (HFIP) was purchased from Aladdin (Shanghai, China). 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT), dimethylsulfoxide (DMSO) and okada acid (OA) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). The Annexin V FITC kit, normal/apoptotic/necrotic cell detection kit, reactive oxygen species assay kit, and the total superoxide dismutase assay kit were obtained from KeyGEN biotech (Nanjing, China). The Caspase 3 activity assay kit, mitochondrial membrane potential detection kit (JC-1), RIPA Buffer and BCA Protein Assay Kit were obtained from Beyotime (Haimen, China). β-Actin rabbit monoclonal antibody was purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA), phospho-Tau pThr205 antibody was purchased from Pierce Biotechnology (Pierce, USA). Anti-Tau (Phospho S396) antibody, anti-Tau (Phospho S202) antibody and HRP-conjugated anti-rabbit IgG polyclonal secondary antibody were purchased from Abcam Ltd. (New Territories, Hong Kong). All chemicals used were of analytical grade.2.2 Purification and identification of FZU-H
Monascus azaphilone rubropunctatin (above 98.5% purity) was purified from red mold rice (Fuzhou Longlixin Biological Product Company) by High-speed Counter-current Chromatography (Zheng et al. 2010). Rubropunctatin was reduced by sodium borohydride to give the compound FZU-H (above 98.5% purity), which was identified by HPLC-MS and NMR spectroscopy. The detailed preparation and purification procedure was as follow: to a solution of Monascus azaphilone rubropunctatin (354 mg, 1 mmol) in CH3OH (10 mL) was added a solution of NaBH4 (112 mg, 3 mmol) in CH3OH (20 mL). The resulting solution was stirred at room temperature for 12 h (the progress of the reaction was monitored by TLC). Then H2O (5 mL) and CHCl3 (15 mL) were added. The combined organic phases were washed with brine, dried (anhyd Na2SO4), filtered, and concentrated. The residue was purified by column chromatography on silica gel, eluting with CH3OH–CHCl3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), to give FZU-H; yield: 249 mg (70.3%). An Agilent 1100 series ion trap LC/MS systems (Agilent, USA) was used to determine the molecular weight. The mass spectrometer was equipped with an electrospray ionization (ESI) source. The ESI conditions were as follows: capillary voltage, 3.5 kV; nebulizer pressure, 40 psi; drying gas flow, 10 mL min−1; temperature, 350 °C. The mass range was from m/z 100 to 400.
:1, v/v), to give FZU-H; yield: 249 mg (70.3%). An Agilent 1100 series ion trap LC/MS systems (Agilent, USA) was used to determine the molecular weight. The mass spectrometer was equipped with an electrospray ionization (ESI) source. The ESI conditions were as follows: capillary voltage, 3.5 kV; nebulizer pressure, 40 psi; drying gas flow, 10 mL min−1; temperature, 350 °C. The mass range was from m/z 100 to 400.
1H and 13C NMR spectra were measured with an Inova-500M NMR spectrometer (Varian, USA) in CDCl3 operating at 500 MHz for 1H NMR and at 125 MHz for 13C NMR. Tetramethylsilane (TMS) was employed as an internal standard.
FZU-H: ESI-MS, m/z 357.1 [M + H]+; 1H NMR (CDCl3, 500 MHz), δ0.87 (3H, t, H-19), 1.304–1.335 (4H, m, H-17-18), 1.55 (3H, s, H-10), 1.61 (2H, m, H-16), 1.93 (3H, d, H-13), 2.14 (1H, brs, O–H), 2.93 (2H, m, H-15), 4.37 (1H, s, H-4), 6.01 (1H, d, H-11), 6.13 (1H, s, H-8), 6.57 (1H, dq, H-12), 6.88 (1H, s, H-9), 6.97 (1H, s, H-5); 13C NMR (CDCl3, 125 MHz), δ13.963 (C-19), 18.735 (C-10), 22.569 (C-13), 23.380 (C-18), 28.283 (C-16), 31.338 (C-17), 41.533 (C-15), 48.928 (C-4), 67.650 (C-3a), 104.173 (C-8), 104.377 (C-4a), 115.560 (C-9), 116.293 (C-11), 118.666 (C-1), 136.345 (C-12), 139.081 (C-5), 141.582 (C-8a), 155.661 (C-7), 169.193 (C-2), 179.257 (C-9a), 190.772 (C-14).
2.3 Cell culture and drug preparation
Neuro-2A mouse neuroblastoma cells were grown and maintained in MEM-EBSS media containing 10% FBS, 2 mM glutamine, 40 U mL−1 gentamicin at 37 °C under 5% CO2. FZU-H was dissolved in sterile water, and stored in the dark at −20 °C until used. Preparation of Aβ(1-42) peptide was performed following the manufacturer recommendations. The Aβ(1-42) peptide was initially dissolved to 1 mM in HFIP and separated into aliquots in sterile microcentrifuge tubes. HFIP was removed under vacuum in a freeze dryer (FDU-1200, Shimadzu Instruments, Japan), and the peptide film was stored dessicated at −80 °C. When used, the peptide was first resuspended in dry DMSO to a concentration of 5 mM, and then 10 mM HCl was added to bring it to a final concentration of 100 μM and incubated for 24 h at 37 °C.2.4 MTT assay for cell viability
Neuro-2A cell viability was measured using the MTT assay. Briefly, cells (1 × 105 cells per mL) were plated into 96-well plates in a total volume of 100 μL of culture medium and then cultured for 12 h at 37 °C, 5% CO2. After exposure to different concentration of FZU-H and Aβ(1-42) for 24 h, 10 μL MTT solution was added to each well and the plates were incubated for additional 4 h at 37 °C. To achieve solubilization of the formazan crystal formed in viable cells, the medium containing MTT was aspirated off and 150 μL of DMSO was added to each well. The absorbance was read at 570 nm using SH-1000 microplate reader (Corona Electric, Hitachinaka, Japan).2.5 Cell survival assay (fluorescent staining)
Neuro-2A cells were analyzed in the presence of acridine orange and ethidium bromide staining (AO/EB staining). Neuro-2A cells were plated into six well plates at a density of 5 × 105 cells per mL and incubated for 12 h. After exposure to different concentration of FZU-H and Aβ(1-42) for 24 h, the cells were harvested by trypsin without EDTA and washed with phosphate-buffered saline (PBS). Dye mixture (1 mL) was mixed with 25 μL of cell suspension (5 × 105 cells per mL) on a clean microscope slide and then immediately examined by fluorescence microscopy (Olympus IX71, Tokyo, Japan) at 400× magnification.2.6 Apoptosis assay
To detect early apoptosis and late apoptosis/necrosis, the cells were stained with FITC-conjugated Annexin V and PI after treatment with different concentration of FZU-H and 10 μM of Aβ(1-42) for 24 h. The cells were harvested by trypsin without EDTA and washed with PBS before being resuspended in 500 μL of binding buffer (1 M Hepes and 2 M NaCl, pH 7.0). 5 μL of Annexin V-FITC and 5 μL PI were added and incubated for 15 min at room temperature in the dark. Neuro-2A cells (1 × 104) were analyzed immediately by a Coulter EPICS XL flow cytometer (Beckman Coulter Co. Ltd., FL, USA).2.7 Measurement of caspase-3 activity
The activity of caspase-3 was determined by using the Caspase-3 activity kit according to the manufacturer's protocol. To evaluate the activity of caspase-3, cell lysates were prepared after their respective treatment. Assays were performed on 96-well plates by incubating 10 μL of cell lysate per sample in 80 μL reaction buffer containing 10 μL caspase-3 substrate acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA, 2 mM). Lysates were incubated at 37 °C for 2 h and then measured with a microplate reader (Corona Electric, Hitachinaka, Japan) at an absorbance of 405 nm.2.8 Determination of mitochondrial membrane potential
Mitochondrial transmembrane potential was measured by using JC-1 dye, a fluorochrome which exclusively emits within the spectrum of green light and accumulates in the mitochondrial matrix under the influence of mitochondrial transmembrane potential. JC-1 easily penetrates cells and healthy mitochondria. A green fluorescent JC-1 probe exists as a monomer at low membrane potentials; however, at higher potentials, JC-1 forms red-fluorescent ‘J-aggregates’. Neuro-2A cells (3 × 105 cells per mL) were cultured in 6-well plates (2 mL per well) overnight and then incubated with different concentration of FZU-H and 10 μM of Aβ(1-42) for 24 h. Neuro-2A cells were collected by trypsinization and incubated with medium containing JC-1 for 20 min at 37 °C. Then the cells were washed with PBS, resuspended in 0.5 mL buffer, and analyzed with Epics XL flow cytometer (Beckman-Coulter, Fullerton, CA) at FL1 (529 nm) for green fluorescence and FL2 (590 nm) for red fluorescence.2.9 Measurement of ROS
Generation of ROS was assessed by DCFH-DA, an oxidation-sensitive fluorescent probe. An increase in fluorescence intensity is used to quantify the generation of net intracellular ROS. After treatment of cells with different concentrations, the cells were incubated with 10 μM DCFH-DA at 37 °C for 30 min and then washed twice with PBS. The fluorescence intensity of DCF was measured by Epics XL flow cytometer. ROS generation was quantified by the median fluorescence intensity of 1 × 104 cells.2.10 Measurement of SOD activity
The activity of SOD was measured following the manufacturer's manual with subtle changes. Briefly, the cells were lysated after their respective treatment. The homogenate was centrifuged at 12000 rpm for 8 min at 4 °C to precipitate insoluble material. The supernatant was assayed for SOD activity following the manufacturer's manual and the absorbance at 550 nm was read with a UV-1800 spectrophotometer (Shimadzu Corporation, Tokyo, Japan).
2.11 Western blotting assay
Cells (1 × 107 cells) in different treatment groups were collected and washed with PBS. After centrifugation, sedimented cells were lysed by vigorous shaking in RIPA Lysis Buffer (Beyotime Institute of Biotechnology, Haimen, China) with PMSF for 20 min at 4 °C, and then centrifuged again at 4 °C, 12000 rpm for 15 min. The supernatant was separated and collected for protein analysis and the protein concentration was determined by using the BCA protein assay kit (Beyotime Institute of Biotechnology, Haimen, China). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was performed by loading equal amounts of proteins per lane. The gels were then transferred to PVDF membranes (Millipore) and blocked with 5% BSA in tris(hydroxymethyl)aminomethane–NaCl–Tween20 (TBST) buffer at 4 °C with slightly shaking for 2 h. Then the membranes were incubated with rabbit anti-Tau(phospho ser396) antibody (1:25000), anti-Tau(phospho ser202) antibody (1:10000), anti-Tau(phospho-thr205) antibody (1:750) or anti-β-actin antibody (1:2000) at 4 °C overnight. The secondary antibodies conjugated with horseradish peroxidase at 1:15000 dilutions for 2 h at room temperature. The detection and quantification of specific bands were performed using a fluorescence scanner (Molecular Imager ChemiDoc XRS System, Bio-Rad, Hercules, CA).
2.12 Statistical analysis
The results were expressed as the mean ± standard deviation (SD) from three independent experiments. Statistical analysis was performed using Student's t test. The differences were considered significant for p < 0.05 (*), and p < 0.01 indicated very significant differences (**).3. Results and discussion
3.1 FZU-H ameliorated Aβ(1-42)-induced neurotoxicity
To assess Aβ(1-42)-induced Neuro-2A cells death, the cells were analyzed on MTT assay. As shown in Fig. 2, when Neuro-2A cells were exposed to Aβ(1-42) at different concentration (1, 5, 10 and 15 μM) for 24 h, 4.2 ± 1.6%, 24.2 ± 3.3%, 34.5 ± 3.0% and 48.8 ± 2.0% of neuronal cell death was observed, respectively. Therefore, for the following experiments, the concentration of 10 μM was used for the determination of Aβ(1-42)-induced Neuro-2A cell damage. | ||
| Fig. 2 Effect of FZU-H on cell viability in Aβ(1-42)-induced cytotoxicity. Each bar represents the mean ± SD from six independent experiments (n = 6, *p < 0.05 and **p < 0.01 versus control group; #p < 0.05 versus the group treated Aβ(1-42) alone). | ||
The cell viability was employed to observe the protective effect of FZU-H on Aβ(1-42)-induced neurotoxicity in Neuro-2A cells. As illustrated in Fig. 2, treatment of FZU-H significantly increased Neuro-2A cell viability in a dose-dependent manner, indicating that azaphilone derivative FZU-H exhibited predominant neuroprotective effect against Aβ(1-42)-induced injury.
3.2 Microscopic analysis and fluorescence microscopy to observe morphological characteristics of Neuro-2A cells
As shown in Fig. 3, Aβ(1-42) treatment caused the significant damage of Neuro-2A cells (Fig. 3B) in comparison to the control (Fig. 3A). While the addition of 50 μM FZU-H can protect Aβ(1-42)-induced damage (Fig. 3C). We next used AO/EB double staining to detect the apoptotic Neuro-2A cells. The results are represented in Fig. 3D and E. The figures showed that the Aβ(1-42) treated group (Fig. 3E) induced apoptosis after 24 h of incubation at 10 μM concentration compared with the control group without Aβ(1-42) treatment (Fig. 3D), and cells showed changes in cellular morphology including chromatin condensation and fragmented nuclei. Cells stained green represented viable cells, whereas yellow staining represented early apoptotic cells, and reddish or orange staining represented late apoptotic cells. As shown in Fig. 3F, Neuro-2A cells treated with 50 μM FZU-H can significantly prevent the changes of cellular morphology induced by Aβ(1-42). | ||
| Fig. 3 Effect of FZU-H on morphological characteristics of Neuro-2A cells. Morphological studies by inverted microscope at actual magnification 400×; (A). Normal control group; (B). Aβ(1-42) treatment group; (C). Aβ(1-42) + 50 μM FZU-H treatment group. Morphological observation with AO/EB staining at actual magnification 100×: (D). Normal control group; (E). Aβ(1-42) treatment group; (F). Aβ(1-42) + 50 μM FZU-H treatment group. | ||
3.3 Protective effect of FZU-H on Aβ(1-42)-induced apoptosis of Neuro-2A cells
To determine the apoptotic response in Neuro-2A cells, we initially detected the effect of FZU-H treatment using Annexin/PI staining by flow cytometry. As shown in Fig. 4, there was a significant difference between the FZU-H treatment groups and the Aβ(1-42) group. The ratio of living cells, as evidenced by Annexin V/PI negativity in the Aβ(1-42) group (73.7%), was significantly lower than that of the control group (95.4%). The percentage of apoptotic cells both control group and Aβ(1-42) treatment group were 3.6% (Fig. 4A) and 19.2% (Fig. 4B). While the percentage of FZU-H treatment group was 11.4% (10 μM, Fig. 4C), 10.8% (50 μM, Fig. 4D) and 6.0% (100 μM, Fig. 4E). The results indicated that each treatment of FZU-H can reduce Aβ(1-42)-induced apoptotic cell death in a dose-dependent manner. | ||
| Fig. 4 Effect of FZU-H on Aβ(1-42)-induced apoptosis by flow cytometry; (A). Normal control group; (B). Aβ(1-42) treatment group; (C). Aβ(1-42) + 10 μM FZU-H treatment group; (D). Aβ(1-42) + 50 μM FZU-H treatment group; (E). Aβ(1-42) + 100 μM FZU-H treatment group. This is a representative experiment of three independent experiments. | ||
3.4 FZU-H inhibits Aβ(1-42)-induced caspase-3 protein activation
Caspase-3 activity were determined by a colorimetric assay based on the ability of caspase-3 to change Ac-DEVD-pNA into a yellow formazan product (p-nitroaniline, pNA). Under the exposure of Aβ(1-42) at 10 μM, caspase-3 activity significantly increased (182.5%), in comparison to the control without Aβ(1-42) treatment (Fig. 5). FZU-H significantly blocked the Aβ(1-42)-induced increase of caspase-3 activity. These results implicated that Aβ(1-42)-induced apoptosis occurs through the activation of common executors of apoptosis such as caspase-3, and FZU-H inhibited caspase-3 activity appeared to participate in preventing Aβ(1-42)-induced apoptosis pathway. | ||
| Fig. 5 Inhibitory effect of FZU-H on Aβ(1-42)-induced caspase-3 expression in cultured Neuro-2A cells. The cells were incubated with 10 μM Aβ(1-42) and different concentration of H (10, 50 and 100 μM) for 24 h. The relative activities of caspase-3 shown are calculated from the average of three experiments. Each value was expressed as the ratio of caspase-3 activation level to control level, and the value of control was set to 100 (n = 3, *p < 0.05 and **p < 0.01 compared with normal control group; #p < 0.05 and ##p < 0.01 compared with Aβ(1-42) treatment group). | ||
3.5 Effect of FZU-H on cellular ROS levels
When Neuro-2A cells were treated with 10 μM Aβ(1-42) for 24 h, cells were harvested to evaluate ROS production and detected by flow cytometry. We found that the fluorescence intensity significantly increased from 6.0 to 22.3 by using DCFH-DA assay. However, treatment with different concentration of FZU-H decreased the ROS production in a dose-dependent manner (Fig. 6). The results suggest that ROS is generated by Aβ(1-42) treatment and FZU-H may act as a potential protective agent to prevent apoptosis by attenuating Aβ(1-42)-induced ROS production. | ||
| Fig. 6 Effect of FZU-H on Aβ(1-42)-induced ROS overproduction. After treatment of cells with different concentration of FZU-H and 10 μM Aβ(1-42) for 24 h, the cells were incubated with 10 μM DCFH-DA at 37 °C for 30 min and then washed twice with PBS. The fluorescence intensity of DCF was measured at an excitation wavelength of 488 nm and an emission wavelength of 525 nm. Relative fluorescence intensity obtained from three independent experiments (n = 3, *p < 0.05 and **p < 0.01 compared with normal control group; #p < 0.05 and ##p < 0.01 compared with Aβ(1-42) treatment group). | ||
3.6 Effect of FZU-H in Aβ(1-42)-induced SOD activity
The activity of SOD in Neuro-2A cells treated with Aβ(1-42) and different concentrations of FZU-H are shown in Fig. 7. The relative activity of SOD in cells treated with 10 μM Aβ(1-42) for 24 h was significantly lower than that of the control. Whereas, after supplemented with FZU-H (10, 50, and 100 μM), an evident elevation of SOD activity was detected in Neuro-2A cells, and the treatment of 100 μM FZU-H displayed a significant increase of 13.3% in comparison to the only Aβ(1-42) treatment group. | ||
| Fig. 7 Effects of FZU-H on the inhibition of Aβ(1-42)-induced oxidative stress (n = 3, *p < 0.05 and **p < 0.01 compared with normal control group; #p < 0.05 and ##p < 0.01 compared with Aβ(1-42) treatment group). | ||
3.7 Effect of FZU-H on mitochondrial membrane potential
The changes of mitochondrial membrane potential induced by FZU-H were determined using JC-1 as a fluorescent probe. At high mitochondrial membrane potential, JC-1 forms aggregates, which exhibits red fluorescence. At low mitochondrial membrane potential, JC-1 forms monomers, which exhibits green fluorescence. The changes of mitochondrial membrane potential are shown in Fig. 8. Results are expressed as a ratio of red/green intensities.61 The decrease of the ratio of red fluorescence and green fluorescence showed the impaired of mitochondrial membrane potential. These results suggested that FZU-H inhibited the loss of mitochondrial membrane potential caused by Aβ(1-42) in Neuro-2A cells.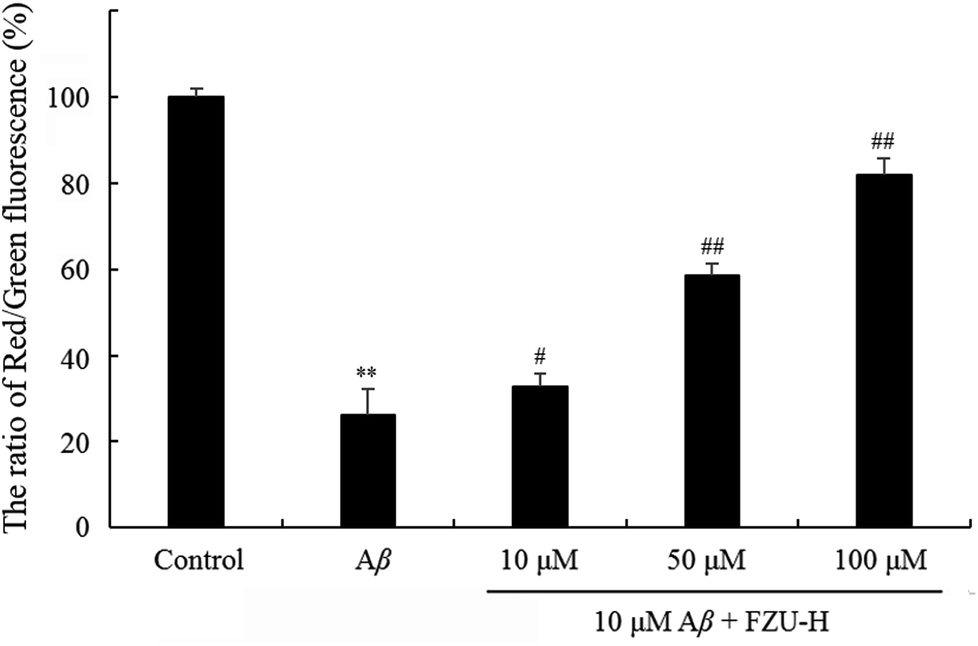 | ||
| Fig. 8 Effect of FZU-H on mitochondrial membrane potential. Mitochondrial transmembrane potential was measured by JC-1 staining. Epics XL flow cytometer at FL1 (529 nm) for green fluorescence and FL2 (590 nm) for red fluorescence (EX = 488 nm). The results were expressed as the mean ± standard deviation (SD) from three independent experiments (*p < 0.05 and **p < 0.01 compared with normal control group; #p < 0.05 and ##p < 0.01 compared with Aβ(1-42) treatment group). | ||
3.8 FZU-H inhibits hyperphosphorylation of Tau caused by okadaic acid in Neuro-2A cells
After OA treatment for 24 h in the presence or absence of FZU-H, we measured Ser396, ser202 and thr205 Tau phosphorylation by Western blot. As shown in Fig. 9, the treatment of Neuro-2A cells with 30 nM OA for 24 h significantly increased the expression of phosphorylated forms of Tau protein compared with the control group. After treatment with different concentration of FZU-H, the relative quantity of Tau exhibited different degree of reduction. In addition, treatment of 100 μM FZU-H significantly reduced Tau phosphorylation induced by OA on these three residues. | ||
| Fig. 9 Western blot analysis of the effects of OA and FZU-H on the expression of Tau in Neuro-2A cells. β-Actin was used as an internal control. The relative quantity obtained by Bio-Rad Image Lab software (n = 3, *p < 0.05 and **p < 0.01 compared with normal control group; #p < 0.05 and ##p < 0.01 compared with Aβ(1-42) treatment group). | ||
4. Discussion
AD is pathologically characterized by the presence of Aβ amyloid plaques and neurofibrillary tangles, as well as the selective loss of neurons and synaptic connections.40 Aβ plays the decisive role in the pathogenesis of AD. It is clear that Aβ-induced neurotoxicity has been attributed to cell necrotic and apoptotic,9,41,42 activation of caspase-3,26 the generation of ROS,43 oxidative stress44 and inflammation with the resultant axon transport dysfunctions and cognitive decline.45 According to this study, treatment of FZU-H was certainly proven to attenuate Aβ(1-42)-induced cell death and apoptosis, redox imbalance and OA-induced Tau phosphorylation.The cell viability and fluorescence staining in this work were employed to observe the protective effect of FZU-H on Aβ(1-42)-induced neurotoxicity in Neuro-2A cells. The results indicated that FZU-H significantly attenuated Aβ(1-42)-induced cell apoptosis and morphology damage. In Annexin/PI staining study, when the cells were treated with 10 μM of Aβ, the percentage of apoptotic cells were increased to 19.2%. After the addition of FZU-H, the apoptosis populations decreased dose-dependently. These results suggest that FZU-H can protect Neuro-2A cells against Aβ(1-42)-induced apoptosis.
Caspases are cysteine-dependent aspartate-specific proteases critically involved in apoptosis.46 Activation of caspase-3 is an important step in the execution phase of apoptosis and inhibition of this pathway blocks cell apoptosis.47 Among the caspase family, consist of more than 10 homologues, caspase-3 has been suggested to play an important role in Aβ-induced apoptosis.48 In this study, we found that treatment of Aβ(1-42) increased caspase-3 activity of Neuro-2A cells, and FZU-H significantly blocked this signaling pathway.
Studies in non-neural cells have suggested that a fall in mitochondrial membrane potential is one of the earliest events in apoptosis.49 Mitochondrial abnormalities have been identified in a large proportion of neurodegenerative diseases.50,51 In this work, we found that the deposit of Aβ protein is thought to be toxic to neurons possibly via induction of mitochondrial membrane potential, our results suggested that FZU-H abolished Aβ-induced cell death and increased mitochondrial membrane potential in Neuro-2A cells, indicating that mitochondrial membrane potential elevation is an associated phenomenon induced by Aβ that can be rescued by FZU-H.
AD is a complex disease that involves a multitude of dysfunctional processes. Oxidative damage in AD may be a direct result of Aβ. Several studies demonstrated that Aβ-induced apoptosis was associated with ROS production,52–54 including the superoxide (O2−), hydroxyl radicals and hydrogen peroxide (H2O2).55,56 Many studies revealed that Aβ-induced cell damage was due to the occurrence of oxidative stress.15,57 In this work, we demonstrated that Aβ significantly increased the ROS production in the Neuro-2A cells and pretreatment with FZU-H ameliorates this change in a dose-dependent manner. In addition, we observed that treatment with FZU-H can decrease the Aβ-induced oxidative damage.
It is worth pointing out that OA, a widely used reagent for Tau protein hyperphosphorylation, has been previously reported as a specific inhibitor of protein phosphatase 2A (PP2A),58–60 which is an important phosphatase responsible for Tau protein phosphorylation in the neurodegenerative progress. Herein, we revealed that FZU-H significantly prevented OA-induced hyperphosphorylation of Tau protein (at Thr205, Ser396 and Ser202 sites).
5. Conclusions
Aβ-induced apoptotic, highly phosphorylated Tau protein and excessive generation of ROS play decisive role in the pathogenesis of AD. Compound FZU-H could ameliorate the apoptosis of Neuro-2A cells induced by Aβ(1-42), and the anti-apoptosis ability enhanced significantly with the increase of drug concentration assayed by caspase 3 activity, mitochondrial membrane potential and Annexin V-FITC/PI staining experiments using flow cytometry. FZU-H significantly prevented OA-induced hyperphosphorylation of Tau protein, alleviated the decrease of SOD activity and inhibited the elevation of intracellular ROS level. Taken together, FZU-H shows promising neuroprotective effects.Conflicts of interest
The authors declare that they have no conflict of interest. This article does not contain any studies with human participants or animals performed by any of the authors. Informed consent was obtained from all individual participants included in the study.Acknowledgements
This work was supported by Natural Science Foundation of Fujian Province of China (No. 2017J01854), Fujian Province Young and Middle-aged Teacher Education Research Project (JAT160655), and the Marine High-tech Industry Development Special Project of Fujian Province of China (Min Marine High-tech [2015]01).References
- D. L. Price, R. E. Tanzi, D. R. Borchelt and S. S. Sisodia, Annu. Rev. Genet., 1998, 32, 461–493 CrossRef PubMed.
- M. Goedert and M. G. Spillantini, Science, 2006, 314, 777–781 CrossRef PubMed.
- O. Klementieva, E. Aso, D. Filippini, N. Benseny-Cases, M. Carmona, S. Juves, D. Appelhans, J. Cladera and I. Ferrer, Biomacromolecules, 2013, 14, 3570–3580 CrossRef PubMed.
- C. S. Casley, J. M. Land, M. A. Sharpe, J. B. Clark, M. R. Duchen and L. Canevari, Neurobiol. Dis., 2002, 10, 258–267 CrossRef PubMed.
- A. Eckert, U. Keil, C. A. Marques, A. Bonert, C. Frey, K. Schussel and W. E. Muller, Biochem. Pharmacol., 2003, 66, 1627–1634 CrossRef PubMed.
- M. T. Lin and M. F. Beal, Nature, 2006, 443, 787–795 CrossRef PubMed.
- D. A. Butterfield, T. Reed, S. F. Newman and R. Sultana, Free Radical Biol. Med., 2007, 43, 658–677 CrossRef PubMed.
- T. Schilling and C. Eder, J. Cell. Physiol., 2011, 226, 3295–3302 CrossRef PubMed.
- D. T. Loo, A. Copani, C. J. Pike, E. R. Whittemore, A. J. Walencewicz and C. W. Cotman, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7951–7955 CrossRef.
- C. Behl, J. B. Davis, F. G. Klier and D. Schubert, Brain research, 1994, 645, 253–264 CrossRef PubMed.
- H. Satoi, H. Tomimoto, R. Ohtani, T. Kitano, T. Kondo, M. Watanabe, N. Oka, I. Akiguchi, S. Furuya, Y. Hirabayashi and T. Okazaki, J. Neurosci., 2005, 130, 657–666 CrossRef PubMed.
- P. Picone, R. Carrotta, G. Montana, M. R. Nobile, P. L. San Biagio and M. Di Carlo, Biophys. J., 2009, 96, 4200–4211 CrossRef PubMed.
- A. Nunomura, G. Perry, G. Aliev, K. Hirai, A. Takeda, E. K. Balraj, P. K. Jones, H. Ghanbari, T. Wataya, S. Shimohama, S. Chiba, C. S. Atwood, R. B. Petersen and M. A. Smith, J. Neuropathol. Exp. Neurol., 2001, 60, 759–767 CrossRef PubMed.
- A. Nunomura, R. J. Castellani, X. Zhu, P. I. Moreira, G. Perry and M. A. Smith, J. Neuropathol. Exp. Neurol., 2006, 65, 631–641 CrossRef PubMed.
- A. Clausen, X. Xu, X. Bi and M. Baudry, J. Alzheimer's Dis., 2012, 30, 183–208 Search PubMed.
- D. C. David, S. Hauptmann, I. Scherping, K. Schuessel, U. Keil, P. Rizzu, R. Ravid, S. Drose, U. Brandt, W. E. Muller, A. Eckert and J. Gotz, J. Biol. Chem., 2005, 280, 23802–23814 CrossRef PubMed.
- C. Ballatore, V. M. Lee and J. Q. Trojanowski, Nat. Rev. Neurosci., 2007, 8, 663–672 CrossRef PubMed.
- T. Fath, J. Eidenmuller and R. Brandt, J. Neurosci., 2002, 22, 9733–9741 CrossRef PubMed.
- N. Shahani, S. Subramaniam, T. Wolf, C. Tackenberg and R. Brandt, J. Neurosci., 2006, 26, 6103–6114 CrossRef PubMed.
- H. L. Li, H. H. Wang, S. J. Liu, Y. Q. Deng, Y. J. Zhang, Q. Tian, X. C. Wang, X. Q. Chen, Y. Yang, J. Y. Zhang, Q. Wang, H. Xu, F. F. Liao and J. Z. Wang, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3591–3596 CrossRef PubMed.
- C. L. Lee, T. Y. Tsai, J. J. Wang and T. M. Pan, Appl. Microbiol. Biotechnol., 2006, 70, 533–540 CrossRef PubMed.
- J. Ma, Y. Li, Q. Ye, J. Li, Y. Hua, D. Ju, D. Zhang, R. Cooper and M. Chang, J. Agric. Food Chem., 2000, 48, 5220–5225 CrossRef PubMed.
- Y. Kohama, S. Matsumoto, T. Mimura, N. Tanabe, A. Inada and T. Nakanishi, Chem. Pharm. Bull., 1987, 35, 2484–2489 CrossRef PubMed.
- J. Shepherd, S. M. Cobbe, I. Ford, C. G. Isles, A. R. Lorimer, P. W. MacFarlane, J. H. McKillop and C. J. Packard, N. Engl. J. Med., 1995, 333, 1301–1307 CrossRef PubMed.
- C. L. Lee, Y. H. Kung, C. L. Wu, Y. W. Hsu and T. M. Pan, J. Agric. Food Chem., 2010, 58, 9013–9019 CrossRef PubMed.
- J. H. Su, M. Zhao, A. J. Anderson, A. Srinivasan and C. W. Cotman, Brain research, 2001, 898, 350–357 CrossRef PubMed.
- Y. C. Shi and T. M. Pan, J. Agric. Food Chem., 2010, 58, 7634–7640 CrossRef PubMed.
- C. L. Lee, T. F. Kuo, C. L. Wu, J. J. Wang and T. M. Pan, J. Agric. Food Chem., 2010, 58, 2230–2238 CrossRef PubMed.
- C. L. Lee, T. F. Kuo, J. J. Wang and T. M. Pan, J. Neurosci. Res., 2007, 85, 3171–3182 CrossRef PubMed.
- D. Wild, G. Toth and H. U. Humpf, J. Agric. Food Chem., 2002, 50, 3999–4002 CrossRef PubMed.
- S. Jongrungruangchok, P. Kittakoop, B. Yongsmith, R. Bavovada, S. Tanasupawat, N. Lartpornmatulee and Y. Thebtaranonth, Phytochemistry, 2004, 65, 2569–2575 CrossRef PubMed.
- Y. Zheng, Y. Xin and Y. Guo, Food Chem., 2009, 113, 705–711 CrossRef.
- T. Akihisa, S. Mafune, M. Ukiya, Y. Kimura, K. Yasukawa, T. Suzuki, H. Tokuda, N. Tanabe and T. Fukuoka, J. Nat. Prod., 2004, 67, 479–480 CrossRef PubMed.
- T. Akihisa, H. Tokuda, K. Yasukawa, M. Ukiya, A. Kiyota, N. Sakamoto, T. Suzuki, N. Tanabe and H. Nishino, J. Agric. Food Chem., 2005, 53, 562–565 CrossRef PubMed.
- M. J. Cheng, M. D. Wu, I. S. Chen and G. F. Yuan, Chem. Pharm. Bull., 2008, 56, 394–397 CrossRef PubMed.
- N. Osmanova, W. Schultze and N. Ayoub, Phytochem. Rev., 2010, 9, 315–342 CrossRef.
- J. M. Gao, S. X. Yang and J. C. Qin, Chem. Rev., 2013, 113, 4755–4811 CrossRef PubMed.
- K. Fassbender, M. Simons, C. Bergmann, M. Stroick, D. Lutjohann, P. Keller, H. Runz, S. Kuhl, T. Bertsch, K. von Bergmann, M. Hennerici, K. Beyreuther and T. Hartmann, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5856–5861 CrossRef PubMed.
- P. Salins, S. Shawesh, Y. He, A. Dibrov, T. Kashour, G. Arthur and F. Amara, Neurosci. Lett., 2007, 412, 211–216 CrossRef PubMed.
- H. C. Huang and Z. F. Jiang, Journal of Alzheimer's disease, 2009, 16, 15–27 CrossRef PubMed.
- B. A. Yankner, Neuron, 1996, 16, 921–932 CrossRef PubMed.
- M. P. Mattson, Nat. Rev. Mol. Cell Biol., 2000, 1, 120–129 CrossRef PubMed.
- A. Gella and N. Durany, Cell Adhes. Migr., 2009, 3, 88–93 CrossRef.
- S. Miranda, C. Opazo, L. F. Larrondo, F. J. Munoz, F. Ruiz, F. Leighton and N. C. Inestrosa, Prog. Neurobiol., 2000, 62, 633–648 CrossRef PubMed.
- A. H. Moore and M. K. O'Banion, Adv. Drug Delivery Rev., 2002, 54, 1627–1656 CrossRef PubMed.
- E. M. Ribe, E. Serrano-Saiz, N. Akpan and C. M. Troy, Biochem. J., 2008, 415, 165–182 CrossRef PubMed.
- I. Budihardjo, H. Oliver, M. Lutter, X. Luo and X. Wang, Annu. Rev. Cell Dev. Biol., 1999, 15, 269–290 CrossRef PubMed.
- J. W. Allen, B. A. Eldadah, X. Huang, S. M. Knoblach and A. I. Faden, J. Neurosci. Res., 2001, 65, 45–53 CrossRef PubMed.
- J. S. Wadia, R. M. Chalmers-Redman, W. J. Ju, G. W. Carlile, J. L. Phillips, A. D. Fraser and W. G. Tatton, J. Neurosci., 1998, 18, 932–947 CrossRef PubMed.
- M. Orth and A. H. Schapira, American journal of medical genetics, 2001, 106, 27–36 CrossRef PubMed.
- K. Hirai, G. Aliev, A. Nunomura, H. Fujioka, R. L. Russell, C. S. Atwood, A. B. Johnson, Y. Kress, H. V. Vinters, M. Tabaton, S. Shimohama, A. D. Cash, S. L. Siedlak, P. L. Harris, P. K. Jones, R. B. Petersen, G. Perry and M. A. Smith, J. Neurosci., 2001, 21, 3017–3023 CrossRef PubMed.
- Y. Morel and R. Barouki, Biochem. J., 1999, 342(Pt 3), 481–496 CrossRef PubMed.
- D. J. Bonda, X. Wang, G. Perry, A. Nunomura, M. Tabaton, X. Zhu and M. A. Smith, Neuropharmacology, 2010, 59, 290–294 CrossRef PubMed.
- S. Chakrabarti, M. Sinha, I. G. Thakurta, P. Banerjee and M. Chattopadhyay, Curr. Med. Chem., 2013, 20, 4648–4664 CrossRef PubMed.
- J. T. Coyle and P. Puttfarcken, Science, 1993, 262, 689–695 Search PubMed.
- V. Contardo-Jara, A. Krueger, H. J. Exner and C. Wiegand, J. Environ. Monit., 2009, 11, 1147–1156 RSC.
- X. Q. Xiao, R. Wang and X. C. Tang, J. Neurosci. Res., 2000, 61, 564–569 CrossRef PubMed.
- P. Cohen, C. F. Holmes and Y. Tsukitani, Trends Biochem. Sci., 1990, 15, 98–102 CrossRef PubMed.
- T. Arendt, M. Holzer, M. K. Bruckner, C. Janke and U. Gartner, J. Neurosci., 1998, 85, 1337–1340 CrossRef.
- L. Amniai, P. Barbier, A. Sillen, J. M. Wieruszeski, V. Peyrot, G. Lippens and I. Landrieu, FASEB J., 2009, 23, 1146–1152 CrossRef PubMed.
- A. Lan, X. Liao, L. Mo, C. Yang, Z. Yang, X. Wang, F. Hu, P. Chen, J. Feng, D. Zheng and L. Xiao, Hydrogen Sulfide Protects against Chemical Hypoxia-Induced Injury by Inhibiting ROS-Activated ERK1/2 and p38MAPK Signaling Pathways in PC12 Cells, PLoS One, 2011, 6, e25921 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |