
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free heterogeneous and mesoporous biogenic graphene-oxide nanoparticle-catalyzed synthesis of bioactive benzylpyrazolyl coumarin derivatives†
T. A. J. Siddiquia,
Balaji G. Ghule b,
Shoyebmohamad Shaikhb,
Pritamkumar V. Shindeb,
Krishna Chaitanya Gunturua,
P. K. Zubaidhaa,
Je Moon Yunc,
Colm O'Dwyer*de,
Rajaram S. Mane*bc and
Kwang Ho Kim*c
b,
Shoyebmohamad Shaikhb,
Pritamkumar V. Shindeb,
Krishna Chaitanya Gunturua,
P. K. Zubaidhaa,
Je Moon Yunc,
Colm O'Dwyer*de,
Rajaram S. Mane*bc and
Kwang Ho Kim*c
aSchool of Chemical Sciences, SRTM University, Nanded, India-431606
bCenter for Nanomaterials & Energy Devices, School of Physical Sciences, SRTM University, Nanded, India-431606. E-mail: rajarammane70@srtmun.ac.in
cGlobal Frontier R&D Center for Hybrid Interface Materials, Pusan National University, San 30 Jangjeon-dong, Geumjeong-gu, Busan, 609-735, Republic of Korea. E-mail: kwhokim@pusan.ac.kr
dSchool of Chemistry, University College Cork, Cork T12 YN60, Ireland. E-mail: c.odwyer@ucc.ie
eMicro-nano Systems Centre, Tyndall National Institute, Lee Maltings, Cork T12 R5CP, Ireland
First published on 14th May 2018
Abstract
We report the preparation of graphene oxide nanoparticles (GONPs), a metal-free, heterogeneous, non-toxic, reusable and mesoporous green-(acid)-catalyst obtained by sugar carbonization through a micro-wave chemical synthesis method for the synthesis of bio-active benzylpyrazolyl coumarin derivatives (BCDs) under thermal conditions (50 °C) in ethanol solvent. The obtained products were purified by re-crystallization from ethanol, assuring usability of GONPs in multicomponent reactions (MCRs) that could find wide application in the synthesis of a variety of biologically potent molecules of therapeutic significance.
Introduction
New facets of conventional heterogeneous catalysts have stimulated extensive efforts in nanochemistry methods for preparing new catalytic materials with greater precision.1 The most widely explored non-noble and platinum group catalytic nanomaterials include transition metals, metal oxides,2 and hybrid materials based on inorganic and enzymes3–7 where the use of ultraviolet-visible irradiation is often obligatory. The development of metal-free green synthesis of fine chemicals and pharmaceuticals8–10 to overcome the shortcomings of transition metal-based catalysts (both heterogeneous and homogenous)11,12 used in the catalytic process finds at niche place in the industrial community.13,14 As such, the search for catalysts that combine the toxicological benefits of a metal-free synthesis with the amenities of heterogeneous work-up, whilst having high activity, is a continuing effort of censorious importance.15,16 Based on abundant elements, finding alternative metal-free hetero or homogeneous, selective or high performance catalysts is alluring and important. The potential of carbon materials (CMs) as heterogeneous catalysts could be profound. The term “carbocatalysis” is used to denote the utilization of metal-free carbon materials as direct/active catalysts rather than inert catalysts. Certain carbonaceous materials have intrinsic physicochemical properties such as exclusive structure, gigantic surface area, good pore interconnectivity and high chemical/thermal stability. The CMs such as activated carbon, fullerene, single and multi-walled carbon nanotubes, and graphene oxide (GO) etc., have been manifested as efficient carbocatalyst. CMs offer new opportunities for the development of advanced carbon-based nanocatalysts with much improved catalytic performance, due to their peculiar molecular structures and optoelectronic properties. Among them, GO is highly preferred due to its low-cost, 2D structure, rich chemical functionality (solid-acid, solid-base, redox and defect sites) and chemical stability and mechanical robustness. To date, several metal-based catalytic organic transformations have been replaced by GO-based carbocatalytic systems.17–21 To the best of our knowledge, the majority of GO sheets reported were obtained by the excruciating methods,22–24 typically carried out by the use of fuming nitric acid, concentrated sulfuric acid, potassium chlorate, potassium permanganate and hydrogen peroxide etc. Furthermore, most of the experimental processes generate waste gas, large amounts of acidic water and a little nitrogen and sulphur as well as metal ion co-doped GO sheets, implying the presence of environmental and health-related issues with processing and use. Therefore, current scenarios require the organic chemist to systematize the research in such a way that it preserves the environment and to develop procedures that are both eco-friendly and economically suitable. One major objective is, therefore, to simplify and accommodate the traditional procedures in a modern way with the aim of minimizing pollution effects to the least.25,26 In continuation of research on the synthesis of highly functionalized BCDs,27–30 here, we report an unveiling protocol for the preparation of the GONPs which has specific area ∼5.15 m2 g−1, mean pore diameter ∼4.6 nm with pore volume of ∼0.0876 cm3 g−1(ref. 31) and their catalytic applications as novel and stable heterogeneous catalyst for the synthesis of BCDs. Environmental prestige, economic soul and recyclability are the advantages of this novel catalyst. In a nutshell, synthesis of microwave-mediated GONPs on market quality sugar carbonation was carried out by irradiating radiations at 400–500 W power for 10–15 min duration. The synthesized product was air-annealed at 450 °C for 2 h and the black-product was obtained i.e. GONPs (Scheme 1). The GONPs were characterized for their structure, morphology, grain-size, phase-purity, porosity, pore-size distribution, elemental composition and catalytic activity. | ||
| Scheme 1 A micro-oven-assisted chemical method used for obtaining GONPs from market quality sugar. | ||
Experimental details
Chemicals
All the reagents were purchased from Merck and Spectrochem and were used without further purification. The reaction was monitored by TLC using 0.2 mm Merck silica gel 60 F254 precoated plates, which were seen with UV light. Melting points were measured by open capillary method.Synthesis of GONPs through market quality sugar
Dry table-sugar was crushed in a mortar to form uniform powder and then transferred into a glass beaker and heated under 400–500 W power radiations for 4–6 times with each of 1–2 min duration in a microwave oven. The formed charcoal was collected and crushed in a mortar pastor to get smooth powder and annealed at 450 °C for 2 h. The sample formed is black in appearance and smooth in nature. This black powder was insoluble in water, ethanol, methanol, and benzene but, partially soluble in dimethyl sulphoxide. The annealed powder was collected and well characterized by the X-ray diffraction (XRD, D8-Discovery Bruker, Cu Kα, 40 kV, 40 mA) pattern, scanned from 20 to 80°. The Raman spectrum was recorded on a WITec system within a Raman shift of 1000–2000 cm−1. Field-emission scanning electron microscopy (FE-SEM, Hitachi, S-4800, 15 kV), energy dispersive X-ray (EDX) analysis, and high-resolution transmission electron microscopy (HR-TEM) along with the selected area electron diffraction (SAED) pattern (Technai F20) were used to observe the surface morphology, grain size, and elemental configuration, respectively. X-ray photoelectron spectroscopy (XPS, VG Scientific ESCALAB250) was utilized to analyze the chemical composition and bonding status. The Brunauer–Emmett–Teller surface area was obtained using a Micrometrics ASAP2010 analyzer in a N2 environment with proper moisture removal from the sample.General procedure for the preparation of benzylpyrazoly coumarin derivatives
To a mixture of 20% of catalyst and 3 ml of ethanol as a solvent was added phenyl hydrazine 1 (1 mmol), ethyl acetoacetate 2 (1 mmol), aromatic aldehyde (1 mmol) and 4-hydroxycoumarin 4 (1 mmol) and heated at 50 °C. The clear reaction mixture was stirred for the stipulated time and the mixture becomes turbid as the reaction proceeds. After completion of the reaction (monitored by TLC), the free-flowing solid was dissolved in dichloromethane to separate the heterogeneous catalyst by simple filtration method. Afterwards, the filtrate was evaporated under reduced pressure to obtain crude products as pale-yellow solids. The products, thus, obtained was recrystallized from hot ethanol to the get pure compounds as white or pale-yellow solids. Yields were calculated after recrystallization (Scheme 2). The isolated compounds were well characterized by 1H NMR, 13C NMR, and Mass spectrum and reported in supportive information. | ||
| Scheme 2 Green synthesis of BCDs. | ||
Results and discussion
Fig. 1a shows the X-ray diffraction pattern of as-prepared GONPs, which is in accordance with (002) and (100) planes of graphitic carbon (JCPDF# 41-1487). The Raman shift spectrum (Fig. 1b) reveals the presence of two characteristic bands of carbon at 1352 cm−1 (D-band) for the sp3 configuration and 1592 cm−1 (G-band) for the sp2 graphitic configuration. The 0.75 intensity ratio of the D-band over the G-band signifies the graphitic behavior of GONPs.32 The Fourier-transform infrared (FTIR) spectrum of GONPs (Fig. 1c) confirms basal planes of sp2 carbon functionalized with hydroxyl (–OH), epoxide (C–O–C) and carboxylic acid (–COOH) groups33,34 qualitatively demonstrating the presence of GO rather other carbonaceous phases such as graphite and graphene. The FTIR spectrum shows broad and strong bands at ∼3390 and 1090 cm−1 which are attributed to stretching vibrations and in plane bending vibration of –OH/H2O. A weak band at ∼2926 cm−1 corresponds to the stretching vibrations of the C–H bond. Presence of the featured bands at ∼1622 cm−1 and 1716 cm−1 confirms the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. A relatively sharp band at ∼1205 cm−1 is due to stretching of –C–O or bending of alkyl groups. Band appeared at 618 cm−1 was assigned to CH deformation. The field-emission scanning electron microscopy image of GONPs exhibits crystallites of irregular dimensions (Fig. 1d). The globular GONPs are typically interconnected to one another at the edges. The 72
O. A relatively sharp band at ∼1205 cm−1 is due to stretching of –C–O or bending of alkyl groups. Band appeared at 618 cm−1 was assigned to CH deformation. The field-emission scanning electron microscopy image of GONPs exhibits crystallites of irregular dimensions (Fig. 1d). The globular GONPs are typically interconnected to one another at the edges. The 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :28 composition ratio for carbon and oxygen elements supports a GO composition (Fig. S1†). Fig. 1e and f presents digital images of normal and high-resolution transmission electron microscopy where, surprisingly, irregular grains of 10–50 nm diameters are found. Selected area electron diffraction pattern in Fig. 1f demonstrates the nanocrystalline nature of GONPs.35,36 Consistent with X-ray diffraction results, the interplanar distances of ∼0.38 nm (inset of Fig. 1f) and ∼0.28 nm for lattice fringe widths of the (002) and (100) planes, respectively, are corroborated.
:28 composition ratio for carbon and oxygen elements supports a GO composition (Fig. S1†). Fig. 1e and f presents digital images of normal and high-resolution transmission electron microscopy where, surprisingly, irregular grains of 10–50 nm diameters are found. Selected area electron diffraction pattern in Fig. 1f demonstrates the nanocrystalline nature of GONPs.35,36 Consistent with X-ray diffraction results, the interplanar distances of ∼0.38 nm (inset of Fig. 1f) and ∼0.28 nm for lattice fringe widths of the (002) and (100) planes, respectively, are corroborated.
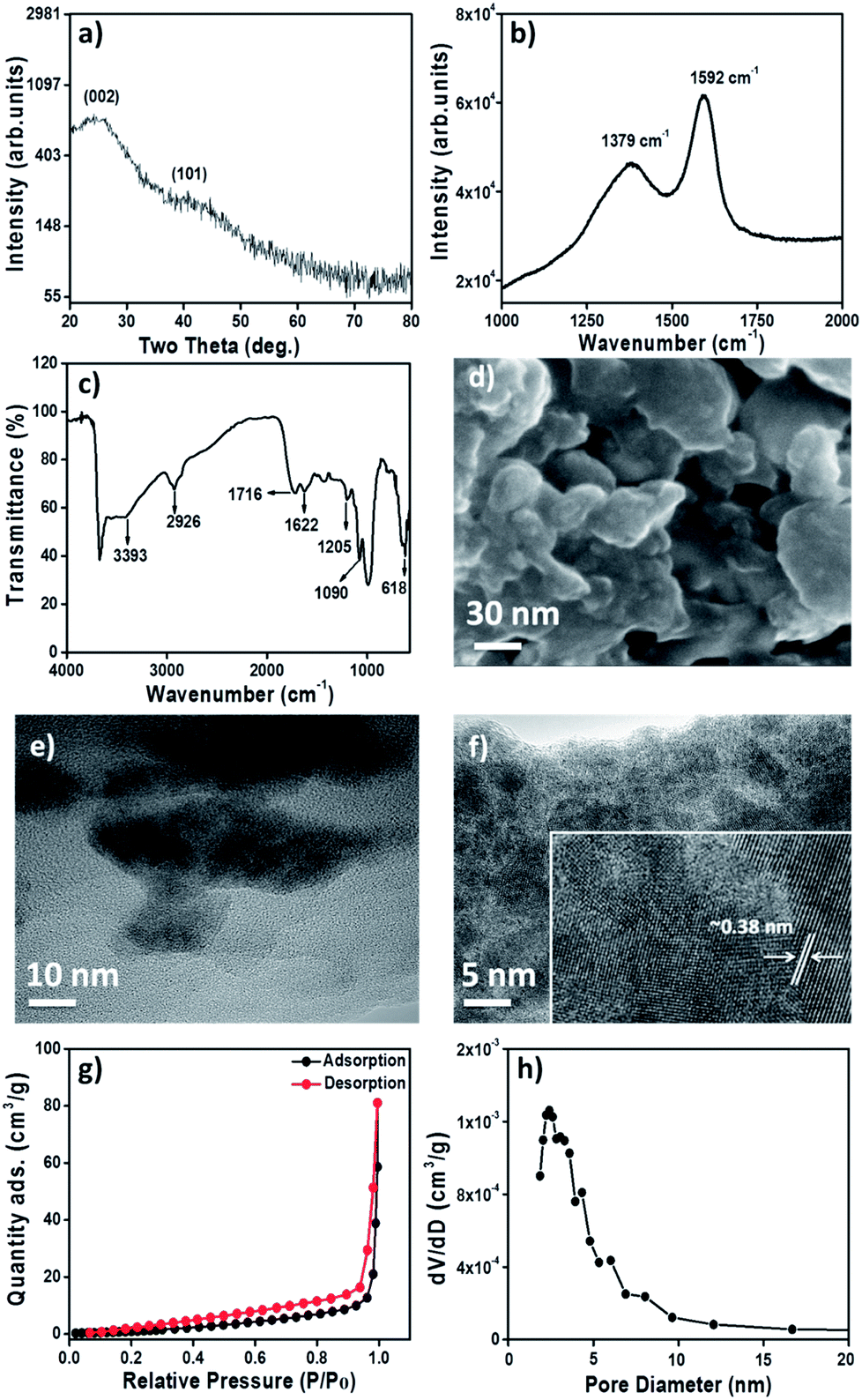 | ||
| Fig. 1 (a) X-ray diffraction pattern, (b) Raman spectrum (concluding graphitic nature), (c) FTIR spectrum, (d) FE-SEM, (e) TEM (showing interconnected and irregular signature), (f) HR-TEM (inset shows 0.38 nm interplanar spacing of (002) plane), (g) adsorption–desorption isotherm, and (h) pore-size distribution curve (confirming the mesoporous character) of GONPs. | ||
A steep-rise in the nitrogen/adsorption curve is observed after a relative pressure (p/po) > 0.3, signifying (Fig. 1g) the mesoporous nature of GONPs. Correspondingly derived surface-area and micro-pore surface-area of GONPs are 5.15 and 33.12 m2 g−1, respectively. The pore-size distribution curve (Fig. 1h) of GONPs reveals a total specific pore-volume and average pore-diameter of 0.0876 cm3 g−1 and 46.13 Å, respectively. The deconvoluted high resolution C1s photoemission peak from GONPs by XPS is shown in Fig. S2.† The calibration of carbon C1s core-level of adventitious was 284.6 eV, corresponding to the sp2 carbon phase. The C1s peaks were fitted with three contributions: sp2 (284.6 eV), C–O (286.8 eV), CO (288.9 eV), respectively.
The amount of oxygen in the GONPs can be explained by bonding of oxygen to the defected carbon bonds. The above-mentioned measurements have provided evidence that our microwave-assisted product is structurally close to GO. Taking into consideration the importance of carbon-based materials,35,36 the catalytic behaviour of the prepared GONPs was studied for the synthesis of BCDs.
Design and development of interweaved molecular scaffolds for the biological applications have fascinated much attention as these moieties exhibit wide-ranging and improved bio-activities. It is anticipated that the integration of the 3-benzyl coumarin and pyrazolone moieties leading to benzylpyrazolyl coumarin scaffolds could be fascinating and beneficial from a biological point of view.27–30 Amongst few earlier reports for this scaffold, the use of glacial acetic acid in water under reflux conditions along with molecular motif of scaffold explored by single X-ray crystallographic analysis27 and metal containing catalysts were explored.28–30 Natural carbon was demonstrated as an alternative to metal and metal oxide-based catalysts due to its worldwide natural abundance, stability, inexpensiveness and non-toxic signature.19,20 As part of our interest in exploring environmentally benign GONPs and other materials as green-catalysts for the synthesis of several biologically important heterocyclic compounds, we report a facile one-pot MCR for the synthesis of various BCDs in ethanol solvent using GONPs derived from natural sugar. The effects of solvent, catalyst amount, reaction duration and molar-ratio of the raw material on the yield of BCDs were studied systematically. The synthesis of BCDs 4-[(4-hydroxy-2-oxo-2H-chromen-3-yl)-phenylmethyl]-5-methyl-2-phenyl-1,2-dihydro-pyrazol-3-one with high percent yield by one-pot four-component condensation reactions is one of the major objectives of this work. These four-components i.e. phenylhydrazine (1), ethyl acetoacetate (2), aromatic aldehyde (3) and 4-hydroxycoumarin (4) react in the presence of GONPs as a heterogeneous acid catalyst by using ethanol solvent at 50 °C. Furthermore, the optimization study of this four-component reaction was studied in different solvents (Table 2). The best results were obtained in ethanol solvent, while water as solvent results in a sticky product with unreacted starting material. The reaction without GONPs catalyst could not provide the product even after 10 h under identical conditions (see entry 1 of Table 2). The optimized yield of the product (5) was obtained in ethanol as a solvent medium using 20% of GONPs as catalyst (see 10 entry of Table 2). As envisaged, the GONPs displayed excellent catalytic properties due to their considerable surface area, abundant active sites and unique chemical/physical properties. The functional groups (hydroxy, epoxide and carboxyl etc.) as seen in FTIR studies endow suitable acidity and oxidizing properties providing high tenability of ethanol solvent towards polar organic substrates and reagents. The ethanol solvent polarity created ambient conditions for good conversion, which was further optimized for the reaction purity, yield and speed. Under optimized conditions in ethanol solvent, as high as 93% yield of BCD was obtained. Having set the reaction conditions, the protocol was studied with a variety of substituted benzaldehydes. Equimolar amounts of substituted of phenyl hydrazine (1), ethyl acetoacetate (2), aromatic aldehyde (3), 4-hydroxycoumarin (4), and 20% of catalyst under thermal condition (50 °C) in solvent ethanol (Scheme 2) have provided a library of BCD (5) in good to excellent yields (86–93%) (Table 1). The reaction proceeded smoothly and tolerated electron withdrawing and electron donating substituted benzaldehydes.
| Sr. no | Aromatic aldehydes | Time (h) | Product (BCDs) | Yield (%) |
|---|---|---|---|---|
| 1 |  |
6 |  |
90 |
| 2 |  |
5 |  |
93 |
| 3 | 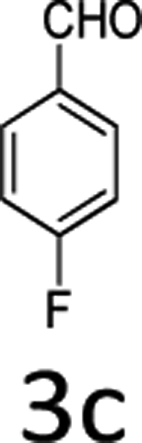 |
8 |  |
87 |
| 4 |  |
3 |  |
90 |
| 5 | 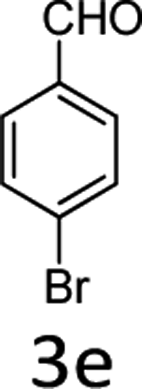 |
4 |  |
93 |
| 6 | 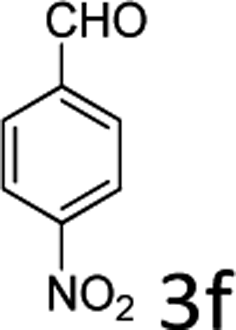 |
10 | 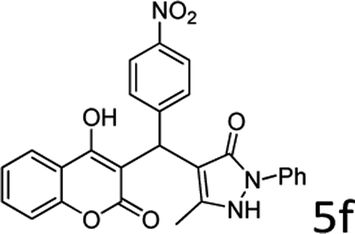 |
93 |
| 7 |  |
8 | 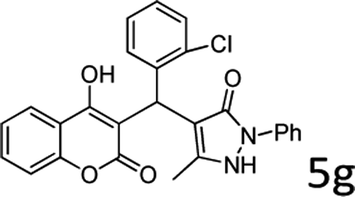 |
86 |
| 8 |  |
10 |  |
87 |
| 9 |  |
7 | 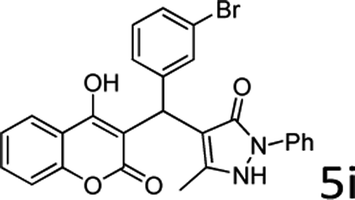 |
86 |
| 10 |  |
10 |  |
88 |
| Sr. no. | Catalyst (%) | Solvent | Temperature (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | No catalyst | EtOH | Reflux | 10 | — |
| 2 | 5 | EtOH | Reflux | 12 | 40 |
| 3 | 10 | MeOH | Reflux | 10 | 30 |
| 4 | 10 | CH3CN | Reflux | 10 | 40 |
| 5 | 10 | DCM | Reflux | 10 | 40 |
| 6 | 10 | CHCl3 | Reflux | 10 | 35 |
| 7 | 10 | DMF | Reflux | 10 | 40 |
| 8 | 10 | H2O | Reflux | 10 | 50 |
| 9 | 10 | EtOH:H2O |
Reflux | 10 | 55 |
| 10 | 20 | EtOH | 50 °C | 7 | 93 |
The reactions were consistently carried out at 1 mmol scale and no change of product yield was observed when scaled up to the 10 mmol scale. It was encouraging to find that the final products (5) could be isolated by simple filtration, avoiding any requirement for chromatographic separation. This optimized reaction condition was applied to different aldehydes to check the general applicability and functional group tolerance. The reaction proceeds well to give excellent yields of the product without any column purification. The catalyst was easily removed from the reaction mixture due to its heterogeneous character. Furthermore, their purity was so high, no preparation in an analytically pure form by single recrystallization using ethanol as a solvent was necessary. The final products were confirmed by elemental analysis, IR, 1H NMR, 13C NMR and HR-MS analytical and spectral techniques (SI-1).
For nanostructured carbon materials like GONPs, the weak adsorption sites are characteristically placed with their basal planes consisting of the hexagonal arrangement of carbon atoms, while the high energy sites are typically associated at the edges, which are often saturated with hydrogen atoms attached to heteroatoms. The edge hydrogen atoms can be replaced by other heteroatoms, e.g. oxygen and/or nitrogen, to provide strong chemical relativities for redox or acid–base reactions.37 The GONPs catalyzed Knoevenagel condensation and Michael addition reaction supports the present mechanistic pathway in the synthesis of BCDs.38,39 As we mentioned before GONPs which has specific area ∼5.15 m2 g−1, mean pore diameter ∼4.6 nm with pore volume of ∼0.0876 cm3 g−1(ref. 31) and this surfaces of the GONPs play an important role in these MCRs. The active sites i.e. hydroxy, carboxyl, epoxide etc.33,34 fulfill the requirement of acidic sites for the reaction to occur in GONPs. A plausible reaction mechanism for the GONPs catalyzed synthesis of BCDs is given in Scheme 3. The carbonyl groups in the ethyl acetoacetate are initially activated by acidic functional groups of the catalyst to react with phenylhydrazine to form pyrazolone (I). Then, the activated aldehydes by acidic functional groups of the catalyst react to form the Knoevenagel product. Subsequently, during the Michael addition step, nucleophilic attack on (II) by tautomeric form of pyrazolone (I) affords the desired product (5) via intermediate (III). Alternatively, there is another possible reaction pathway for the reaction, via the formation of intermediate (IV) followed by reaction with 4-hydroxycoumarin to afford product (5) (see Scheme 2). It was revealed that intermediates (II) and (IV) were very reactive towards the subsequent reaction with pyrazolone and 4-hydroxycoumarin respectively.27
 | ||
| Scheme 3 Plausible mechanism for the formation of BCDs. | ||
In the multicomponent reactions proceeded through intermediate (II) or (IV) could not be measurably detected in current study. After completion of the reaction, tautomerisation occurred in the pyrazolone ring of product (5). This may exist in three tautomeric forms viz. the CH, OH and NH.27–30,40,41 The GONPs catalyst has both non-toxic and reusable features. Thus, the present protocol fulfills the criteria for green-synthesis and reusability. The robustness of GONPs for the synthesis of BCDs was tested by scrutinizing the effect of the phase and its reusability.17–19 When the synthesis of BCDs was carried out using the phases and active sites, the GONPs play significant role in catalytic activity. Finally, the GONPs, being a heterogeneous catalyst, can be easily separated from the reaction mixture and reused five times successfully (Fig. 2), by reuse of GONPs for the model reaction.
 | ||
| Fig. 2 Reproducibility test of GONPs for BCDs synthesis. | ||
Conclusion
In conclusion, the present protocol has several features such as; (i) the prepared GONPs are nanocrystalline and mesoporous with acidic functional groups, (ii) the GONPs demonstrate a superior catalytic performance with as high as 93–82% isolated product yields, (iii) unveiling report of the biologically important highly functionalized BCDs using a solid, metal-free, nontoxic and reusable heterogeneous green/eco-friendly acid catalyst. A facile one-pot multi-component protocol has been demonstrated for the synthesis of biologically important, highly functionalized BCDs. The reaction was performed in ethanol solvent at 50 °C using GONPs as a heterogeneous acid catalyst. The products were purified by recrystallization from ethanol and chromatography-free separation methods, where the use of volatile and hazardous solvents, generally preferred, is avoided. These GONPs should find several applications in MCRs leading to biologically potent molecules in the near future, which is a topic of ongoing work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by; (a) the Global Frontier Program through the Global Frontier Hybrid Interface Materials (GFHIM) of the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning 10 (2013M3A6B1078874), (b) Science Foundation Ireland under grant no. 14/IA/2581 and by the Irish Research Council under award GOIPD/2016/575, and (c) SFS would like to thank University Grants Commission, New Delhi for awarding D. S. Kothari Post-Doctoral Fellowship scheme (F.4-2/2006(BSR)/CH/16-17/0015).Notes and references
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef PubMed.
- D. Pla and M. Gómez, ACS Catal., 2016, 6, 3537–3552 CrossRef.
- L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 20, 1825–1837 CrossRef PubMed.
- H. Song, Acc. Chem. Res., 2015, 48, 491–499 CrossRef PubMed.
- J. A. Torres, F. G. E. Nogueira, M. C. Silva, J. H. Lopes, T. S. Tavares, T. C. Ramalho and A. D. Corrêa, RSC Adv., 2017, 7, 16460–16466 RSC.
- Y. Yong, R. Su, X. Liu, W. Xu, Y. Zhang, R. Wang, P. Ouyang, J. Wu, J. Ge and Z. Liu, Biochem. Eng. J., 2018, 129, 26–32 CrossRef.
- Q. Husain, J. Nanosci. Nanotechnol., 2018, 18, 486–499 CrossRef.
- J. B. Bharate, S. B. Bharate and R. A. Vishwakarma, ACS Comb. Sci., 2014, 16, 624–630 CrossRef PubMed.
- D. R. Dreyer, H. P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6813–6816 Search PubMed.
- C. Su, M. Acik, K. Takai, J. Lu, S. Hao, Y. Zheng, P. Wu, Q. Bao, T. Enoki, Y. J. Chabal and K. P. Loh, Nat. Commun., 2012, 3, 1298–1307 CrossRef PubMed.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef PubMed.
- D. L. Trimm, Appl. Catal., A, 2001, 212, 153–160 CrossRef.
- Y. Li and W. Shen, Chem. Soc. Rev., 2014, 43, 1543–1574 RSC.
- I. Fechete, Y. Wang and J. C. Vedrine, Catal. Today, 2012, 189, 2–27 CrossRef.
- G. V. Smith and F. Notheisz, Heterogenous Catalysis in Organic Chemistry, Academic Press, New York, 1999 Search PubMed.
- F. Hu, M. Patel, F. Luo, C. Flach, R. Mendelsohn, E. Garfunkel, H. He and M. Szostak, J. Am. Chem. Soc., 2015, 137, 14473–14480 CrossRef PubMed.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef PubMed.
- D. Deng, L. Xiao, I. Chung, I. S. Kim and M. Gopiraman, ACS Sustainable Chem. Eng., 2017, 5, 1253–1259 CrossRef.
- C. L. Su and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275–2285 CrossRef PubMed.
- D. S. Su, J. Zhang, B. Frank, A. Thomas, X. Wang, J. Paraknowitsch and R. Schlogl, ChemSusChem, 2010, 3, 169–180 CrossRef PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- A. Lerf, H. Y. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef.
- T. Nakajima and Y. Matsuo, Carbon, 1994, 32, 469–475 CrossRef.
- J. H. Clark, Acc. Chem. Res., 2002, 35, 791–797 CrossRef PubMed.
- R. A. Sheldon, Green Chem., 2005, 7, 267–278 RSC.
- P. P. Ghosh, G. Pal, S. Pal and A. R. Das, Green Chem., 2012, 14, 2691–2698 RSC.
- A. Saha, S. Payra and S. Banerjee, Green Chem., 2015, 17, 2859–2866 RSC.
- B. Karami, K. Eskandari, S. Khodabakhshi, S. S. Hoseini and F. Hashemian, RSC Adv., 2013, 3, 23335–23342 RSC.
- S. Yaragorla, A. Pareek and R. Dada, Tetrahedron Lett., 2015, 56, 4770–4774 CrossRef.
- B. G. Ghule, S. Shaikh, S. U. Ekar, U. T. Nakate, K. C. Gunturu, N. M. Shinde, M. Naushad, K. H. Kim, C. O'Dwyer and R. S. Mane, ACS Appl. Mater. Interfaces, 2017, 9, 43051–43060 Search PubMed.
- Y. Li, Y. Hu, H. Li, L. Chen and X. J. Huang, J. Mater. Chem. A, 2016, 4, 96–104 Search PubMed.
- J. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef PubMed.
- B. Garg, T. Bisht and Y. C. Ling, Molecules, 2014, 19, 14582–14614 CrossRef PubMed.
- M. Patel, W. Feng, K. Savaram, M. R. Khoshi, R. Huang, J. Sun, E. Rabie, C. Flach, R. Mendelsohn, E. Garfunkel and H. He, Small, 2015, 11, 3358–3368 CrossRef PubMed.
- P. L. Chiu, D. D. T. Mastrogiovanni, D. Wei, C. Louis, M. Jeong, G. Yu, P. Saad, C. R. Flach, R. Mendelsohn, E. Garfunkel and H. He, J. Am. Chem. Soc., 2012, 134(13), 5850–5856 CrossRef PubMed.
- L. R. Radovic and F. Rodriguez-Reinso, Carbon Materials in Catalysis. Chemistry and Physics of Carbon: A Series of Advances, Marcel Dekker, Inc., New York, 1997 Search PubMed.
- S. Verma, H. P. Mungse, N. Kumar, S. Choudhary, S. L. Jain, B. Sain and O. P. Khatri, Chem. Commun., 2011, 47, 12673–12675 RSC.
- S. M. Islam, A. S. Roy, R. C. Dey and S. Paul, J. Mol. Catal. A: Chem., 2014, 394, 66–73 CrossRef.
- V. I. Minkin, L. P. Olekhnovic and Y. A. Zhdanov, Molecular Design of Tautomeric Compounds, Reidel, Dordrecht, The Netherlands, 1988 Search PubMed.
- D. H. Barton, Comprehensive Organic Chemistry: The Synthesis and Reactions of Organic Compounds, Pergamon Press, Oxford, U.K., 1979, vol. 4 Heterocyclic Compounds, 2698 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Quantitative analysis data of energy-dispersive X-ray spectroscopy spectrums, deconvolution of GONPs XPS C1s peak, structural analysis data of 1H NMR, 13C NMR and mass spectra and their respective spectra. See DOI: 10.1039/c7ra12550j |
| This journal is © The Royal Society of Chemistry 2018 |