
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Promotion of autophagosome–lysosome fusion via salvianolic acid A-mediated SIRT1 up-regulation ameliorates alcoholic liver disease†
Xue Shi‡
ab,
Ruimin Sun‡a,
Yan Zhao‡a,
Rong Fua,
Ruiwen Wanga,
Huanyu Zhaoa,
Zhecheng Wanga,
Fan Tanga,
Ning Zhangc,
Xiaofeng Tiand and
Jihong Yao *a
*a
aDepartment of Pharmacology, Dalian Medical University, Dalian, 116044, China. E-mail: yaojihong65@hotmail.com; Fax: +86-0411-86110010; Tel: +86-0411-8611-0410
bDepartment of Pharmacy, First Affiliated Hospital of Xinxiang Medical University, Xinxiang, 453100, China
cDepartment of Pharmacy, Second Hospital of Dalian Medical University, Dalian, 116027, China
dDepartment of General Surgery, Second Affiliated Hospital, Dalian Medical University, Dalian, 116023, China
First published on 5th June 2018
Abstract
Salvianolic acid A (SalA) is a water-soluble phenolic carboxylic acid extracted from Salvia miltiorrhiza that has extensive pharmacological activities and plays an essential role in liver disease treatment. However, the mechanism of SalA in treating alcoholic liver disease (ALD) remains unclear. Here, we studied the protective effects of SalA on chronic ethanol-induced liver injury involving Sirtuin 1 (SIRT1)-mediated autophagy activation. The results showed that SalA pretreatment reduced the levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglyceride (TG) and cholesterol (TC) in vivo and enhanced hepatic cell viability while mitigating apoptosis and hepatic steatosis in vitro. Furthermore, SalA protected against chronic ethanol-induced liver injury by restoring autophagosome–lysosome fusion, as indicated by the increased expression levels of LC3-II, cathepsin B, lysosomal-associated membrane protein 2 (LAMP-2), and RAB7 and the decreased expression of SQSTM1. More importantly, pretreatment with SalA significantly up-regulated the expression of SIRT1, an NAD+-dependent deacetylase. These increased levels of SIRT1 stimulated autophagy under conditions of chronic ethanol exposure. Interestingly, SIRT1 siRNA abrogated SalA-induced autophagosome–lysosome fusion. This finding indicates that the protective effects of SalA are associated with SIRT1 activation. Collectively, our study demonstrates that SalA pretreatment protects against chronic ethanol-induced liver injury via the SIRT1-mediated restoration of autophagosome–lysosome fusion.
Introduction
Alcoholic liver disease (ALD) due to excess alcohol consumption is a major cause of morbidity and mortality worldwide.1 ALD is a complex condition that involves a wide spectrum of hepatic lesions, including simple steatosis, alcoholic hepatitis, hepatic fibrosis, and cirrhosis.2–4 Chronic ethanol consumption causes dramatic metabolic changes, lipid homeostasis disruption, mitochondrial damage, reactive oxygen species (ROS) accumulation, and hepatocyte cell death; these metabolic changes accelerate the hepatotoxicity of ethanol and ultimately cause liver injury.5–8 Although much progress has been made in the past few decades, effective approaches for regulating ALD progression remain limited.Salvia miltiorrhiza, commonly called “Danshen”, is an important medicinal plant that has been used in China for thousands of years. Salvianolic acid A (SalA) is one of the primary active, water-soluble components of Salvia miltiorrhiza.9 SalA has extensive bioactivities, including antioxidative, anti-inflammatory, anticirrhotic effects.10 SalA can prevent a variety of diseases, including coronary artery diseases, cerebral ischemia, and diabetes mellitus.11–13 Salvia miltiorrhiza Bunge and its active component cryptotanshinone protect hepatocytes from acute ethanol-induced cytotoxicity and lipid accumulation.14 In addition, we have demonstrated that SalA has hepatoprotective effects against high-fat diet (HFD)-induced nonalcoholic fatty liver disease and concanavalin A-induced liver injury.15,16 However, the effects of SalA on ALD and potential molecular mechanisms remain unknown.
Autophagy is a self-degradation process in which dysfunctional or damaged components are incorporated into double-membrane vesicles called autophagic vacuoles (AVs) or autophagosomes, which fuse with lysosomes to be digested by resident hydrolases; this entire process is called autophagy flux.17,18 In addition, AV fusion with lysosomes to form autolysosomes is a crucial step in maintaining proper autophagy flux.19,20 A recent study shows that autophagy is important in limiting ethanol-induced liver injury and hepatocyte apoptosis because it removes damaged mitochondria and lipid droplets that have accumulated.21 Chronic alcohol consumption can suppress autophagy because alcohol damages the lysosomes, suppresses lysosomal proteolysis activity, and blocks autolysosome formation.22,23 Furthermore, the pharmacological modulation of autophagy in the liver can alleviate hepatic steatosis and liver injury caused by excessive alcohol intake; this information suggests that autophagy may be a potential therapeutic target for treating ALD.24 Therefore, we investigated whether autophagy is involved in the protective role of SalA in ALD.
Sirtuin 1 (SIRT1) is currently the most studied member of the sirtuin family; SIRT1 is involved in many biological processes, including lipid metabolism, oxidation, inflammation, and apoptosis.25–28 We have demonstrated that SIRT1 participates in the alleviation of chronic alcoholic liver injury by preventing fat accumulation, decreasing ROS production, and reducing inflammation and cell death.29,30 Interestingly, SIRT1 deacetylates a number of transcription factors, including histone H4, NF-κB, and p53.31,32 The SIRT1/FoxO pathway accelerates AV development, augments autolysosome formation, and further stimulates autophagy flux.33 These effects suggest that SIRT1 may be a positive regulator of autophagy.
In this study, we investigated the effects of SalA on chronic ethanol-induced liver injury and assessed the involvement of the SIRT1/autophagy pathway. The present study may offer new insight into chronic ethanol-induced liver injury treatment and provide diverse therapeutic targets for treating ALD.
Materials and methods
Reagents
SalA (>98% purity) was purchased from Shanghai Winherb Medical Science Co., Ltd. (Shanghai, China) and dissolved in pathogen-free saline. Ethanol (>99% purity) was obtained from Sigma Co., Ltd. (St. Louis, MO, USA).Animals and treatments
Male Sprague-Dawley rats weighing 180 to 220 g were obtained from the Experimental Animal Center of Dalian Medical University (Dalian, China). All animal procedures were conducted according to the guidelines of the Institutional Animal Care and Use Committee of Dalian Medical University and experiments were approved by the Institutional Ethics Committee of Dalian Medical University. Sixty rats were maintained under standard laboratory conditions for one week. Then, the rats were randomly divided into five groups: (1) normal nonethanol diet (ND); (2) ND + SalA (16 mg kg−1 d−1); (3) Lieber–DeCarli ethanol diet (ED); (4) ED + SalA (8 mg kg−1 d−1), and (5) ED + SalA (16 mg kg−1 d−1). Rats were fed a Lieber–DeCarli ethanol diet or a normal nonethanol diet as previously described;34 these diets contained the same number of calories. The rats were intragastrically administered SalA (8 or 16 mg kg−1 d−1) or the same volume of normal saline for 8 weeks.Measurement of serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglyceride (TG), and cholesterol (TC)
Rat blood samples were acquited from the abdominal aorta and centrifuged at 3000 rpm for 15 min to separate serum. The serum levels of ALT, AST, TG and TC were measured using enzymatic assay kits according to the manufacturer's instructions (Nanjing Jiancheng Corp., Nanjing, China).Liver histopathologic examination
Liver tissues were fixed, paraffin-embedded, and sliced into 5 μm sections. Then, the slides were stained with hematoxylin and eosin (H&E) and examined under a light microscope.Cell culture and cell viability assay
The mouse AML-12 hepatocyte cell line was purchased from the American Type Culture Collection (ATCC, USA). AML-12 cells were cultured in 90% 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Dulbecco's modified Eagle's medium:Ham's F12 medium (DMEM:F12) with 1% insulin–transferrin–selenium (ITS) (Gibco, CA, USA), 40 ng mL−1 dexamethasone (Gibco, CA, USA), and 10% (v/v) fetal bovine serum (FBS) (Gibco, CA, USA). The cells were maintained in a humidified incubator with 5% CO2 at 37 °C. To determine cell survival rates, CCK-8 assays (Selleck Chemicals, Shanghai, China) were used according to the manufacturer's instructions.
:1 Dulbecco's modified Eagle's medium:Ham's F12 medium (DMEM:F12) with 1% insulin–transferrin–selenium (ITS) (Gibco, CA, USA), 40 ng mL−1 dexamethasone (Gibco, CA, USA), and 10% (v/v) fetal bovine serum (FBS) (Gibco, CA, USA). The cells were maintained in a humidified incubator with 5% CO2 at 37 °C. To determine cell survival rates, CCK-8 assays (Selleck Chemicals, Shanghai, China) were used according to the manufacturer's instructions.
Transmission electron microscopy (TEM)
Samples were fixed in a 0.1 M sodium cacodylate buffer (pH 7.4) and 2.5% glutaraldehyde solution for 4 h. Then, the samples were rinsed (three times, 10 min) with 0.1 M sodium cacodylate buffer (pH 7.4) and 7.5% sucrose and postfixed in a 1% OsO4 solution for 1 h. After dehydration using an ethanol gradient (70% ethanol (20 min), 96% ethanol (20 min), and 100% ethanol (two times, 20 min)), the samples were embedded in Durcupan ACM. Ultrathin sections were stained with uranyl acetate and lead citrate. Sections were examined under a JEOL JEM-2000EX microscope (JEOL, Tokyo, Japan) at 60 kV.Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
TUNEL staining was performed using an In Situ Cell Death Detection Kit (Roche, NJ, USA) according to the manufacturer's instructions. DAPI solution was used to stain the nuclei.35Nile red staining
Nile red is a selective fluorescent stain for intracellular lipid droplets. AML-12 cells were fixed in 4% paraformaldehyde and stained with Nile red solution (1 μg mL−1) (Sigma, no. 19123) at 37 °C for 10 min in the dark. Lipid-bound Nile red was observed under a fluorescence microscope.
Western blotting analysis
Total proteins were extracted from liver tissues by a protein extraction kit (KeyGen Biotech, Nanjing, China), and cultured cells were lysed in RIPA buffer according to the manufacturer's instructions. Protein concentrations were determined using a BCA protein assay kit (Beyotime Biotech, Shanghai, China). The same amount of protein from each sample was separated by 10–15% SDS-PAGE (Bio-Rad, Hercules, CA, USA). Blots were incubated overnight at 4 °C with the following primary antibodies: SIRT1 (Abcam Ltd, Cambridge, UK); LC3, SQSTM1, lysosomal-associated membrane protein 2 (LAMP-2), and RAB7 (Bioworld Technology, Inc., St Louis Park, MN, USA); and β-actin and cathepsin B (Proteintech Group Inc., Wuhan, China). After subsequent washing with TTBS, the blots were immunostained with secondary antibodies at 37 °C for 2 h. The membranes were exposed to Enhanced Chemiluminescence Plus reagent (Beyotime Institute of Biotechnology, Shanghai, China). Images were captured by a BioSpectrum 410 multispectral imaging system with a Chemi 410 HR camera and analyzed with Gel-Pro Analyzer Version 4.0 (Media Cybernetics, MD, USA).RNA interference
AML-12 cells were transfected with specific siRNA or nonbinding control siRNA (100 nM) using Lipofectamine 3000 (Invitrogen, Karlsruhe, Germany) according to the manufacturer's instructions. The small interfering RNA (siRNA) sequence targeting SIRT1 was as follows: sense 5′-CCCUGUAAAGCUUUCAGAAdtdt-3′ and antisense 5′-UUCUGAAAGCUUUACAGGGdtdt-3′. Both specific and control siRNAs were obtained from Genepharma (Shanghai, China). After siRNA transfection for 48 h, the cells were treated with 50 μM SalA for an additional 6 h. Then, the proteins were extracted for western blotting.Adenoviral vectors
AML-12 cells were plated in 6-well plates and allowed to reach 50–70% confluence for transfection. MRFP-GFP-LC3 adenoviral vectors were purchased from HanBio Technology (Shanghai, China), and the adenoviral infection was performed according to the manufacturer's instructions. In the green and red merged images, the red and yellow puncta represent autolysosomes and autophagosomes, respectively. LC3 puncta were examined with a Leica TCS SP5 II confocal microscope (Leica, Wetzlar, Germany).Results
SalA protects against chronic ethanol-induced liver injury
First, we evaluated whether SalA could protect rats against chronic ethanol-induced liver injury. After 8 weeks, the ALT, AST, TC, and TG levels were clearly higher in the rats fed a diet with ethanol compared with the ND group (Table 1). However, compared to ethanol treatment alone, SalA pretreatment abrogated these increases in ALT, AST, TG, and TC levels. Liver injury caused by chronic ethanol consumption and the protective effects of SalA were further evaluated by histological assays. As shown in Fig. 1a, chronic ethanol administration caused the formation of a large number of fat droplets, increased cellular size, and induced hepatocellular macrovesicular steatosis in the liver; these effects were significantly reduced by pretreatment with SalA. In vitro, SalA pretreatment (0.5, 5, or 50 μM) increased the cell viability, which inhibited by ethanol consumption, in a dose-dependent manner (Fig. 1b). Furthermore, the ethanol-induced increase in ALT, AST, TC, and TG levels was significantly prevented by SalA pretreatment in AML-12 cells (Table 2). These results indicate that SalA effectively protected against chronic ethanol-induced liver injury in vivo and in vitro.| Group | ALT (IU L−1) | AST (IU L−1) | TC (mmol L−1) | TG (mmol L−1) |
|---|---|---|---|---|
| a The data are presented as the mean ± S.D. (n = 6). **p < 0.01 versus ND group, &p < 0.05 versus ED group, ##p < 0.01 versus ED group. | ||||
| ND | 9.69 ± 1.14 | 18.74 ± 3.36 | 2.12 ± 0.15 | 0.99 ± 0.12 |
| ND + SalA (16 mg kg−1) | 9.41 ± 1.11 | 18.66 ± 1.88 | 2.03 ± 0.17 | 1.02 ± 0.19 |
| ED | 15.13 ± 2.17** | 44.08 ± 8.99** | 3.00 ± 0.30** | 2.16 ± 0.44** |
| ED + SalA (8 mg kg−1) | 12.31 ± 1.63## | 30.12 ± 9.43## | 2.66 ± 0.33& | 1.56 ± 0.37## |
| ED + SalA (16 mg k−1) | 10.72 ± 1.33## | 23.51 ± 4.90## | 2.22 ± 0.16## | 1.38 ± 0.29## |
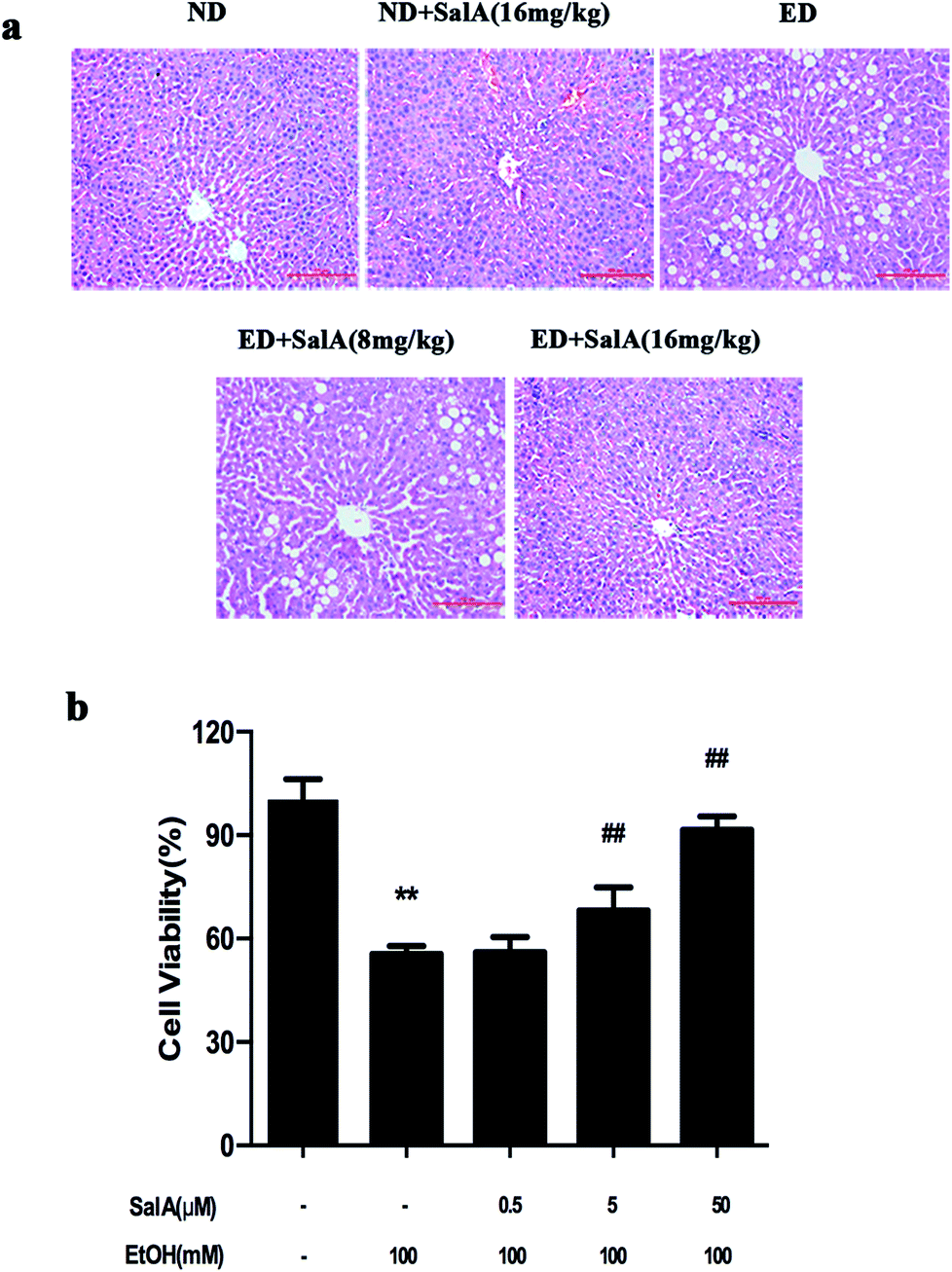 | ||
| Fig. 1 SalA pretreatment attenuates chronic ethanol-induced liver injury. (a) H&E staining. The experimental groups subjected to H&E staining were as follows: ND; ND + SalA (16 mg kg−1); ED; ED + SalA (8 mg kg−1); and ED + SalA (16 mg kg−1). H&E-stained sections were photographed at 200× magnification. (b) Cell viability. AML-12 cells were pretreated with SalA (0.5, 5 or 50 μM) for 6 h and then treated with ethanol for 24 h. Cell viability was assessed using CCK8 assays. The data are presented as the mean ± S.D. (n ≥ 8). **P < 0.01 versus the control group, ##P < 0.01 versus the ethanol group. | ||
| Group | ALT (IU L−1) | AST (IU L−1) | TC (mmol L−1) | TG (mmol L−1) |
|---|---|---|---|---|
| a The data are presented as the mean ± S.D. (n = 6). **p < 0.01 versus control group. ##p < 0.01 versus ethanol group. | ||||
| Control | 20.59 ± 4.65 | 21.93 ± 2.02 | 54.15 ± 4.59 | 43.44 ± 5.11 |
| Control + SalA (50 μM) | 18.84 ± 2.33 | 20.58 ± 2.34 | 50.19 ± 2.24 | 38.10 ± 2.53 |
| Ethanol (100 mM) | 42.57 ± 6.61** | 47.01 ± 4.48** | 135.98 ± 6.50** | 121.98 ± 6.24** |
| Ethanol + SalA (50 μM) | 26.97 ± 0.94## | 28.93 ± 2.87## | 72.36 ± 10.89## | 59.03 ± 5.26## |
SalA prevents chronic ethanol-induced liver injury by activating autophagy
The induction of autophagy can effectively alleviate alcoholic liver injury.24 Therefore, we investigated whether autophagy activation was associated with the protective effects of SalA against chronic ethanol-induced liver injury. A recent study demonstrated that excessive AV accumulation in hepatic steatosis does not represent autophagy activation but is a result of lipid overload-induced AV-lysosome fusion impairment.36 According to our results (Fig. 2a), after ethanol consumption, AVs were unambiguously observed by TEM. These findings suggest that chronic ethanol treatment induced AV accumulation in the liver. To study the entire autophagy process in the chronic ethanol model, we used adenoviruses to infect AML-12 cells and measure autophagy flux. Ethanol exposure increased AV formation, yet autolysosomes were rarely observed. These results indicate that chronic ethanol administration reduced autolysosome formation. However, the number of autolysosomes was increased by SalA pretreatment, which suggests that SalA can activate autophagy under conditions of chronic ethanol consumption (Fig. 2b). Next, we investigated whether SalA exerted this protective role by activating autophagy. It is well known that ethanol can lead to steatosis6 and hepatocyte apoptosis.37 Accordingly, lipid accumulation, apoptosis, and cell death were up-regulated by ethanol consumption, whereas SalA treatment considerably attenuated this process. Ethanol exposure aggravated lipid accumulation, apoptosis, and cell death in AML-12 cells treated with both SalA and the autophagy-specific inhibitor chloroquine (CQ) (Fig. 3a, b and c). The protective effects of autophagy during chronic ethanol consumption were further confirmed using the autophagy activator Earle's balanced salt solution (EBSS).38 SalA and EBSS treatment reversed ethanol-induced cell injury (Fig. 3c). These results indicate that activating autophagy by SalA could attenuate chronic ethanol-induced liver injury.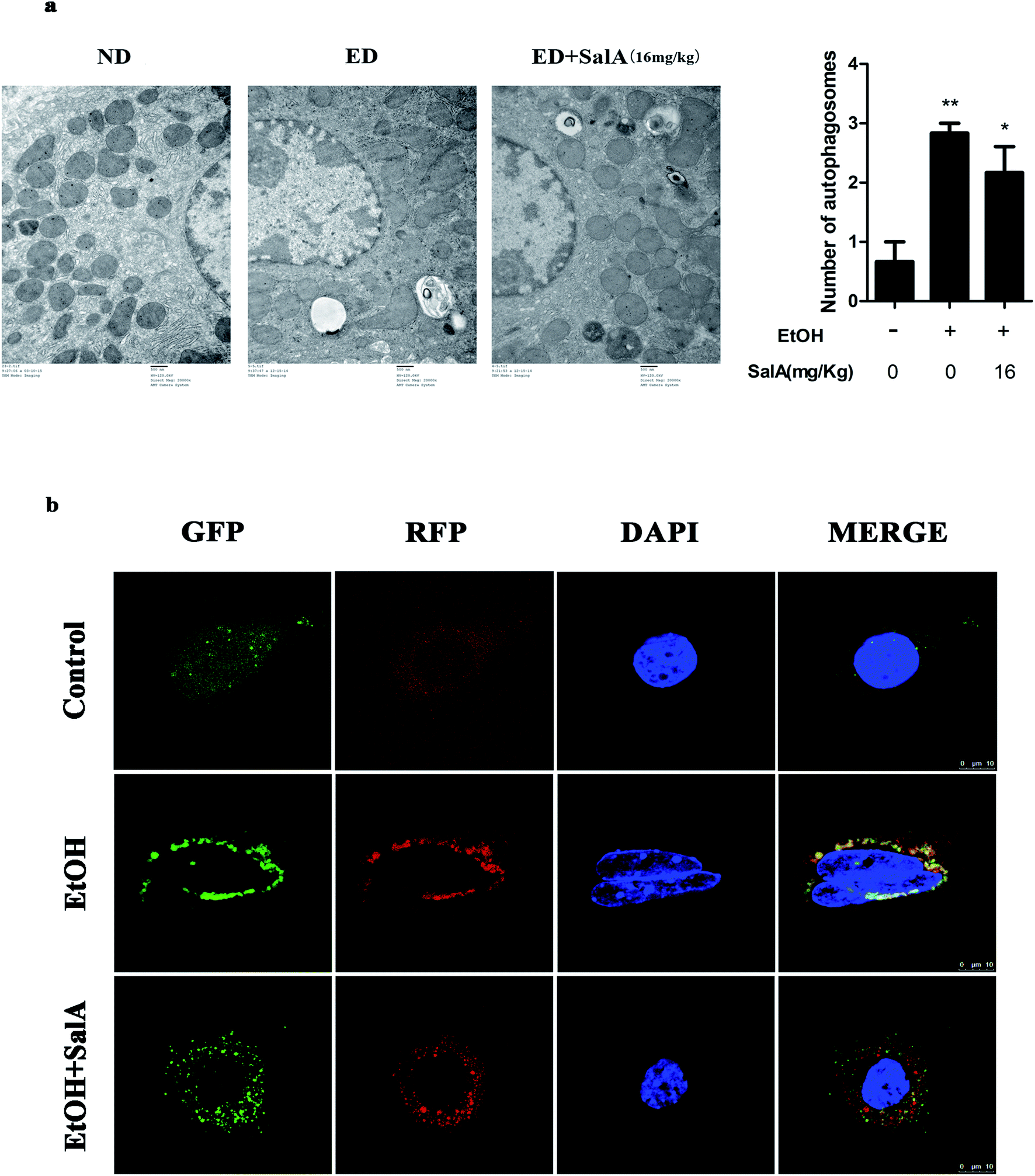 | ||
| Fig. 2 SalA mediates autophagy activation. (a) Effects of SalA on AV formation in ALD liver were assessed using the TEM assay (1500×), and autophagosomes were quantified. The data are presented as the mean ± S.D. *P < 0.05 versus the ND group, **P < 0.01 versus the ND group (n = 3). (b) Autophagy flux visualized through confocal microscopy. AML-12 cells were infected with adenoviruses as described in Materials and methods. Autophagy flux was increased when the numbers of both yellow and red puncta were increased in cells, whereas autophagy flux was blocked when the number of only yellow puncta was increased in AML-12 cells. | ||
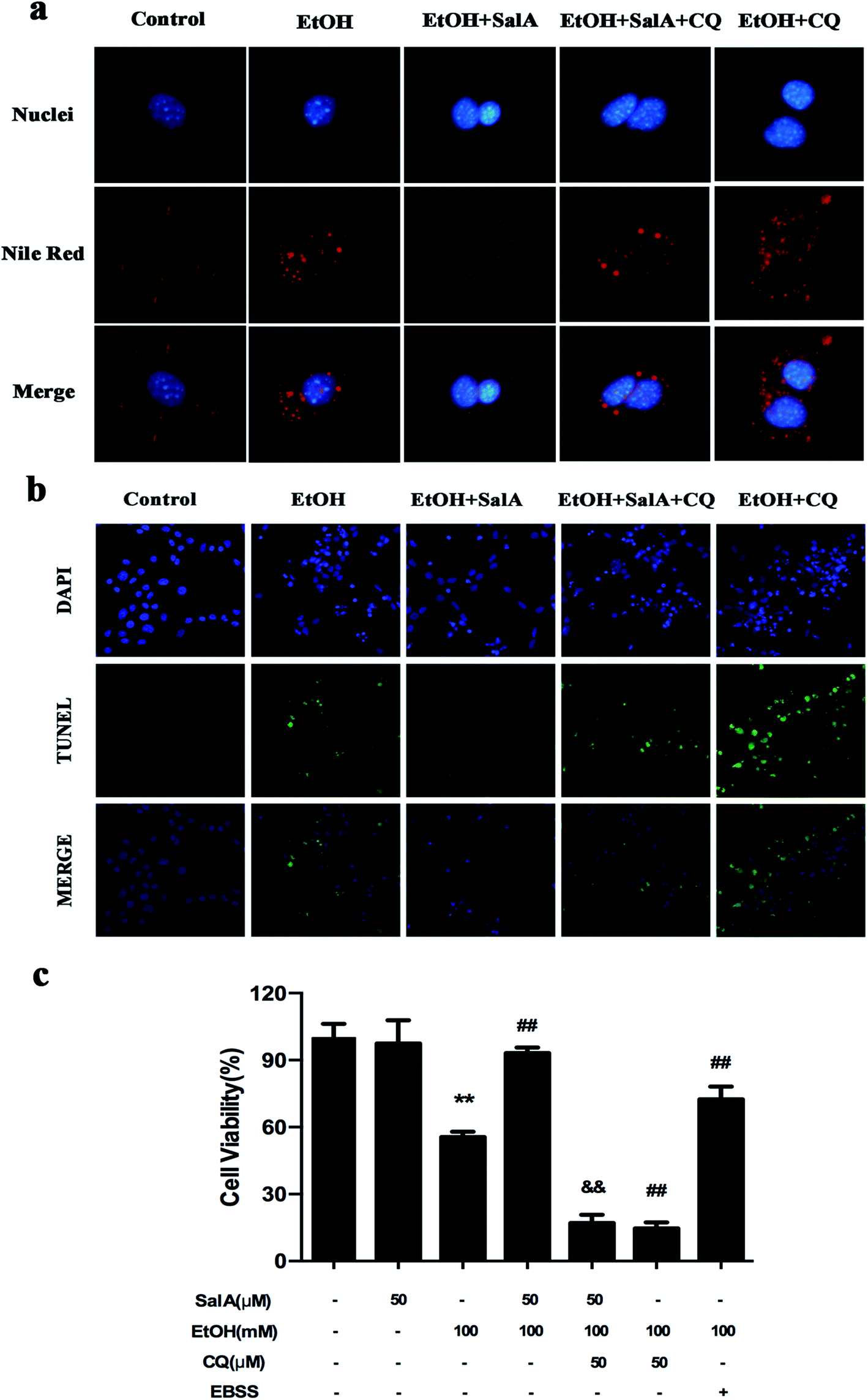 | ||
| Fig. 3 Autophagy activation by SalA exerts a protective effect against chronic ethanol-induced hepatocyte injury. (a and b) SalA-induced autophagy activation decreases lipid accumulation and apoptosis. AML-12 cells were pretreated with SalA (50 μM) or CQ (50 μM) for 6 h before exposure to ethanol (100 mM) for another 24 h. (a) Nile red staining (400×). (b) TUNEL staining (400×). (c) Cell viability. AML-12 cells were pretreated with SalA (50 μM), CQ (50 μM), or EBSS for 6 h and then treated with ethanol for 24 h; afterwards, cell viability was determined. The data are presented as the mean ± S.D. **P < 0.01 versus the control group, ##P < 0.01 versus the ethanol group, &&P < 0.01 versus the ethanol + SalA group (n = 8). | ||
SalA activates autophagy which is inhibited by chronic ethanol consumption via restoring AV-lysosome fusion
To further assess whether SalA exerted its protective effects against ALD by regulating autophagy, we measured the expression levels of LC3-II and SQSTM1, which are marker proteins of autophagy and degradation in the lysosomes.39,40 The protein levels of LC3-II and SQSTM1 were higher in the ethanol group than those in the control group (Fig. 4a); however, LC3-II levels were even higher and SQSTM1 levels were lower in the SalA group than those in the ethanol group. Furthermore, we measured the protein levels of lysosomal proteins, including the mature form of cathepsin B, LAMP-2, and RAB7 (Fig. 4b). After chronic ethanol consumption, LAMP-2, RAB7 and the mature form of cathepsin B levels were decreased. However, pretreatment with SalA prior to ethanol exposure resulted in increased levels of LAMP-2, RAB7, and cathepsin B. Thus, the variable expression levels of autophagy-related proteins indicate that chronic ethanol consumption damaged AV-lysosome fusion and that SalA pretreatment could restore autolysosome formation and activate autophagy.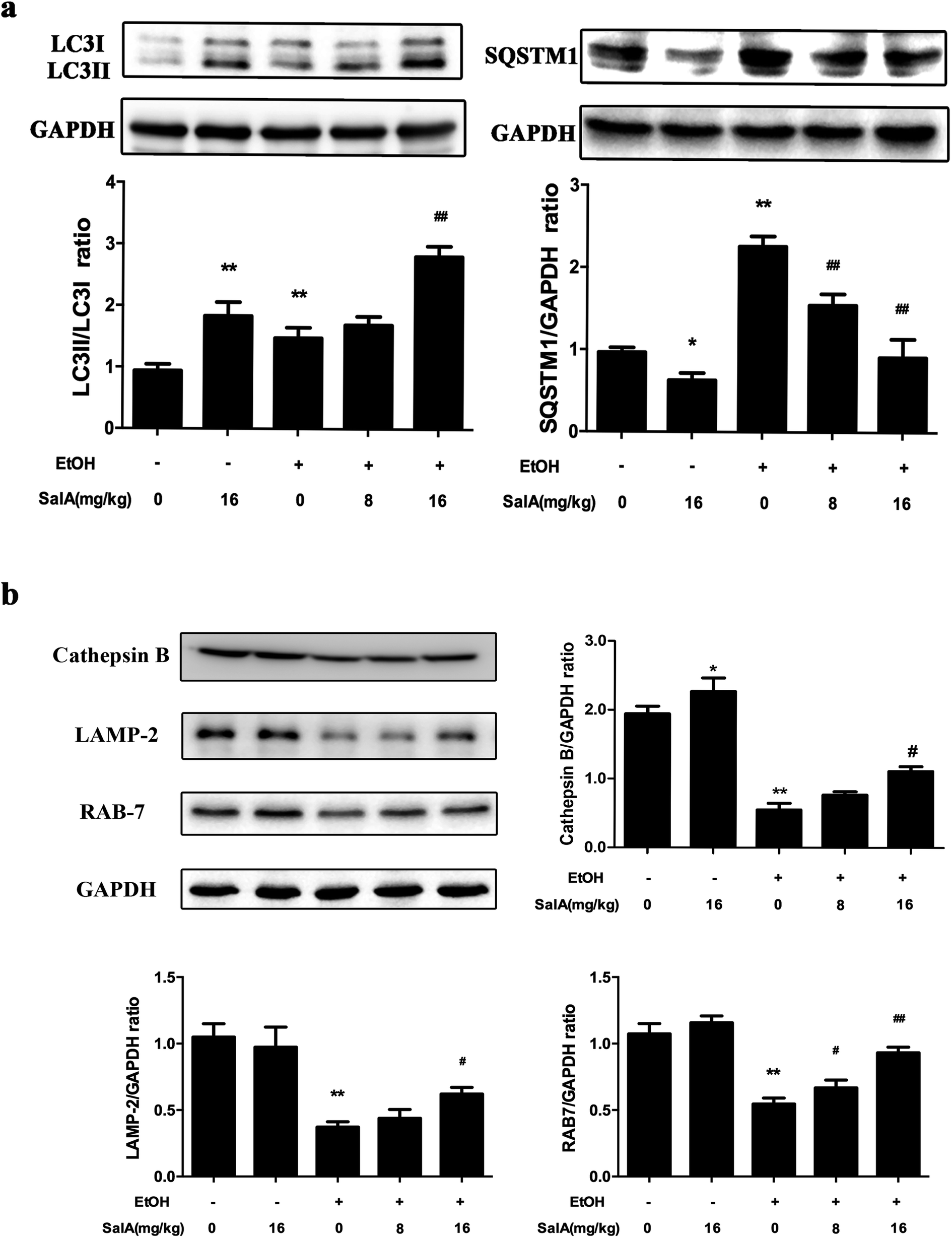 | ||
| Fig. 4 SalA promotes autophagy activation to prevent chronic ethanol-induced AV-lysosome fusion damage. (a and b) The expression levels of LC3-II, SQSTM1, the mature form of cathepsin B, RAB7, and LAMP-2 in the liver were evaluated by western blotting. The data are presented as the mean ± S.D. (n = 3). *P < 0.05 versus the ND group, **P < 0.01 versus the ND group, #P < 0.05 versus the ED group, ##P < 0.01 versus the ED group. | ||
SalA promotes AV-lysosome fusion in a SIRT1-dependent manner
SIRT1 is an NAD+-dependent protein deacetylase that regulates autophagy by modifying histones and transcription factors to activate autophagy flux.31,33 We thus examined whether the SalA-mediated promotion of AV-lysosome fusion in response to chronic ethanol exposure was regulated by SIRT1. As shown in Fig. 5a, compared with ethanol treatment alone, SalA pretreatment increased the SIRT1, RAB7, and LAMP-2 expression levels remarkably. We then evaluated the effects of SalA on AV-lysosome fusion after SIRT1 siRNA treatment in AML-12 cells. Compared with the control siRNA, SIRT1 siRNA treatment resulted in significantly decreased levels of RAB7 and LAMP-2 (ESI Fig. 1†). After chronic ethanol consumption, the SalA-induced increased levels of RAB7 and LAMP-2 were mostly abolished in the presence of SIRT1 siRNA (Fig. 5a). In addition, SIRT1 siRNA abrogated the SalA-induced activation of AV-lysosome fusion (Fig. 5b). These data suggest that SalA-mediated SIRT1 induction activated autophagy by promoting AV-lysosome fusion. | ||
| Fig. 5 SIRT1 mediates the restoration of AV-lysosome fusion by SalA in ethanol-treated AML-12 cells. (a) AML-12 cells were transfected with SIRT1-specific siRNA or control siRNA for 48 h before treatment with SalA (50 μM). Then, the transfected cells were exposed to ethanol (100 mM) for another 24 h. SIRT1, LAMP-2, and RAB7 protein levels in cellular lysates were evaluated by western blotting. The data are presented as the mean ± S.D. **P < 0.01 versus the si-control + ethanol group (n = 3). (b) Autophagy flux visualized using confocal microscopy. AML-12 cells were infected with adenoviruses as described in Materials and methods. | ||
Discussion
Alcohol-induced liver injury has become a worldwide disease. Although many treatments for ALD are currently being used, few are very effective. Thus, novel therapeutic approaches are required. The present research aimed to determine (1) whether SalA conferred protection against chronic ethanol-induced liver injury and (2) whether the protective effects of SalA against ALD were associated with autophagy activation caused by SIRT1-induced AV-lysosome fusion.SalA is one of the primary active components that can be isolated from Salvia miltiorrhiza. Studies have reported that SalA exerts protective effects on the liver; it can prevent HFD-induced hepatic fibrosis, scavenge ROS, and protect against concanavalin A (ConA)-induced hepatitis.13,15,41 Our study aimed to demonstrate that SalA plays a protective role in chronic ethanol-induced liver injury. As expected, SalA protected the liver from chronic ethanol exposure, decreased high serum transaminase levels, attenuated histological liver damage, and prevented ethanol-induced liver cell injury in vitro (Tables 1, 2, and Fig. 1). We further explored the molecular mechanisms involved in the protective effects of SalA against ALD.
Studies have demonstrated that autophagy is an essential catabolic pathway that degrades cellular components within the lysosome. Moreover, autophagy is important for maintaining cellular functions and controlling lipid metabolism in the liver. Interestingly, chronic alcohol consumption suppresses autophagy; this finding indicates that decreased autophagy may underlie the pathogenesis of ALD.42 Alcohol induces AV formation but decreases the number of autolysosomes.43,44 These data are consistent with our results. However, SalA treatment markedly reversed this trend. This finding suggests that SalA may have the same effects as an autophagy activator in protecting against ALD (Fig. 2). We tested this possibility in the AML-12 cell line. As expected, SalA increased cell viability and elicited the same effects as EBSS (a specific activator of autophagy) after ethanol exposure (Fig. 3c). Ample evidence has indicated that the important role of autophagy in reducing ALD is likely due to the selective removal of lipid droplets and damaged mitochondria.24,45,46 Similarly, genetically increasing autophagy by overexpressing the autophagy gene Atg7 ameliorated hepatic steatosis and insulin resistance in ob/ob and HFD-fed mice.47 Growing research also indicates that the cytoprotective function of autophagy is mediated in many circumstances by the negative modulation of apoptosis. Under certain circumstances, autophagy constitutes a stress adaptation that suppresses apoptosis by regulating the BCl2-family proteins BAX and BAK and caspases to attenuate diseases such as Alzheimer's disease and hepatocellular carcinoma.48–50 In ALD, the activation of autophagy alleviates ethanol-induced apoptosis mediated by the AMPK/FoxO3A axis.51 In our study, the lipid accumulation and apoptosis induced by ethanol were diminished by SalA; however, SalA pretreatment was no longer protective after treatment with the autophagy inhibitor CQ (Fig. 3a and b). Collectively, these results indicate that SalA plays a protective role in chronic ethanol-induced liver injury via activating autophagy.
To investigate the protective role of SalA in chronic ethanol-induced autophagy inhibition, we measured the protein levels of LC3-II and SQSTM1. The lipidation of LC3-I to LC3-II is a very important step for AV development and maturation.20 However, the accumulation of LC3-II could represent either the induction of autophagy or the impairment of autophagy flux by blocking lysosomal fusion and degradation.40 Consistent with these findings, chronic ethanol or SalA treatment increased the expression levels of LC3-II. These data indicate that chronic ethanol or SalA treatment induced AV formation. However, increased expression levels of the autophagy substrate SQSTM1 suggest that chronic ethanol consumption inhibited the degradation of SQSTM1, whereas SalA pretreatment rescued this process (Fig. 4a). These results demonstrate that SalA pretreatment induced AV formation and the degradation of the autophagy substrate. Furthermore, we assessed whether SalA activated autophagy by promoting AV-lysosome fusion. Recent findings have suggested that chronic ethanol consumption diminishes the proteolytic capacity of liver lysosomes, decreases cathepsin B levels, blocks AV-lysosome fusion, and inhibits protein degradation.23,40,52 Previous reports have indicated that knocking down the lysosomal markers RAB7 and LAMP-2 inhibits AV-lysosome fusion;33,53,54 in addition, RAB7 is a central regulator that is important for regulating hepatic lipid droplet (LD)-specific autophagy (lipophagy).55,56 As expected, chronic ethanol exposure significantly decreased the levels of LAMP-2, RAB7, and the mature form of cathepsin B. These results indicate that AV-lysosome fusion was impaired, but pretreatment or co-treatment of SalA could both prevented these impairments (Fig. 4b, ESI Fig. 2†), consistent with the protective effect of Torin1, a known autophagy inducer57 (ESI Fig. 3†). Therefore, variation in the levels of these autophagy-related proteins illustrates that restoring AV-lysosome fusion and autophagy were required for SalA to alleviate chronic ethanol-induced liver injury.
SIRT1, an NAD+-dependent deacetylase, has important effects on metabolic diseases, including ALD. Chronic ethanol accumulation decreases SIRT1 expression levels, whereas SIRT1-specific transgenic mice are protected from hepatic steatosis caused by ethanol consumption.58 In our study, SIRT1 protein levels were significantly decreased after chronic ethanol exposure, whereas SalA treatment prevented this effect. These results suggest that SalA may confer protection against chronic ethanol-induced ALD by increasing SIRT1 expression (ESI Fig. 4 and 5a†). SIRT1 is well recognized as a primary regulator of autophagy through its deacetylation and activation of major components of the autophagy induction network; these components include ATGs and transcriptional regulators, such as ATG5, ATG7, CREB, and PPARα.31 Additionally, SIRT1 mediates the effect of adipose triglyceride lipase (ATGL) in promoting lipophagy to control hepatic lipid metabolism.59 Moreover, autophagy flux is damaged, and autophagy is inhibited in liver-specific SIRT1−/− mice.32,60 A recent study revealed that SIRT1 protects against ischemia/reperfusion injury in alcoholic fatty liver through the activation of autophagy.61 However, this study did not determine whether SIRT1 regulated AV-lysosome fusion. We found that after chronic ethanol consumption, SIRT1 up-regulation via SalA increased LAMP-2 and RAB7 protein levels in AML-12 cells; however, SIRT1 knockdown notably inhibited this increase, suggesting that autophagosome–lysosome fusion promoted by SalA was mostly abrogated in the presence of SIRT1 siRNA (Fig. 5a). Furthermore, by using confocal microscopy, we found that SIRT1 knockdown increased the number of AVs while decreasing the number of autolysosomes; SalA treatment, however, could not reverse this trend (Fig. 5b). Together, these results suggest that the SalA-mediated recovery of AV-lysosome fusion and autophagy is regulated by SIRT1.
Conclusions
In summary, the present study revealed that SalA had a protective effect against chronic ethanol-induced liver injury. The protective effect of SalA was associated with autophagy activation. The results indicate that SalA conferred protection against chronic ethanol-induced ALD, at least partly through the SIRT1-mediated promotion of AV-lysosome fusion and autophagy. Our study presents an attractive pharmacological target for the development of new drugs to prevent ALD progression.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81473266).References
- A. Louvet and P. Mathurin, Alcoholic liver disease: mechanisms of injury and targeted treatment, Nat. Rev. Gastroenterol. Hepatol., 2015, 12, 231–242 CrossRef PubMed.
- C. L. Vuittonet, M. Halse, L. Leggio, S. B. Fricchione, M. Brickley, C. L. Haass-Koffler, T. Tavares, R. M. Swift and G. A. Kenna, Pharmacotherapy for alcoholic patients with alcoholic liver disease, Am. J. Health-Syst. Pharm., 2014, 71, 1265–1276 CrossRef PubMed.
- R. Bruha, K. Dvorak and J. Petrtyl, Alcoholic liver disease, World J Hepatol., 2012, 4, 81–90 CrossRef PubMed.
- R. S. O'Shea, S. Dasarathy and A. J. McCullough, Practice Guideline Committee of the American Association for the Study of Liver and Practice Parameters Committee of the American College of, Alcoholic liver disease, Hepatology, 2010, 51, 307–328 CrossRef PubMed.
- S. M. Bailey and C. C. Cunningham, Contribution of mitochondria to oxidative stress associated with alcoholic liver disease, Free Radical Biol. Med., 2002, 32, 11–16 CrossRef PubMed.
- C. S. Lieber, Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis, Alcohol, 2004, 34, 9–19 CrossRef PubMed.
- S. Zakhari and T. K. Li, Determinants of alcohol use and abuse: impact of quantity and frequency patterns on liver disease, Hepatology, 2007, 46, 2032–2039 CrossRef PubMed.
- W. X. Ding, S. Manley and H. M. Ni, The emerging role of autophagy in alcoholic liver disease, Exp. Biol. Med., 2011, 236, 546–556 CrossRef PubMed.
- P. Xie, Y. Duan, X. Guo, L. Hu and M. Yu, SalA attenuates hypoxia-induced endothelial endoplasmic reticulum stress and apoptosis via down-regulation of VLDL receptor expression, Cell. Physiol. Biochem., 2015, 35, 17–28 CrossRef PubMed.
- X. Zheng, S. Chen, Q. Yang, J. Cai, W. Zhang, H. You, J. Xing and Y. Dong, Salvianolic acid A reverses the paclitaxel resistance and inhibits the migration and invasion abilities of human breast cancer cells by inactivating transgelin 2, Cancer Biol. Ther., 2015, 16, 1407–1414 CrossRef PubMed.
- T. Zhang, J. Xu, D. Li, J. Chen, X. Shen, F. Xu, F. Teng, Y. Deng, H. Ma, L. Zhang, G. Zhang, Z. Zhang, W. Wu, X. Liu, M. Yang, B. Jiang and D. Guo, Salvianolic acid A, a matrix metalloproteinase-9 inhibitor of Salvia miltiorrhiza, attenuates aortic aneurysm formation in apolipoprotein E-deficient mice, Phytomedicine, 2014, 21, 1137–1145 CrossRef PubMed.
- Y. Guo, X. Xu, Q. Li, Z. Li and F. Du, Anti-inflammation effects of picroside 2 in cerebral ischemic injury rats, Behav. Brain Funct., 2010, 6, 43 CrossRef PubMed.
- G. Qiang, X. Yang, Q. Xuan, L. Shi, H. Zhang, B. Chen, X. Li, M. Zu, D. Zhou, J. Guo, H. Yang, L. Zhang and G. Du, Salvianolic acid a prevents the pathological progression of hepatic fibrosis in high-fat diet-fed and streptozotocin-induced diabetic rats, Am. J. Chin. Med., 2014, 42, 1183–1198 CrossRef PubMed.
- H. Q. Yin, Y. J. Choi, Y. C. Kim, D. H. Sohn, S. Y. Ryu and B. H. Lee, Salvia miltiorrhiza Bunge and its active component cryptotanshinone protects primary cultured rat hepatocytes from acute ethanol-induced cytotoxicity and fatty infiltration, Food Chem. Toxicol., 2009, 47, 98–103 CrossRef PubMed.
- X. Xu, Y. Hu, X. Zhai, M. Lin, Z. Chen, X. Tian, F. Zhang, D. Gao, X. Ma, L. Lv and J. Yao, Salvianolic acid A preconditioning confers protection against concanavalin A-induced liver injury through SIRT1-mediated repression of p66shc in mice, Toxicol. Appl. Pharmacol., 2013, 273, 68–76 CrossRef PubMed.
- C. Ding, Y. Zhao, X. Shi, N. Zhang, G. Zu, Z. Li, J. Zhou, D. Gao, L. Lv, X. Tian and J. Yao, New insights into salvianolic acid A action: regulation of the TXNIP/NLRP3 and TXNIP/ChREBP pathways ameliorates HFD-induced NAFLD in rats, Sci. Rep., 2016, 6, 28734 CrossRef PubMed.
- C. He and D. J. Klionsky, Regulation mechanisms and signaling pathways of autophagy, Annu. Rev. Genet., 2009, 43, 67–93 CrossRef PubMed.
- P. Chandra, S. Ghanwat, S. K. Matta, S. S. Yadav, M. Mehta, Z. Siddiqui, A. Singh and D. Kumar, Mycobacterium tuberculosis Inhibits RAB7 Recruitment to Selectively Modulate Autophagy Flux in Macrophages, Sci. Rep., 2015, 5, 16320 CrossRef PubMed.
- D. Hu, J. Wu, W. Wang, M. Mu, R. Zhao, X. Xu, Z. Chen, J. Xiao, F. Hu, Y. Yang and R. Zhang, Autophagy regulation revealed by SapM-induced block of autophagosome-lysosome fusion via binding RAB7, Biochem. Biophys. Res. Commun., 2015, 461, 401–407 CrossRef PubMed.
- B. Ding, G. Zhang, X. Yang, S. Zhang, L. Chen, Q. Yan, M. Xu, A. K. Banerjee and M. Chen, Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome–lysosome fusion to increase virus production, Cell Host Microbe, 2014, 15, 564–577 Search PubMed.
- W. X. Ding, M. Li and X. M. Yin, Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets, Autophagy, 2011, 7, 248–249 CrossRef PubMed.
- K. K. Kharbanda, D. L. McVicker, R. K. Zetterman and T. M. Donohue, Jr, Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes, Biochim. Biophys. Acta, 1995, 1245, 421–429 CrossRef.
- N. A. Osna, P. G. Thomes and T. M. Donohue, Jr, Involvement of autophagy in alcoholic liver injury and hepatitis C pathogenesis, World J. Gastroenterol., 2011, 17, 2507–2514 CrossRef PubMed.
- C. W. Lin, H. Zhang, M. Li, X. Xiong, X. Chen, X. Chen, X. C. Dong and X. M. Yin, Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice, J. Hepatol., 2013, 58, 993–999 CrossRef PubMed.
- A. Purushotham, T. T. Schug, Q. Xu, S. Surapureddi, X. Guo and X. Li, Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation, Cell Metab., 2009, 9, 327–338 CrossRef PubMed.
- Y. Cao, Y. Xue, L. Xue, X. Jiang, X. Wang, Z. Zhang, J. Yang, J. Lu, C. Zhang, W. Wang and G. Ning, Hepatic menin recruits SIRT1 to control liver steatosis through histone deacetylation, J. Hepatol., 2013, 59, 1299–1306 CrossRef PubMed.
- M. Li, Y. Lu, Y. Hu, X. Zhai, W. Xu, H. Jing, X. Tian, Y. Lin, D. Gao and J. Yao, Salvianolic acid B protects against acute ethanol-induced liver injury through SIRT1-mediated deacetylation of p53 in rats, Toxicol. Lett., 2014, 228, 67–74 CrossRef PubMed.
- R. R. Alcendor, S. Gao, P. Zhai, D. Zablocki, E. Holle, X. Yu, B. Tian, T. Wagner, S. F. Vatner and J. Sadoshima, Sirt1 regulates aging and resistance to oxidative stress in the heart, Circ. Res., 2007, 100, 1512–1521 CrossRef PubMed.
- L. Gao, W. Shan, W. Zeng, Y. Hu, G. Wang, X. Tian, N. Zhang, X. Shi, Y. Zhao, C. Ding, F. Zhang, K. Liu and J. Yao, Carnosic acid alleviates chronic alcoholic liver injury by regulating the SIRT1/ChREBP and SIRT1/p66shc pathways in rats, Mol. Nutr. Food Res., 2016, 60, 1902–1911 Search PubMed.
- N. Zhang, Y. Hu, C. Ding, W. Zeng, W. Shan, H. Fan, Y. Zhao, X. Shi, L. Gao, T. Xu, R. Wang, D. Gao and J. Yao, Salvianolic acid B protects against chronic alcoholic liver injury via SIRT1-mediated inhibition of CRP and ChREBP in rats, Toxicol. Lett., 2017, 267, 1–10 CrossRef PubMed.
- G. Qiu, X. Li, X. Che, C. Wei, S. He, J. Lu, Z. Jia, K. Pang and L. Fan, SIRT1 is a regulator of autophagy: implications in gastric cancer progression and treatment, FEBS Lett., 2015, 589, 2034–2042 CrossRef PubMed.
- I. H. Lee, L. Cao, R. Mostoslavsky, D. B. Lombard, J. Liu, N. E. Bruns, M. Tsokos, F. W. Alt and T. Finkel, A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3374–3379 CrossRef PubMed.
- N. Hariharan, Y. Maejima, J. Nakae, J. Paik, R. A. Depinho and J. Sadoshima, Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes, Circ. Res., 2010, 107, 1470–1482 CrossRef PubMed.
- C. S. Lieber and L. M. DeCarli, Animal models of chronic ethanol toxicity, Methods Enzymol., 1994, 233, 585–594 Search PubMed.
- R. Wu, W. W. Chaung, M. Zhou, Y. Ji, W. Dong, Z. Wang, X. Qiang and P. Wang, Milk fat globule EGF factor 8 attenuates sepsis-induced apoptosis and organ injury in alcohol-intoxicated rats, Alcohol.: Clin. Exp. Res., 2010, 34, 1625–1633 CrossRef PubMed.
- K. Miyagawa, S. Oe, Y. Honma, H. Izumi, R. Baba and M. Harada, Lipid-Induced Endoplasmic Reticulum Stress Impairs Selective Autophagy at the Step of Autophagosome–Lysosome Fusion in Hepatocytes, Am. J. Pathol., 2016, 186, 1861–1873 CrossRef PubMed.
- M. A. Abdelmegeed, A. Banerjee, S. Jang, S. H. Yoo, J. W. Yun, F. J. Gonzalez, A. Keshavarzian and B. J. Song, CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis, Free Radical Biol. Med., 2013, 65, 1238–1245 CrossRef PubMed.
- Y. Min, W. Xu, D. Liu, H. Shen, Y. Xu, S. Zhang, L. Zhang and H. Wang, Earle's balanced salts solution and rapamycin differentially regulate the Bacillus Calmette-Guerin-induced maturation of human dendritic cells, Acta Biochim. Biophys. Sin., 2013, 45, 162–169 CrossRef PubMed.
- H. S. Lee, B. H. Daniels, E. Salas, A. W. Bollen, J. Debnath and M. Margeta, Clinical utility of LC3 and p62 immunohistochemistry in diagnosis of drug-induced autophagic vacuolar myopathies: a case-control study, PLoS One, 2012, 7, e36221 Search PubMed.
- H. I. Cho, J. W. Choi and S. M. Lee, Impairment of autophagosome–lysosome fusion contributes to chronic ethanol-induced liver injury, Alcohol, 2014, 48, 717–725 CrossRef PubMed.
- X. J. Wang, Z. B. Wang and J. X. Xu, Effect of salvianic acid A on lipid peroxidation and membrane permeability in mitochondria, J. Ethnopharmacol., 2005, 97, 441–445 CrossRef PubMed.
- P. E. Rautou, A. Mansouri, D. Lebrec, F. Durand, D. Valla and R. Moreau, Autophagy in liver diseases, J. Hepatol., 2010, 53, 1123–1134 CrossRef PubMed.
- L. Murrow and J. Debnath, Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease, Annu. Rev. Pathol.: Mech. Dis., 2013, 8, 105–137 CrossRef PubMed.
- D. Wu, X. Wang, R. Zhou, L. Yang and A. I. Cederbaum, Alcohol steatosis and cytotoxicity: the role of cytochrome P4502E1 and autophagy, Free Radical Biol. Med., 2012, 53, 1346–1357 CrossRef PubMed.
- R. Guo, X. Xu, S. A. Babcock, Y. Zhang and J. Ren, Aldehyde dehydrogenase-2 plays a beneficial role in ameliorating chronic alcohol-induced hepatic steatosis and inflammation through regulation of autophagy, J. Hepatol., 2015, 62, 647–656 CrossRef PubMed.
- X. Guo, R. Cui, J. Zhao, R. Mo, L. Peng and M. Yan, Corosolic acid protects hepatocytes against ethanol-induced damage by modulating mitogen-activated protein kinases and activating autophagy, Eur. J. Pharmacol., 2016, 791, 578–588 CrossRef PubMed.
- L. Yang, P. Li, S. Fu, E. S. Calay and G. S. Hotamisligil, Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance, Cell Metab., 2010, 11, 467–478 CrossRef PubMed.
- M. C. Maiuri, E. Zalckvar, A. Kimchi and G. Kroemer, Self-eating and self-killing: crosstalk between autophagy and apoptosis, Nat. Rev. Mol. Cell Biol., 2007, 8, 741–752 CrossRef PubMed.
- Y. Wang, C. Han, L. Lu, S. Magliato and T. Wu, Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells, Hepatology, 2013, 58, 995–1010 CrossRef PubMed.
- A. Salminen, K. Kaarniranta, A. Kauppinen, J. Ojala, A. Haapasalo, H. Soininen and M. Hiltunen, Impaired autophagy and APP processing in Alzheimer's disease: the potential role of Beclin 1 interactome, Prog. Neurobiol., 2013, 106–107, 33–54 CrossRef PubMed.
- S. Nepal and P. H. Park, Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in HepG2 cells: involvement of AMPK/FoxO3A axis, Biochim. Biophys. Acta, 2013, 1833, 2111–2125 CrossRef PubMed.
- A. Dolganiuc, P. G. Thomes, W. X. Ding, J. J. Lemasters and T. M. Donohue, Jr., Autophagy in alcohol-induced liver diseases, Alcohol.: Clin. Exp. Res., 2012, 36, 1301–1308 CrossRef PubMed.
- F. Fortunato, H. Burgers, F. Bergmann, P. Rieger, M. W. Buchler, G. Kroemer and J. Werner, Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis, Gastroenterology, 2009, 137, 350–360 CrossRef PubMed.
- O. A. Mareninova, K. Hermann, S. W. French, M. S. O'Konski, S. J. Pandol, P. Webster, A. H. Erickson, N. Katunuma, F. S. Gorelick, I. Gukovsky and A. S. Gukovskaya, Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis, J. Clin. Invest., 2009, 119, 3340–3355 Search PubMed.
- B. Schroeder, R. J. Schulze, S. G. Weller, A. C. Sletten, C. A. Casey and M. A. McNiven, The small GTPase Rab7 as a central regulator of hepatocellular lipophagy, Hepatology, 2015, 61, 1896–1907 CrossRef PubMed.
- L. Wang, J. Zhou, S. Yan, G. Lei, C. H. Lee and X. M. Yin, Ethanol-triggered Lipophagy Requires SQSTM1 in AML12 Hepatic Cells, Sci. Rep., 2017, 7, 12307 CrossRef PubMed.
- M. Hosel, A. Huber, S. Bohlen, J. Lucifora, G. Ronzitti, F. Puzzo, F. Boisgerault, U. T. Hacker, W. J. Kwanten, N. Kloting, M. Bluher, A. Gluschko, M. Schramm, O. Utermohlen, W. Bloch, F. Mingozzi, O. Krut and H. Buning, Autophagy determines efficiency of liver-directed gene therapy with adeno-associated viral vectors, Hepatology, 2017, 66, 252–265 CrossRef PubMed.
- J. H. Yu, S. J. Song, A. Kim, Y. Choi, J. W. Seok, H. J. Kim, Y. J. Lee, K. S. Lee and J. W. Kim, Suppression of PPARgamma-mediated monoacylglycerol O-acyltransferase 1 expression ameliorates alcoholic hepatic steatosis, Sci. Rep., 2016, 6, 29352 CrossRef PubMed.
- A. Sathyanarayan, M. T. Mashek and D. G. Mashek, ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism, Cell Rep., 2017, 19, 1–9 CrossRef PubMed.
- S. Dong, C. Jia, S. Zhang, G. Fan, Y. Li, P. Shan, L. Sun, W. Xiao, L. Li, Y. Zheng, J. Liu, H. Wei, C. Hu, W. Zhang, Y. E. Chin, Q. Zhai, Q. Li, J. Liu, F. Jia, Q. Mo, D. P. Edwards, S. Huang, L. Chan, B. W. O'Malley, X. Li and C. Wang, The REGgamma proteasome regulates hepatic lipid metabolism through inhibition of autophagy, Cell Metab., 2013, 18, 380–391 CrossRef PubMed.
- H. I. Cho, M. J. Seo and S. M. Lee, 2-Methoxyestradiol protects against ischemia/reperfusion injury in alcoholic fatty liver by enhancing sirtuin 1-mediated autophagy, Biochem. Pharmacol., 2017, 131, 40–51 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00798e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |