DOI:
10.1039/C8RA00138C
(Paper)
RSC Adv., 2018,
8, 8259-8268
Synthesis of an unusual quinazoline alkaloid: theoretical and experimental investigations of its structural, electronic, molecular and biological properties†
Received
6th January 2018
, Accepted 16th February 2018
First published on 23rd February 2018
Abstract
An unusual quinazoline alkaloid (1) was obtained when 2-aminobenzaldehyde was refluxed with pyrrolidine in ethanol for 12 h. The synthesized compound was characterized using spectral data analysis augmented with X-ray and literature precedent. Single crystal analysis depicted four conformations differing slightly in bond angles and bond lengths. Compound 1 crystallizes in a triclinic crystal system with a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group having two molecules within the unit cell. The experimentally obtained parameters were compared to those obtained theoretically, which depicted a good agreement. Using the DFT/B3LYP/6-31G (d,p) level of theory, HOMO–LUMO energy gap, molecular electrostatic potential (MEP), vibrational (IR) and NMR analyses were carried out. The HOMO–LUMO energy gap allowed the calculation of chemical hardness, chemical inertness, electronegativity and the electrophilicity index of the molecule, which depicted its potential kinetic stability and reactivity. Prediction of activity spectra of the target compound revealed that compound 1 possesses notable antineoplastic activity with Pa = 0.884. The molecule was therefore evaluated against various cancerous cell lines in an in vitro SRB assay which depicted that compound 1 possesses the highest growth inhibition activity against THP-1 cell lines with an IC50 of 7 μM.
space group having two molecules within the unit cell. The experimentally obtained parameters were compared to those obtained theoretically, which depicted a good agreement. Using the DFT/B3LYP/6-31G (d,p) level of theory, HOMO–LUMO energy gap, molecular electrostatic potential (MEP), vibrational (IR) and NMR analyses were carried out. The HOMO–LUMO energy gap allowed the calculation of chemical hardness, chemical inertness, electronegativity and the electrophilicity index of the molecule, which depicted its potential kinetic stability and reactivity. Prediction of activity spectra of the target compound revealed that compound 1 possesses notable antineoplastic activity with Pa = 0.884. The molecule was therefore evaluated against various cancerous cell lines in an in vitro SRB assay which depicted that compound 1 possesses the highest growth inhibition activity against THP-1 cell lines with an IC50 of 7 μM.
Introduction
Alkaloids are a very important class of compounds as far as their role in metabolism and activity is concerned.1 They are among the oldest drugs used for the treatment of diseases2 and are still used as promising therapeutic agents.3,4 Deep sea environments are nowadays the focus of research for discovering newer alkaloids with potential therapeutic activities.5–7 New alkaloids are also being produced via chemical synthesis and natural product modifications with some of them being more active than the naturally obtained ones.8,9 In general alkaloids possess a range of biological activities which include analgesic, antiviral, antimalarial, antineoplastic, antimicrobial, anti-inflammatory, antioxidant antifungal, antibacterial, hemoglobinization agents of human leukemia cells, effects on the CNS, estrogenic effects etc.10–24 Based on these literature reports of alkaloids and our previous work on the isolation, and synthesis of bioactive compounds,25–37 we carried out the synthesis of an unusual self-condensation product of 2-aminobenzaldehyde by a method reported by Seidal and coworkers.38 In order to get deeper insights in to the molecular, structural and biological properties of the synthesized compound, we resorted to spectral, X-ray and DFT analysis. The experimentally obtained parameters were in close agreement to those obtained theoretically. Molecular electrostatic potential and frontier molecular orbital analysis were performed which permitted the calculation of HOMO–LUMO energy gap and related parameters. Based on PASS results, the molecule was evaluated against four different human cancer cell lines to explore its biological potential.
Results and discussion
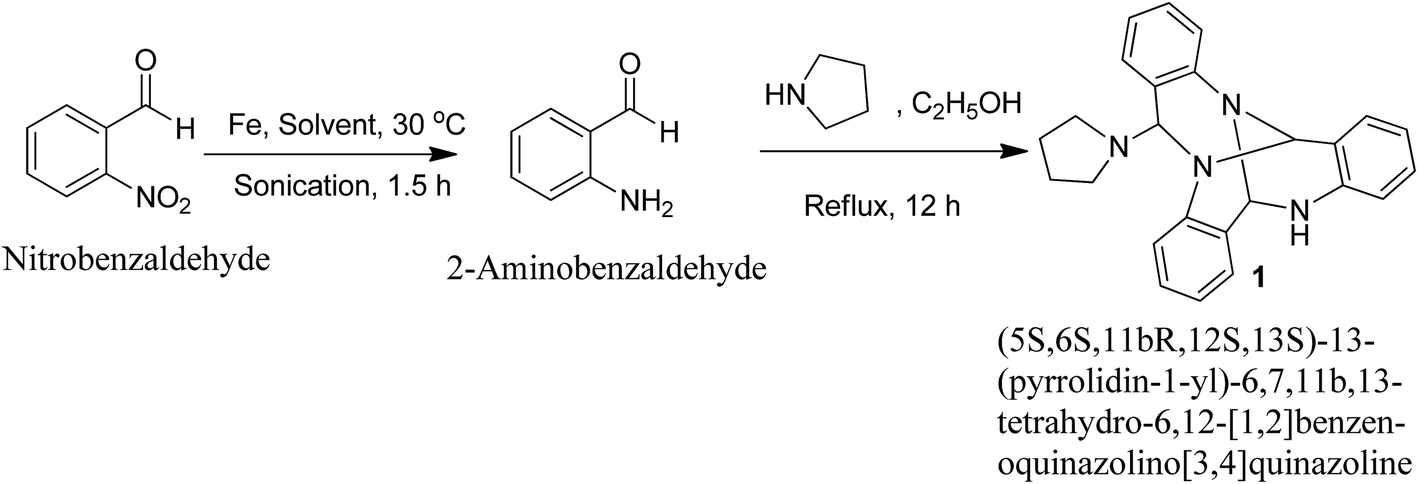
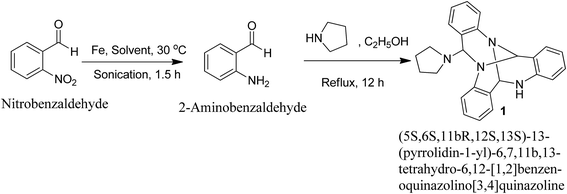
Reduction of 2-nitrobenzaldehyde using iron powder in glacial acetic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol:water system (2:2:1) under ultrasonic irradiation conditions for 1.5 h at 30 °C yielded the corresponding 2-aminobenzaldehyde. Refluxing 2-aminobenzaldehyde in ethanol with pyrrolidine yielded a reaction mixture containing an unusual self-condensation quinazoline alkaloid (1) (Fig. 1).
:ethanol:water system (2:2:1) under ultrasonic irradiation conditions for 1.5 h at 30 °C yielded the corresponding 2-aminobenzaldehyde. Refluxing 2-aminobenzaldehyde in ethanol with pyrrolidine yielded a reaction mixture containing an unusual self-condensation quinazoline alkaloid (1) (Fig. 1).
 |
| | Fig. 1 Synthesis of an unusual quinazoline alkaloid (1). | |
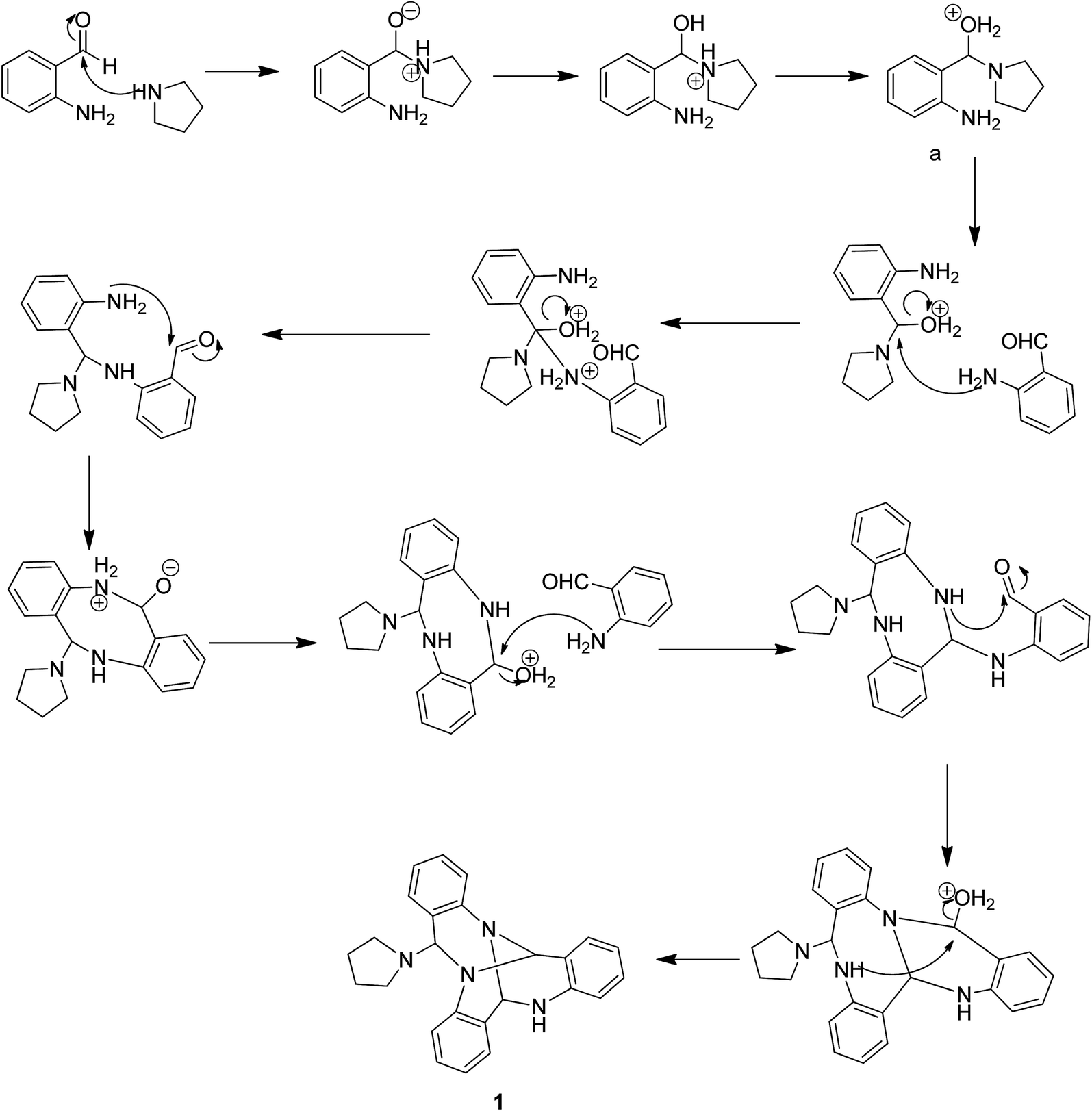
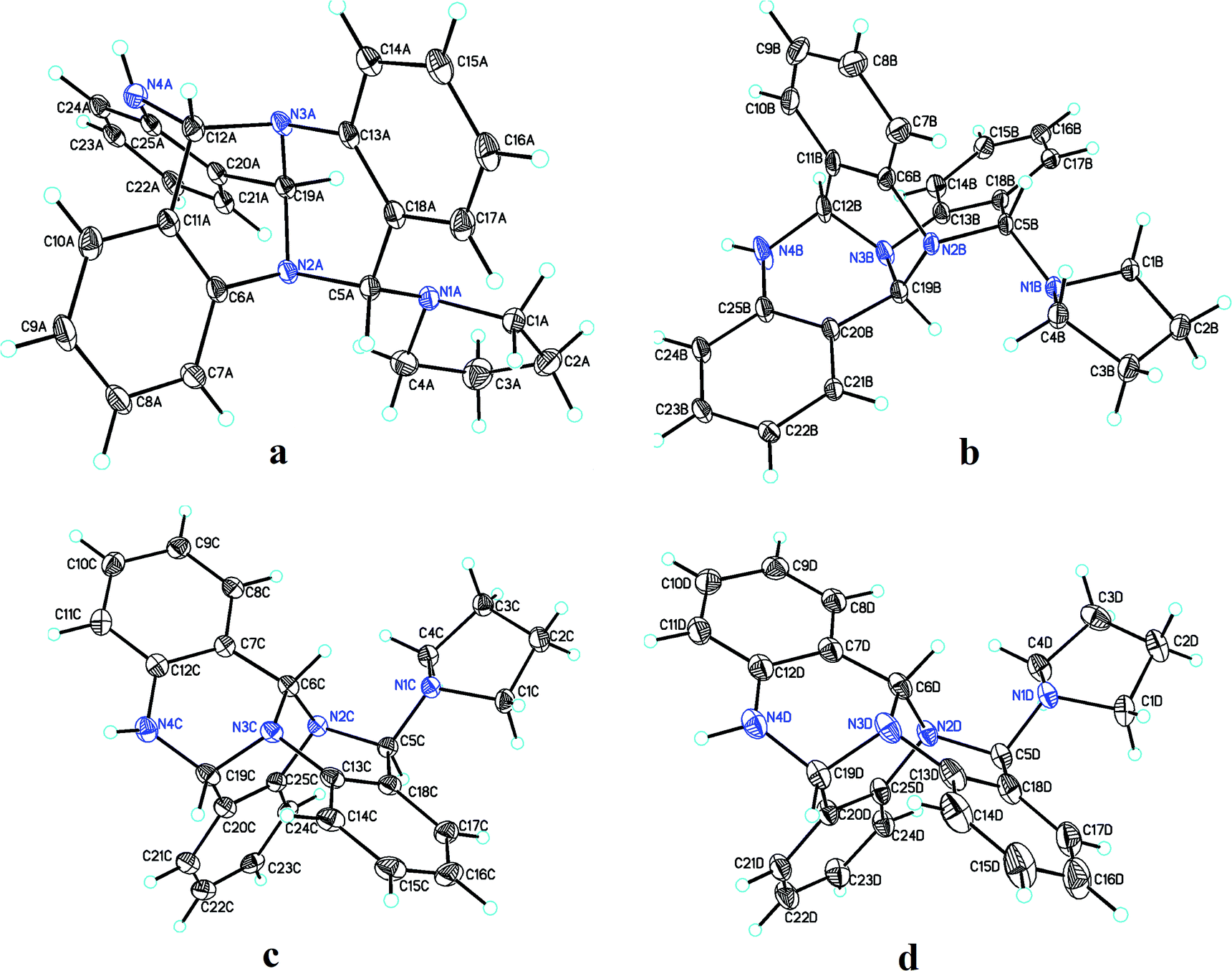
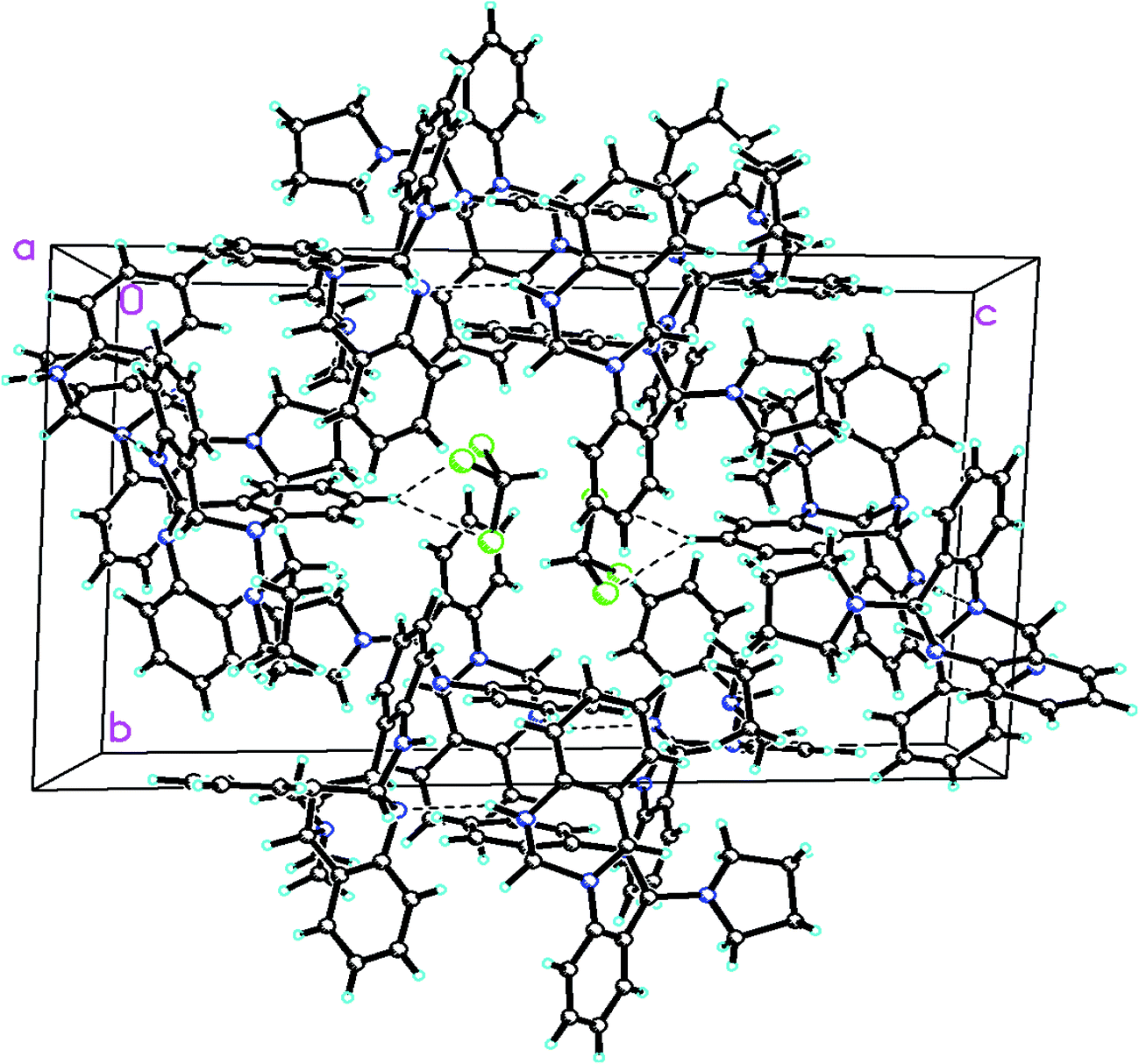
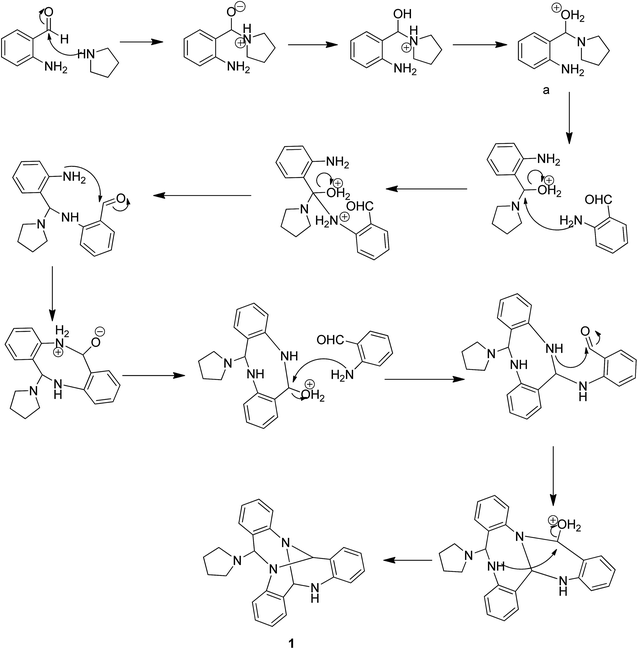
Pure 1 was obtained when the reaction mixture was subjected to normal phase silica gel column chromatography. It's structure was elucidated while analyzing the spectral data in light of literature. The reaction proceeds via a series of nucleophilic additions of basic nitrogen of pyrrolidine ring on the highly electrophilic aldehyde group of 2-aminobenzaldehyde to give an intermediate which again condenses with two molecules of 2-aminobenzaldehyde to yield compound 1 (Fig. 2). The product was characterized using LC-MS, IR and NMR data analysis. LC-MS depicted m/z at 381 assignable to [M + H]+ corresponding to its molecular formula C25H24N4. 1H NMR depicted the presence of 12 aromatic protons assignable to three aromatic rings in the molecule whose presence was further supported by the appearance of 18 13C signals in the aromatic region of the molecule. Three downfield 1H NMR singlets at δ 5.96, 5.29 and 4.52 ppm correspond to 3 CH groups present in the molecule which was supported by three carbon NMR resonances at δ 84, 71 and 64 ppm in its DEPT-NMR. Presence of upfield resonances at 51 and 23 ppm on the downside in DEPT NMR were assignable to four CH2 groups of the pyrrolidine moiety which was supported by upfield resonances corresponding to 8 protons in 1H NMR (see ESI†). The actual structure was however arrived at using single crystal X-ray analysis (Fig. 3). Pure crystals of 1 were obtained by its crystallization from chloroform and ethanol at room temperature. The molecule crystallizes in a triclinic system with P space group having 2-units in one unit cell. An X-ray ortep structure of compound 1 depicted four different conformations as shown in (Fig. 3). The crystal refinement of the molecule is shown in Table 1. The four conformers have slight variations among bond lengths and bond angles. Only a few significant differences with regard to bond lengths have been mentioned in Table 2. The characteristic feature of crystal packing view (Fig. 4) is the hydrogen bonding network in the form of N⋯H–N, Cl⋯H–C. The molecules are interconnected by means of these hydrogen bonds. Another interesting hydrogen bond is the one involving Cl⋯H–C (Table 3). Since crystallization of the molecule has been carried out in CHCl3:EtOH solvent, it is likely that one of CHCl3 molecule per unit cell has intruded inside during the process of crystallization providing additional stability to the unit cell.
 |
| | Fig. 2 Mechanism involved in the formation of quinazoline (1). | |
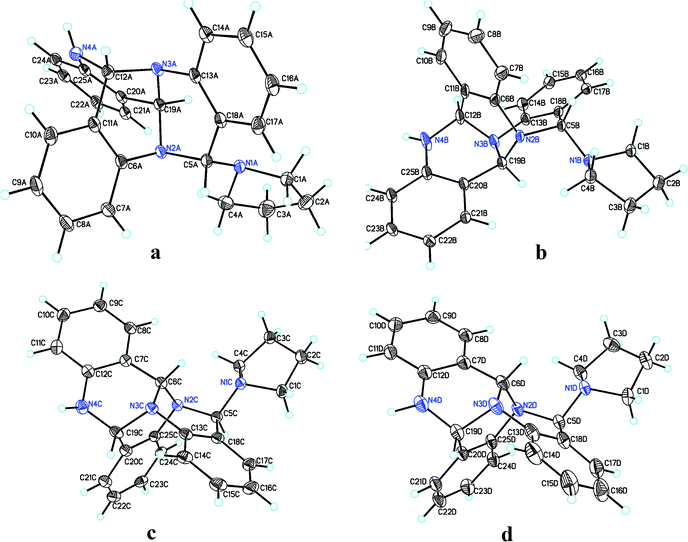 |
| | Fig. 3 Four different conformations (a–d) of compound 1 as shown by single crystal X-ray analysis. | |
Table 1 Crystal data and structure refinement of compound 1
| Identification code |
Shelx |
| Empirical formula |
C101H97Cl3N16 |
| Formula weight |
1641.29 |
| Temperature |
153 (2) K |
| Wavelength |
1.54184 Å |
| Crystal system |
Triclinic |
| Space group |
P |
| Unit cell dimensions |
a = 12.8967 (4) Å |
| b = 13.9278 (3) Å |
| c = 23.2246 (7) Å |
| Volume |
4093.1 (2) Å3 |
| Z |
2 |
| Density (calculated) |
1.332 mg m−3 |
| Absorption coefficient |
1.497 mm−1 |
| F(000) |
1732 |
| Crystal size |
0.345 × 0.275 × 0.233 mm3 |
| Theta range for data collection |
3.492 to 76.684° |
| Index ranges |
−16 ≤ h ≤ 16, −11 ≤ k ≤ 17, −29 ≤ l ≤ 28 |
| Reflections collected |
29828 |
| Independent reflections |
16636 [R(int) = 0.0333] |
| Completeness to theta = 67.684° |
99.8% |
| Absorption correction |
Gaussian |
| Max. and min. transmission |
1.000 and 0.322 |
| Refinement method |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
16636/12/1111 |
| Goodness-of-fit on F2 |
1.045 |
Table 2 Comparative analysis of experimentally obtained bond lengths of four conformers with that obtained theoretically (notable ones have been shown, for all refer to ESI file)
| Bond |
Exp. bond length (Å) of Conf. A |
Exp. bond length (Å) of Conf. B |
Exp. bond length (Å) of Conf. C |
Exp. bond length (Å) of Conf. D |
Theo. bond length (Å) |
| N(4)–H(4) |
0.80 |
0.88 |
0.86 |
0.96 |
1.0138 |
| C(2)–C(3) |
1.496 |
1.531 |
1.527 |
1.566 |
1.5385 |
| C(3)–C(4) |
1.518 |
1.520 |
1.523 |
1.485 |
1.5335 |
| C(6)–C(7) |
1.399 |
1.383 |
1.511 |
1.512 |
1.420 |
| C(11)–C(12) |
1.525 |
1.514 |
1.399 |
1.399 |
1.487 |
| C(21)–C(22) |
1.388 |
1.385 |
1.377 |
1.386 |
1.398 |
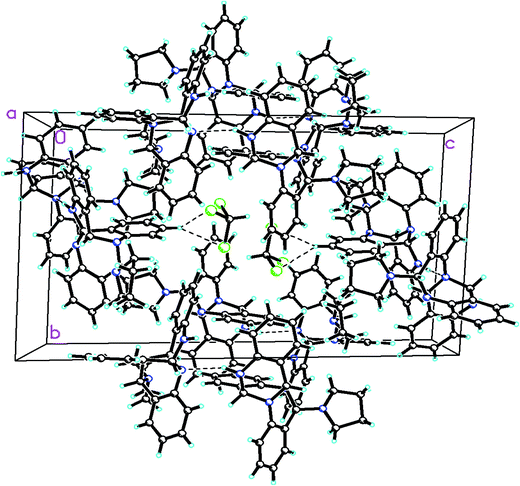 |
| | Fig. 4 A perspective view of the crystal packing in the compound 1. | |
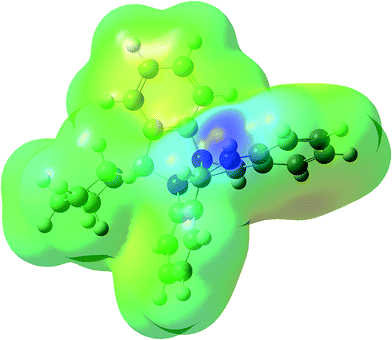 |
| | Fig. 5 MEP plot of compound 1 calculated using DFT/B3LYP/6-31G (d,p) level of theory. | |
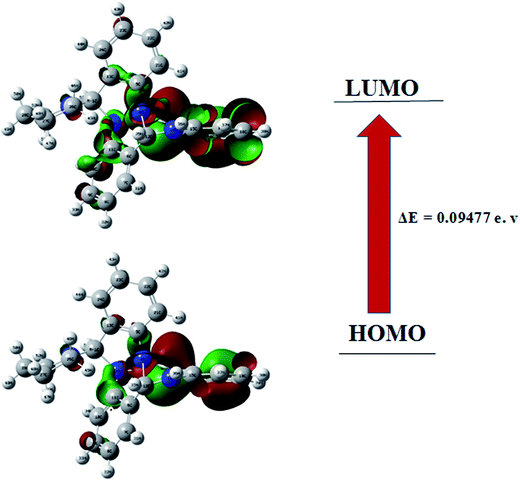 |
| | Fig. 6 HOMO–LUMO plots of compound 1. | |
Table 3 Hydrogen bonds for compound 1 [Å and °]
| D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
∠(DHA) |
| N(4B)–H(4B)⋯N(3A) |
0.88(5) |
2.34(5) |
3.143(4) |
153(4) |
| N(4D)–H(4D)⋯N(3C) |
0.96(6) |
2.25(6) |
3.125(4) |
151(4) |
| C(5D)–H(5DA)⋯Cl(1T) |
1.00 |
2.96 |
3.718(7) |
133.1 |
DFT studies
Theoretical calculations using DFT were performed. This computational method is emerging as an important quantum, chemical tool for studying various chemical problems and is because this versatile method determines energy based on electron density instead of wave-function. The DFT calculations were carried out with a hybrid functional B3LYP (Becke's three parameter hybrid functional) at 6-31G (d,p) level of theory using Gaussian 09 package. The geometries were optimized without imposing any molecular symmetry constraints.
Optimized structures
The computational chemistry studies in this work aimed at determination of geometric parameters for compound 1 and the same were compared with that of the experimentally obtained X-ray data. Table 2 summarizes the selected experimentally obtained (single crystal X-ray analysis) and theoretically calculated parameters (selected bond lengths) using DFT methods. A good approximation of bond lengths while doing optimization using DFT was observed to the experimentally obtained values.
Molecular electrostatic potential surface analysis
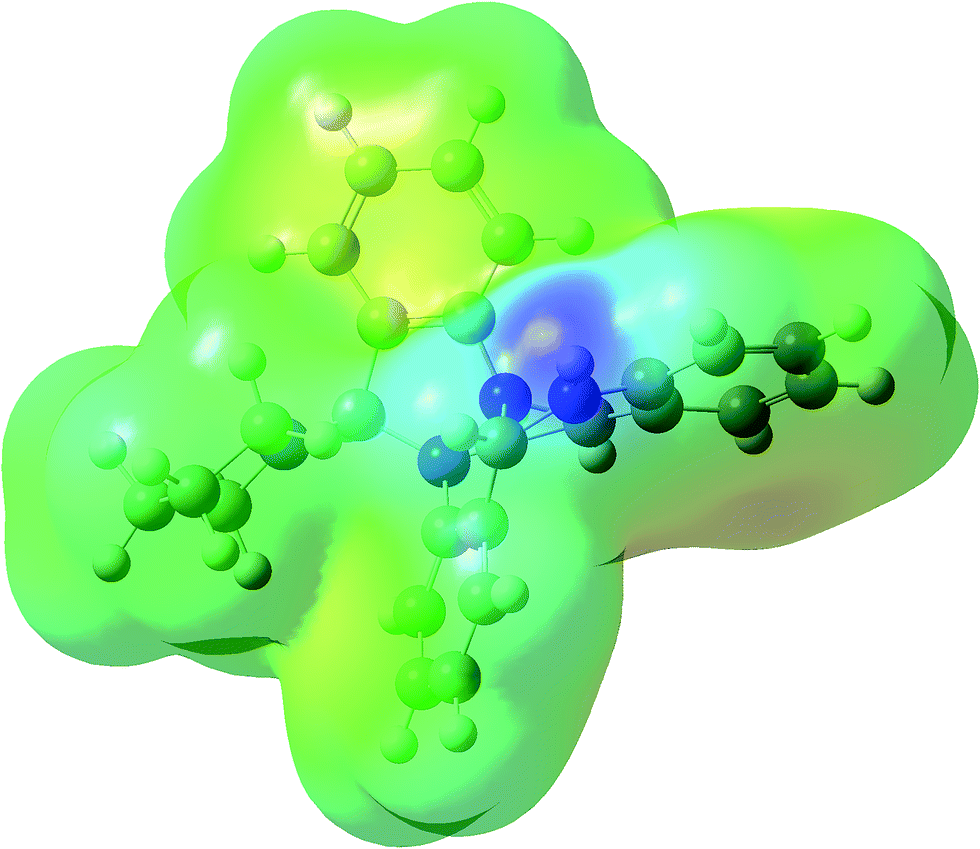
MEPS is a plot of electrostatic potential mapped on to constant potential electron density surface. These surfaces help in predicting reactivity sites towards positively and negatively charged reactants, in studies of biological recognition of one molecule by another as in drug receptor and enzyme substrate interactions as well as hydrogen-bonding interactions. MEP surfaces reveal the size shape and variation of electron density, electronegativity, partial charges and the sites of chemical reactivity with in the molecule.39–43 Fig. 5 shows MEP surface for compound 1, calculated using DFT/B3LYP/6-31G (d,p) level of theory. The pictorial representation with rainbow colour scheme of electrostatic potential for compound 1 lies in the range of −5.422 a.u to 5.422 a.u. The darkest red regions indicate regions with high electron density (negative potential) which invite electrophilic attacks while as the darkest blue regions indicate regions with low electron density (positive potential) which allow nucleophiles to approach easily. As can be seen in the MEP for compound 1 the region surrounding the secondary N-atom seems to the darkest blue that is one with lowest electron density, a site more vulnerable to nucleophilic attack. A plausible explanation in this regard is that the lone pair of electrons on N-atom enters in conjugation with the phenyl moiety thereby decreasing the electron density around that N-atom.
Frontier orbital analysis
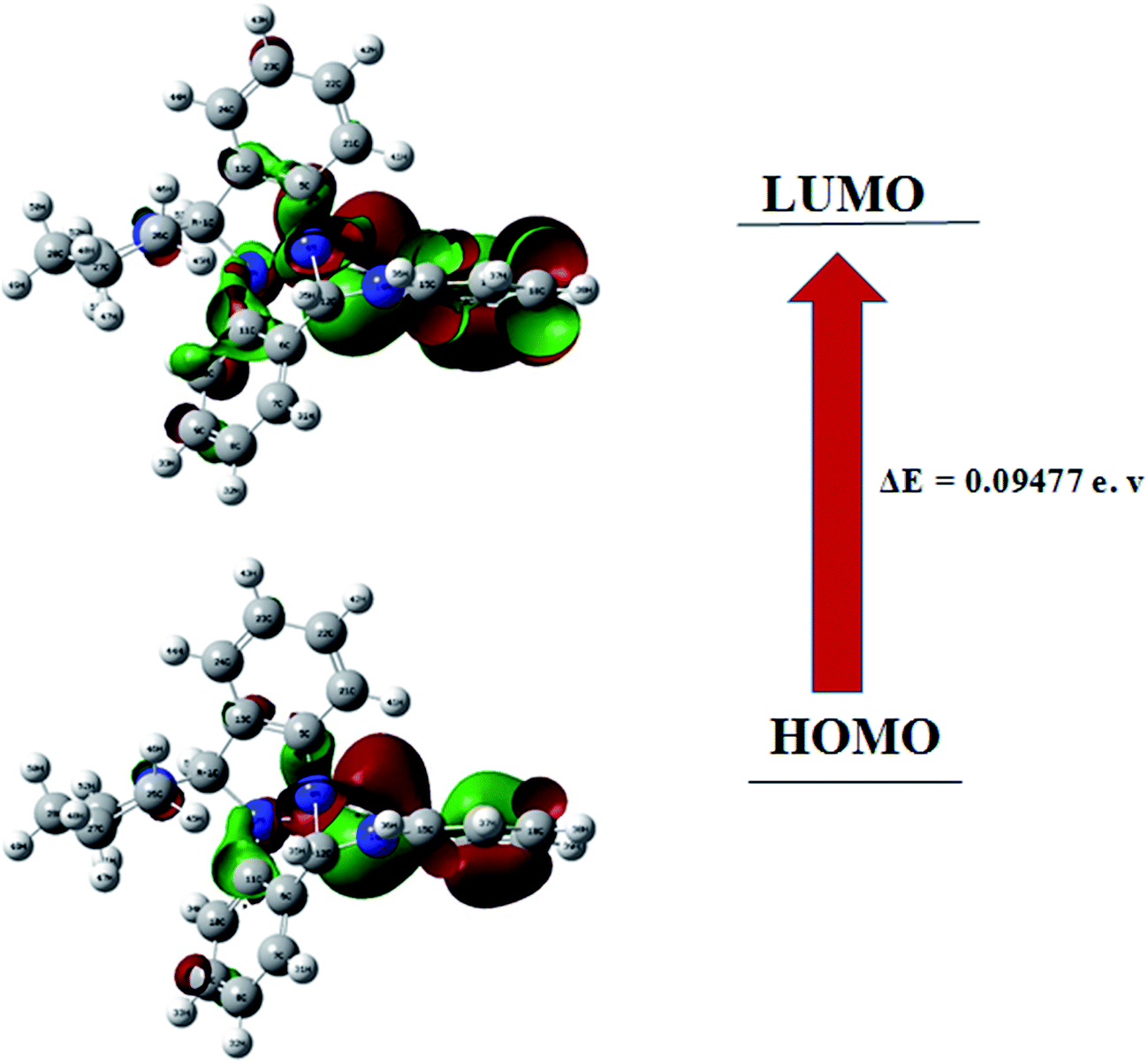
Frontier molecular orbitals play important role in the determination of electric and optical properties electronic transitions, kinetic stability.44 FMO's of compound 1 were calculated using the Gaussian 09 package at the B3LYP/6-31G (d,p) level of theory. The results are shown in Fig. 6. As can be seen from the figures HOMO and LUMO are located or spread over the regions containing the N-atoms and delocalised over one of the benzene ring with adjacent NH moiety. In the target compound 1, HOMO–LUMO energy gap is 0.094 eV. Using HOMO–LUMO energy gap important relative reactivity descriptors like hardness (η), chemical potential (μ), electronegativity (χ), ionization energy (I) and electron affinity (A) were calculated and are defined as follows:45–49
where A and I represent electron affinity and ionization potential of the molecule, which are in turn obtained from HOMO and LUMO energies as I = −EHOMO and A = −ELUMO as per Janak theorem50 and Perdew et al.51 As can be seen the HOMO–LUMO energy gap in compound 1 is just 0.094 eV which indicates its highly reactive nature. Another important factor is the global electrophilicity index (ω), a global reactivity index that is related to chemical hardness and chemical potential, introduced by Parr et al.49 represented as ω = μ2/2η and was found to be 0.012 eV indicating a poor electrophilic nature of 1. All the above parameters have been calculated for compound 1 using B3LYP/6-31G (d,p) level of theory and are presented in Table 4 as shown:
Table 4 Calculated energy values for compound 1 using B3LYP/6-31G (d,p) level of theory
| Parameter |
Compound 1 |
| Energy (au) |
−1186.014 |
| Dipole moment (debye) |
3.499 |
| EHOMO (eV) |
−0.139 |
| ELUMO (eV) |
−0.045 |
| EHOMO–LUMO (eV) |
0.094 |
| EHOMO−1 (eV) |
−0.175 |
| ELUMO+1 (eV) |
−0.005 |
| E(HOMO−1)–(LUMO+1) (eV) |
0.169 |
| Hardness (η) |
0.047 |
| Chemical potential (μ) |
0.092 |
| Electronegativity (χ) |
−0.092 |
| Electrophilicity index (ω) |
0.012 |
NMR analysis
Experimentally 1H and 13C NMR of the title compound was evaluated in CDCl3 solvent using TMS as an internal standard. Theoretically it was calculated in polarisable continuum model with CHCl3 as solvent using GIAO method52 involving B3LYP/6-31 G (d,p) level of theory. Both theoretical and experimental 1H and 13C NMR spectral data are presented in Table 5. Experimental 13C NMR of 1 revealed the presence of 23 resonances assignable to its 25 carbons atoms because of symmetrical nature of pyrrolidine ring. However its theoretical 13C NMR spectrum displayed all the 25 resonances for 25 carbon atoms. Similarly 1H NMR revealed the presence of 24 protons out of which 12 correspond to aromatic protons of three phenyl rings, three protons as CH groups, one as NH and 8 protons assignable to the pyrrolidine moiety. In 1H NMR three CH protons appeared at 4.52, 5.29 and 5.96 ppm and calculated using DFT at 5.4, 5.8 and 6.2 ppm respectively. Similarly eight protons of the pyrrolidine ring were observed in the range 1.25–3.32 ppm and calculated at 1.37–4.1 ppm for DFT which again revealed a fair agreement. The remaining 12 aromatic protons observed in the range 6.6–7.30 ppm were calculated at 6.6–8.2 ppm for DFT.
Table 5 Comparsion of theoretical and experimental NMR data of compound 1
| 1H NMR |
13C NMR |
| Experimental (δ ppm) |
Theoretical |
Experimental (δ ppm) |
Theoretical |
| 7.30–6.70 (m, 12H) |
8.10 (s, 1H) |
146.08 |
133.86 |
| 5.96 (s, 1H) |
7.60 (s, 1H) |
144.23 |
132.90 |
| 5.28 (d, J = 1.5 Hz, 1H) |
7.38 (s, 2H) |
140.08 |
118.62 |
| 4.82 (br s, 1H) |
7.19 (s, 2H) |
130.20 |
114.61 |
| 4.52 (s, 1H) |
7.10 (s, 2H) |
129.59 |
113.18 |
| 3.32 (m, 2H) |
6.98 (s, 1H) |
129.12 |
112.86 |
| 2.85 (m, 2H) |
6.78 (s, 1H) |
128.73 |
112.63 |
| 2.05–1.92 (m 4H) |
6.72 (s, 1H) |
128.48 |
111.98 |
| |
6.65 (s, 1H) |
128.31 |
110.86 |
| |
6.59 (s, 1H) |
127.88 |
110.58 |
| |
6.17 (s, 1H) |
124.97 |
109.86 |
| |
5.83 (s, 1H) |
124.74 |
108.72 |
| |
5.39 (s, 1H) |
124.56 |
108.11 |
| |
4.10 (s, 1H) |
123.93 |
107.89 |
| |
3.53 (s, 1H) |
123.32 |
106.40 |
| |
3.06 (s, 1H) |
123.09 |
105.42 |
| |
2.62 (s, 1H) |
119.98 |
104.73 |
| |
1.89 (s, 1H) |
117.44 |
97.81 |
| |
1.71 (s, 1H) |
84 |
94.84 |
| |
1.66 (s, 1H) |
71.04 |
88.20 |
| |
1.38 (s, 1H) |
64.12 |
76.45 |
| |
|
51.43 |
45.55 |
| |
|
51.43 |
36.62 |
| |
|
23.76 |
19.07 |
| |
|
23.76 |
18.70 |
IR analysis
Theoretically a total of 153 normal modes of vibration were obtained for compound 1. However in the desired region between 500 and 4000 cm−1 only 130 such modes were observed. The calculations have been performed within the harmonic approximation for vibrations. Comparision of selected theoretical and experimental modes is presented in Table 6. A fiar agreement is observed between theoretical and experimental IR data. Experimental FT-IR displayed bands in the region 3400–2800 cm−1, 1700–1000 and 1000–500 cm−1. Bands in the region 3400–2800 cm−1 were assignable to N–H stretchings, C–H stretchings of all the aromatic rings, C–H stretchings in which C-atoms are connected to N-atoms and CH2 stretchings of the pyrrolidine moiety. The bands between 1620–1000 cm−1 were assignable to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations in aromatic rings, H–CC aromatic ring bend modes, CH2 scissorings, CH2 twistings and N–C stretching vibrations. However bands between 900-500 mostly correspond to the torsion modes i.e. distortions in dihedral angles mostly of HCCC, CCCC, NCCC and CNCC type.
C stretching vibrations in aromatic rings, H–CC aromatic ring bend modes, CH2 scissorings, CH2 twistings and N–C stretching vibrations. However bands between 900-500 mostly correspond to the torsion modes i.e. distortions in dihedral angles mostly of HCCC, CCCC, NCCC and CNCC type.
Table 6 Comparsion of theoretical and experimental IR (cm−1) data of compound 1 (selected modes only)
| Experimental (cm−1) |
Theoretical (cm−1) |
Experimental (cm−1) |
Theoretical (cm−1) |
Experimental (cm−1) |
Theoretical (cm−1) |
| 3401 |
3224 |
1465 |
1472 |
960 |
967 |
| 3062 |
3061 |
1341 |
1336 |
856 |
857 |
| 2980 |
3028 |
1301 |
1314 |
795 |
792 |
| 2830 |
3006 |
1253 |
1257 |
750 |
753 |
| 2800 |
2986 |
1222 |
1225 |
685 |
689 |
| 1620 |
1613 |
1120 |
1126 |
620 |
617 |
| 1560 |
1555 |
1036 |
1031 |
580 |
577 |
| 1500 |
1501 |
1000 |
987 |
534 |
535 |
| 1490 |
1475 |
982 |
979 |
512 |
521 |
PASS and biological evaluation
After having successfully optimized and calculated various reactivity descriptors for compound 1, using DFT employing the well-known B3LYP/6-31G (d,p) level of theory, we next studied its bioevaluation against various human cancer cell lines. Before carrying out the actual analysis, it was envisaged to predict the pharmacological properties of the target compound (1), using an available online PASS.53 PASS is an important tool that evaluates the biological activity of a molecule in relation to its structure. It assesses the druglikeness and toxicities of molecules like teratogenicity, carcinogenicity embryogenecity etc. The average accuracy of prediction is about 95% according to leave-one-out-cross validation (LOOCV) estimation and the probabilities, Pa (probable activity) and Pi (probable inactivity), are values that vary from 0.000 to 1.000, and generally Pa + Pi ≠ 1 as these probabilities are calculated independently.54 PASS analysis results for the title compound 1 have been enlisted in Table 7. Among the range of activities predicted, the most notable is the antineoplastic activity with Pa of 0.884. To validate the pass results, the title compound was evaluated using sulphorhodamine B (SRB) assay against human-leukemia (THP-1), colon (HCT-116), prostate (PC-3) and lung (A-549) cancer cell lines. It was observed that the title compound possesses a broad spectrum anticancer effect on the cancerous cells with THP-1 cell line as its most easy and soft target. Compound 1 displayed IC50 of 07, 12, 10, and 17 μM against THP-1, colon (HCT-116), prostate (PC-3) and human lung (A-549) cancer cell lines respectively. Since the lowest IC50 is observed against human leukemia cell lines (THP-1) cell-lines, it implies that the usual target of this compound may be leukemia cell lines. However it is equally true that leukemia cell lines are more-sensitive.
Table 7 PASS prediction for the activity spectrum of compound 1 with Pa > 0.5
| Pa |
Pi |
Activity name |
| 0.884 |
0.003 |
Antineoplastic enhancer |
| 0.851 |
0.009 |
5 Hydroxytryptamine release stimulant |
| 0.769 |
0.014 |
Nicotinic alpha-2-beta-2-receptor antagonist |
| 0.728 |
0.026 |
Nicotinic alpha-6-beta-3-beta-4-alpha-5-receptor antagonist |
| 0.707 |
0.007 |
Antihypoxic |
| 0.676 |
0.014 |
Thioredoxin inhibitor |
| 0.690 |
0.080 |
Phobic disorders treatment |
| 0.605 |
0.003 |
Poly(ADP-ribose) polymerase inhibitor |
| 0.637 |
0.052 |
Fibrinolytic |
| 0.591 |
0.009 |
Atherosclerosis treatment |
| 0.585 |
0.007 |
Stroke treatment |
| 0.576 |
0.010 |
Octopamine antagonist |
| 0.617 |
0.065 |
Antineurotic |
| 0.576 |
0.027 |
(R)-6-Hydroxynicotine oxidase inhibitor |
| 0.567 |
0.021 |
(S)-6-Hydroxynicotine oxidase inhibitor |
| 0.559 |
0.030 |
1,4-Lactonase inhibitor |
| 0.563 |
0.034 |
Chloride peroxidase inhibitor |
| 0.579 |
0.054 |
Pseudolysin inhibitor |
| 0.589 |
0.068 |
Glycosylphosphatidylinositol phospholipase D inhibitor |
| 0.550 |
0.033 |
Alopecia treatment |
| 0.594 |
0.089 |
Antieczematic |
| 0.580 |
0.098 |
Testosterone 17 beta-dehydrogenase (NADP+) inhibitor |
| 0.503 |
0.077 |
Calcium channel (voltage-sensitive) activator |
| 0.528 |
0.114 |
Nootropic |
| 0.516 |
0.065 |
Phthalate 4,5-dioxygenase inhibitor |
| 0.526 |
0.081 |
Kidney function stimulant |
Conclusion
An unusual quinazoline alkaloid (1) was synthesized and characterized using IR and NMR spectral techniques. Single crystal analysis depicted four conformations differing slightly in bond angles and bond lengths. Compound 1 crystallizes in the triclinic crystal system with P-1 space group having two molecules within the unit cell. The experimentally obtained parameters were compared to that obtained theoretically which depicted a good agreement. Using DFT/B3LYP/6-31G (d,p) level of theory, HOMO–LUMO energy gap, molecular electrostatic potential surface (MEPS), vibrational and NMR analysis were carried out. HOMO–LUMO energy gap was established to carry out the calculation of chemical hardness, chemical inertness, electronegativity and electrophilicity index of the molecule, which depicted the potential kinetic stability and reactivity of the molecule. Prediction of activity spectra of the target compound was carried out which revealed that compound 1 possesses notable antineoplastic activity. The pass results were validated by evaluating the molecule against colon (HCT-116), leukemia (THP-1), prostate (PC-3) and human lung (A-549) cancer cell lines respectively in an SRB assay. It was observed that compound 1 possesses remarkable potency against THP-1 cell lines with IC50 of 7 μM.
Experimental
General considerations
2-Nitrobenzaldehyde, iron powder, pyrrolidine etc were procured from Sigma-Aldrich (USA) and used as received. The FT-IR spectra of the compound was recorded on Agilent FT-IR spectrometer in KBr discs (4000–400 cm−1). 1H, 13C NMR spectra were recorded on a Bruker Spectrospin DPX-400 NMR spectrometer at 400.13 and 100.47 respectively using TMS as an internal standard. Mass analysis was carried out using Nexera UHPLC at 130 MPa with SIL-30 AC Nexera autosampler coupled to an LC-MS 8030 tandem mass spectrometer manufactured by Shimadzu Corporation, Kyoto, Japan. The software SADABS was used for absorption correction and SHELXTL for space group, structure determination and refinements. All non-hydrogen atoms were refined anisotropically. All the computations have been carried out at DFT/B3LYP/6-31G (d,p) level of theory using GAUSSIAN 09 software.
Synthesis of 2-aminobenzaldehyde
2-Aminobenzaldehyde was synthesized by a procedure reported by Gamble et al.55 To a suspension of 2-nitrobenzaldehyde (0.200 g, 1.32 mmol) in a mixture of glacial acetic acid (2 mL), ethanol (2 mL) and water (1 mL), reduced iron powder (0.280 g, 5.00 mmol) was added. The reaction mixture was exposed to ultrasonic irradiation for 1–2 h at room temperature with continuous TLC monitoring for the completion of reaction. The mixture was filtered to remove the iron residue. The filtrate was partitioned with 2 M KOH and the basic layer was further extracted with ethyl acetate (3 × 25 mL). The combined organic extracts were washed with brine and water and dried under reduced pressure. The crude residue was then chromatographed over silica gel column chromatography using 30% EtOAc:Hexane as eluent to give 2-aminobenzaldehyde in 80% yield.
Synthesis of 1
A 100 mL round bottomed flask was charged with 2-aminobenzaldehyde (120 mg, 1 mmol),ethanol (2.5 mL) and pyrrolidine (0.027 mL, 0.33 mmol) and stirred under reflux conditions and the reaction monitored using TLC analysis till it was complete. After the completion of reaction the solvent of the mixture was evaporated under reduced pressure. The solid residue so obtained, was subjected to silica gel column chromatography and purified using 65:45 ethyl acetate:hexane as eluent. Compound 1 was obtained as pure yellowish solid.
Yield: 60%, mp: 183 °C, IR (KBr cm−1): 3401, 3062, 2980, 2830, 2800, 1620, 1560, 1500, 1490, 1465, 1380, 1341, 1301, 1253, 1222, 1120, 1036, 1000, 982, 960, 856, 795, 750, 685, 620, 580, 534, 512, 500. 1H NMR (400 MHz, CDCl3) 7.30–6.70 (m, 12H), 5.96 (s, 1H), 5.28 (d, J = 1.5 Hz, 1H), 4.82 (br s, 1H), 4.52 (s, 1H), 3.32 (m, 2H), 2.85 (m, 2H), 2.05–1.92 (m 4H), 13C NMR (100 MHz, CDCl3) δ 146.08, 144.23, 140.08, 130.20, 129.59, 129.12, 128.73, 128.48, 128.31, 127.88, 124.97, 124.74, 124.56, 123.93, 123.32, 123.09, 119.98, 117.44, 84.00, 71.04, 64.12, 51.43 (2C), 23.76 (2C). (ESI-MS) at m/z = 381 for [M + H]+.
X-ray diffraction study
Pure rod shaped crystals were selected after recrystallization of the final product (1) from CHCl3 and ethanol solvent. The diffraction data on single crystal of compound 1 was collected on a Bruker AXS SMART Apex CCD diffractometer using Mo-Kα (0.71073 Å) radiations at 298 K. The software SADABS56 was used for absorption correction and SHELXTL for space group, structure determination and refinements.57,58 All non-hydrogen atoms were refined anisotropically.
Density functional theory study
All the computations are carried out using GAUSSIAN 09 software.59 The DFT modeling method, using the hybrid B3LYP60 functional was used to calculate theoretical parameters for compound 1 with the basis set combination 6-31 G (d,p).61 Geometry optimization was carried out until global minima were achieved. The results of the optimized structure for compound 1 are reported in Table 3 and are compared with X-ray diffraction results.
SRB cytotoxic assay
SRB cytotoxic assay was used to screen the compounds for cell cytotoxicity as per the reported procedures.62,63 Various human cancer cell lines, human leukemia (THP-1, at a density of 7 × 103), human lung carcinoma cell line (A-549, at a density of 8 × 103 cells per mL per 100 μL per well), human prostate cancer cell line (PC-3, at a density of 8 × 103 cells per mL per 100 μL per well) and human colon cancer cell line (HCT-116, at a density of 1 × 104 cells per mL per 100 μL per well) used in this study were purchased from European collection of cell culture (ECACC) (USA) and seeded in flat-bottomed 96-well plates.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
SHL thanks SERB India for the financial support under NPDF scheme (File no: PDF/2016/001690/CS). Salman Jameel thanks CSIR for the Gate Fellowship (Budget Head: P-81101).
References
- T. Aniszewski, Sci. Legumes, 1994, 1, 1–24 CAS.
- M. Wink, A short history of alkaloids, in. Alkaloids: Biochemistry, Ecology, and Medicinal Applications, ed. Roberts M. F., and Wink M., Plenum Press, New York, London, 1998, pp. 11–44 Search PubMed.
- S. Bhattacharya, C. Haertel, A. Maelicke and D. Montag, PLoS One, 2014, 9, 89454 Search PubMed.
- Z. Li, Y. N. Geng, J. D. Jiang and W. J. Kong, J. Evidence-Based Complementary Altern. Med., 2014, 2014, 289264 Search PubMed.
- J. Peng, X. Y. Zhang, Z. C. Tu, X. Y. Xu and S. H. Qi, J. Nat. Prod., 2013, 76(5), 983–987 CrossRef CAS PubMed.
- B. Yang, H. M. Tao, X. F. Zhou, X. P. Lin and Y. H. Liu, Nat. Prod. Res., 2013, 27, 433–437 CrossRef CAS PubMed.
- X. W. Yang, G. Y. Zhang, J. X. Ying, B. Yang, X. F. Zhou and A. Steinmetz, Mar. Drugs, 2013, 11(1), 33–39 CAS.
- X. Guo, Y. Li, C. F. Li, H. M. Luo, L. Z. Wang and J. Qian, Gene, 2013, 527(1), 131–138 CrossRef CAS PubMed.
- R. H. Pouwer, S. M. Deydier, P. V. Le, B. D. Schwartz, N. C. Franken and R. A. Davis, ACS Med. Chem. Lett., 2014, 5(2), 178–182 CrossRef CAS PubMed.
- C. Agyare, G. A. Koffuor, Y. D. Boakye and K. B. Mensah, Pharm. Biol., 2013, 51(4), 418–425 CrossRef CAS PubMed.
- A. A. Boligon, T. F. Kubica, D. N. Mario, T. F. de Brum, M. Piana and R. Weiblen, Acta Physiol. Plant., 2013, 35(7), 2229–2239 CrossRef.
- E. V. Costa, P. E. O. da Cruz, C. C. de Lourenco, V. R. D. Moraes, P. C. D. Nogueira and M. J. Salvador, Nat. Prod. Res., 2013, 27(11), 1002–1006 CrossRef CAS PubMed.
- J. Hu, X. D. Shi, J. G. Chen, X. Mao, L. Zhu and L. Yu, Food Chem., 2014, 148, 437–444 CrossRef CAS PubMed.
- J. F. Wang, X. P. Lin, C. Qin, S. R. Liao, J. T. Wan and T. Y. Zhang, J. Antibiot., 2014, 67(8), 581–583 CrossRef CAS PubMed.
- Y. Yang, W. J. Zuo, Y. X. Zhao, W. H. Dong, W. L. Mei and H. F. Dai, Planta Med., 2012, 78(17), 1881–1884 CrossRef CAS PubMed.
- C. Iannello, J. Bastida, F. Bonvicini, F. Antognoni, G. A. Gentilomi and F. Poli, Nat. Prod. Res., 2014, 28(10), 704–710 CrossRef CAS PubMed.
- L. Zhang, Z. Hua, Y. Song and C. Feng, Fitoterapia, 2014, 97, 142–147 CrossRef CAS PubMed.
- A. O. Adeoye, B. O. Oguntimein, A. M. Clark and C. D. Hufford, J. Nat. Prod., 1986, 49, 534–537 CrossRef CAS.
- M. K. Ray, C. Balachandran, V. Duraipandiyan, P. Agastian, S. Ignacimuthu and A. Vijayakumar, Med. Chem. Res., 2013, 22, 823–830 Search PubMed.
- C. Dupont, E. Couillerot, R. Gillet, C. Caron, M. Zeches-Hanrot and J. F. Riou, Planta Med., 2005, 71, 489–494 CrossRef CAS PubMed.
- S. S. Nazrullaev, I. A. Bessonova and K. S. Akhmed khodzhaeva, Chem. Nat. Compd., 2001, 37, 551–555 CrossRef CAS.
- M. Goyal and D. Sasmal, J. Ethnopharmacol., 2014, 151, 536–542 CrossRef PubMed.
- N. Wang, K. J. Wicht, E. Shaban and T. A. Ngoc, MedChemComm, 2014, 5(7), 927–931 RSC.
- M. Taki, K. Niitu, Y. Omiya, M. Fukuchi, M. Aburada and M. Okada, Planta Med., 2003, 69, 800–803 CrossRef CAS PubMed.
- S. H. Lone, K. A. Bhat, S. Rehman, R. Majeed, A. Hamid and M. A. Khuroo, Bioorg. Med. Chem. Lett., 2013, 3, 4931–4934 CrossRef PubMed.
- S. Naseer, S. H. Lone, J. A. Lone, M. A. Khuroo and K. A. Bhat, J. Chromatogr. B: Biomed. Sci. Appl., 2015, 989, 62–70 CrossRef CAS PubMed.
- S. H. Lone, K. A. Bhat, R. Majeed, A. Hamid and M. A. Khuroo, Bioorg. Med. Chem. Lett., 2014, 24, 1047–1051 CrossRef CAS PubMed.
- S. H. Lone and K. A. Bhat, Tetrahedron Lett., 2015, 56, 1908–1910 CrossRef CAS.
- I. Ayoob, S. H. Lone, M. Rahman, O. A. Zargar, R. Bashir, S. Rehman, M. A. Khuroo and K. A. Bhat, ChemistrySelect, 2017, 2, 10153–10156 CrossRef CAS.
- S. Rehman, K. A. Bhat, S. H. Lone and F. A. Malik, Arabian J. Chem., 2015 DOI:10.1016/j.arabjc.2015.10.009.
- S. Rehman, B. Rah, S. H. Lone, R. Rasool, S. Farooq, D. Nayak, N. A. Chikan, S. Chakraborty, A. Behl, D. M. Mondhe, A. Goswami and K. A. Bhat, J. Med. Chem., 2015, 58, 3432–3444 CrossRef CAS PubMed.
- S. H. Lone, K. A. Bhat, F. A. Malik and M. A. Khuroo, Planta Medica International Open, 2016, 3, 51–54 CrossRef.
- S. H. Lone, S. Rehman and K. A. Bhat, Drug Res., 2017, 67, 111–118 CAS.
- B. A. Dar, S. H. Lone, W. A. Shah and K. A. Bhat, Drug Res., 2016, 66, 427–431 CrossRef CAS PubMed.
- I. Ayoob, Y. M. Hazari, S. H. Lone, S. Rehman, M. A. Khuroo, K. M. Fazili and K. A. Bhat, ChemistrySelect, 2017, 2, 2965–2968 CrossRef CAS.
- S. H. Lone, K. A. Bhat, S. Naseer, R. A. Rather, M. A. Khuroo and S. A. Tasaduq, J. Chromatogr. B: Biomed. Sci. Appl., 2013, 940, 135–141 CrossRef CAS PubMed.
- S. H. Lone and K. A. Bhat, Steroids, 2015, 96, 164–168 CrossRef CAS PubMed.
- M. T. Richers, I. Deb, A. Y. Platonova, C. Zhang and D. Seidel, Synthesis, 2013, 45(13), 1430–1748 Search PubMed.
- K. Abhishek, K. S. Ambrish, G. Shashi, M. Neeraj, M. Avijit and B. Goutam, J. Mol. Des., 2015, 1096, 94–101 Search PubMed.
- M. N. Arshad, A. Bibi, T. Mahmood, A. M. Asiri and K. Ayub, Molecules, 2015, 20, 5851–5874 CrossRef CAS PubMed.
- H. Zhou and J. Skolnick, Mol. Pharm., 2012, 9, 1775–1784 CrossRef CAS PubMed.
- E. Scrocco and J. Tomasi, Adv. Quantum Chem., 1979, 11, 115–193 CrossRef.
- N. Okulik and A. H. Jubert, J. Mol. Des, 2005, 4, 17–30 CAS.
- K. Fukui, T. Yonezawa and H. J. Shingu, J. Chem. Phys., 1952, 20, 722–725 CrossRef CAS.
- R. A. Costa, P. O. Pitt, M. L. B. Pinheiro, K. M. T. Oliveira, K. S. Salome, A. Barison and E. V. Costa, Spectrochim. Acta, Part A, 2017, 174, 94–104 CrossRef CAS PubMed.
- R. A. Costa, K. M. T. Oliveira, E. V. Costa and M. L. B. Pinheiro, J. Mol. Struct., 2017, 1145, 254–267 CrossRef CAS.
- N. Özdemir, S. Dayan, O. Dayan, M. Dinçer and N. Kalaycıoglu, J. Atom. Mol. Phys., 2013, 11(6), 707–723 CrossRef.
- R. Parr, Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- R. Parr, L. Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- J. F. Janak, Phys. Rev. B, 1978, 18, 7165 CrossRef CAS.
- J. P. Perdew, R. G. Parr, M. Levy and J. L. Balduz Jr, Phys. Rev. Lett., 1982, 49, 1691–1694 CrossRef CAS.
- R. Ditchfield, J. Chem. Phys., 1972, 56, 5688–5691 CrossRef CAS.
- A. Lagunin, A. Stepanchikova, D. Filimonov and V. Poroikov, Bioinformatics, 2000, 16, 747–748 CrossRef CAS PubMed.
- R. Pramely and T. L. S. Raj, J. Biochem. Technol., 2012, 3, 375–379 CAS.
- A. B. Gamble, J. Garner, C. P. Gordon, S. M. J. O'Conner and P. A. Keller, Synth. Commun., 2007, 37, 2777–2786 CrossRef CAS.
- Brucker SAINT Brucker AXS Inc., Madison Wisconsin, USA, 2002.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, B. 01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1378 CrossRef CAS;
(b) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem. B, 1994, 98, 11623 CrossRef CAS.
-
(a) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS;
(b) K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef;
(c) J. P. Blaudeau, M. P. McGrath, L. A. Curtiss and L. Radom, J. Chem. Phys., 1997, 107, 5016 CrossRef CAS;
(d) A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033 CrossRef CAS;
(e) P. J. Hay, J. Chem. Phys., 1977, 66, 4377 CrossRef CAS;
(f) K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062 CrossRef;
(g) R. C. Binning Jr and L. A. Curtiss, J. Comput. Chem., 1990, 11, 1206 CrossRef;
(h) M. P. McGrath and L. Radom, J. Chem. Phys., 1991, 94, 511 CrossRef CAS;
(i) L. A. Curtiss, M. P. McGrath, J. P. Blaudeau, N. E. Davis, R. C. Binning Jr and L. Radom, J. Chem. Phys., 1995, 103, 6104 CrossRef CAS.
- R. Majeed, M. V. Reddy, P. K. Chinthakindi, P. L. Sangwan, A. Hamid, G. Chashoo, A. K. Saxena and S. Koul, Eur. J. Med. Chem., 2012, 49, 55 CrossRef CAS PubMed.
- G. Chashoo, S. K. Singh, D. M. Mondhe, P. R. Sharma, S. S. Andotra, B. A. Shah, S. C. Taneja and A. K. Saxena, Eur. J. Pharmacol., 2011, 668, 390 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1814904 contains the supplementary crystallographic data for 1. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra00138c |
| ‡ Both the authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Salman Jameel‡
b,
Muzzaffar A. Bhat‡c,
Rayees A. Loneb,
Ray J. Butcherd and
Khursheed A. Bhat*b
*a,
Salman Jameel‡
b,
Muzzaffar A. Bhat‡c,
Rayees A. Loneb,
Ray J. Butcherd and
Khursheed A. Bhat*b