
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation on converting 1-butene and ethylene into propene via metathesis reaction over W-based catalysts†
Guangzheng Zuo,
Yuebing Xu,
Jiao Zheng,
Feng Jiang and
Xiaohao Liu *
*
Department of Chemical Engineering, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China. E-mail: liuxh@jiangnan.edu.cn
First published on 22nd February 2018
Abstract
Supported W catalysts were extensively investigated for the conversion of 1-butene and ethylene into propene by metathesis reaction. The performance of catalysts was compared by using unsupported WO3, pure SBA-15, supported W/SBA-15 with different W loadings, varied calcination temperatures, and by changing the pretreatment gas atmosphere. The above catalytic results could be employed to deduce the reaction mechanism combined with characterization techniques such as BET, XRD, UV-vis DRS, Raman, pyridine-IR, XPS, and H2-TPR. In this study, over the investigated W/SBA-15 catalysts, the results showed that the silanol group (Si–OH) in SBA-15 could act as a weak Brønsted acid site for 1-butene isomerization. However, the metathesis reaction was catalyzed by W-carbene species. The initially formed W-carbenes (W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH3) as active sites were derived from the partially reduced isolated tetrahedral WOx species which contained WO or W–OH bonds in W5+ species as corresponding Lewis or Brønsted acid sites. Furthermore, the W/SBA-15 being pretreated by H2O led to a complete loss of the metathesis activity. This was mainly due to the sintering of isolated WOx species to form an inactive crystalline WO3 phase as demonstrated by XRD patterns. On the other hand, the reduction of WOx species remarkably suppressed by H2O pretreatment was also responsible for the metathesis deactivation. This study provides molecular level mechanisms for the several steps involved in the propene production, including 1-butene isomerization, W-carbene formation, and metathesis reaction.
CH–CH3) as active sites were derived from the partially reduced isolated tetrahedral WOx species which contained WO or W–OH bonds in W5+ species as corresponding Lewis or Brønsted acid sites. Furthermore, the W/SBA-15 being pretreated by H2O led to a complete loss of the metathesis activity. This was mainly due to the sintering of isolated WOx species to form an inactive crystalline WO3 phase as demonstrated by XRD patterns. On the other hand, the reduction of WOx species remarkably suppressed by H2O pretreatment was also responsible for the metathesis deactivation. This study provides molecular level mechanisms for the several steps involved in the propene production, including 1-butene isomerization, W-carbene formation, and metathesis reaction.
1. Introduction
Propene is one of the fastest growing raw materials as a key building block in the chemical industry, driven primarily by the high growth rate of polypropene consumption.1,2 Also, propene is used in the production of propene oxide which is a chemical precursor for the synthesis of propene glycol and polyols.3 The rest of propene is consumed for the synthesis of acrylonitrile, oxo-alcohol, cumene, and many other industrially relevant chemicals.4 The market for propene has been continuously growing at average rates of 4–5% per year. More than 50 million tons of additional capacity will be needed by the year of 2020. Currently, approximately 42% of propene is primarily derived from steam cracking of naphtha and about 39% of it is recovered from fluid catalytic cracking (FCC).5–8 In recent years, dehydrogenation of propane (PDH),9 methanol-to-olefins (MTO),10 and Fischer–Tropsch to olefins (FTO)11 processes for the production of propene have been blooming in view of the availability of cheap propane in shale gas and the availability of coal-, natural gas-, and biomass-derived syngas. Especially, FTO route can provide a reliable solution for meeting the demand for propene by utilizing the abovementioned alternative carbon-containing resources via syngas conversion.12Compared with the highly selective production of propene via PDH route, the FTO route generally produces the mixed light α-olefins such as ethylene, propene, and 1-butene.13,14 In spite of the high selectivity to total light α-olefins, the selectivity to propene is usually lower than 40%. Therefore, it is necessary for us to search for an efficient approach to significantly enhance the propene fraction in light α-olefins in order to meet its growing demand. Olefin metathesis is a highly versatile process for interconverting olefinic hydrocarbons. It is an efficient approach for us to remarkably enhance the selectivity to propene by using the metathesis reaction of 1-butene and ethylene from FTO route.
Various supported transition metal oxides have been used as heterogeneous catalysts for metathesis reaction, including tungsten (W),15–17 molybdenum (Mo),18,19 and rhenium (Re).20,21 Among these catalysts, supported W catalysts obtain the largest number of commercial applications because of its benefits such as relatively lower price, better stability, and better resistance to poisoning.22 Despite that numerous literatures have reported the olefin metathesis by the supported W catalysts, most of them focus on conversion of 2-butene and ethylene into propene,23–25 and its reverse process.26,27 Self-metathesis of 1-butene28–30 or 2-butene,31 2-pentene metathesis,32 and metathesis of 1-butene and ethylene33–35 have also been paid attention to. However, for converting FTO-based α-olefin products such 1-butene and ethylene into propene by metathesis reaction, the detailed investigation on the reaction mechanism is still insufficient over past studies. Therefore, the molecular level insights into the nature of surface active tungsten oxide (WOx) sites, catalyst activation, and reaction mechanism still need to be taken in order to guide the rational design of advanced catalysts.
It is generally accepted that the high dispersion of WOx and its existing state on support surface play crucial roles in the catalyst efficiency. Especially, the isolated WOx species might be responsible for metathesis activity.36 And these factors depend on the properties of supports and the preparation methods. Therefore, we have designed a series of supported W catalysts with different physicochemical properties and varied preparation conditions to regulate the WOx species state. In this study, several supports such as SiO2, γ-Al2O3, TiO2, and SBA-15 are selected for the preparation of W catalysts, and unsupported WO3 catalyst is also prepared as a reference. The as-prepared W catalysts and pure support are used for the metathesis reaction of 1-butene and ethylene; this can provide important information for speculation on what species in catalysts contribute to the 1-butene isomerization and the olefin metathesis for the formation of propene. Furthermore, the calcination temperature and W loading in catalyst may affect the aggregation of WOx species to lead to the changing ratio of isolated WOx species to crystalline WO3, which can confirm the importance of the high dispersion of WOx phase.
In addition, prior to metathesis reaction, the different pretreatment gas, such as the oxidizing gas (O2 and H2O), reducing gas (H2), and inert gas (N2), can regulate the initially existing state of WOx species. The induction period for the formation of propene over gas-pretreated supported W catalysts could be helpful for us to discern that either oxidative or reductive state of WOx species is responsible for metathesis reaction. It should be noted that H2O is used to pretreat the supported W catalysts before reaction, which is based on the following considerations. On the one hand, the pretreatment with H2O might result in the oxidation of WOx species; on the other hand, the pretreatment with H2O might lead to more crystalline WO3 phase due to the sintering. The designed pretreatment methods might make obvious transformation between oxidative and reductive WOx species, or between isolated and crystalline WOx phases. The metathesis activity over H2O-pretreated W catalysts could provide further verification on the proposed mechanism as reflected by abovementioned a series of investigations. In order to accurately correlate the relationship between the structure of supported W catalysts and catalytic performance, various characterization methods such as BET, XRD, UV-vis DRS, Raman, pyridine-IR, XPS, and H2-TPR have been applied to measuring the physicochemical properties of catalysts. Finally, the influences of WHSV and reaction temperature on metathesis reaction are also investigated to obtain the optimal catalytic activity and propene selectivity. And the mechanisms for 1-butene isomerization, W-carbene formation, and metathesis reaction on SBA-15 supported W catalysts have been proposed and discussed in detail.
2. Experimental
2.1 Catalyst preparation
The supported tungsten catalysts were prepared by an incipient wetness impregnation (IWI) method as described in literature.37 The SBA-15 support was purchased from Jiaxing Tanli New Materials Development Co., Ltd (China). Ammonium metatungstate (AMT, (NH4)6H2W12O40·xH2O, 99.5%, Aladdin) was used as tungsten precursor. Briefly, prior to impregnation, SBA-15 was dried in an oven at 120 °C for 2 h. Then, as-calculated amount of AMT aqueous solution was added into the dried SBA-15. After impregnation, the sample was dried at 120 °C overnight and followed by calcination in air at 550 °C for 4 h with a heating rate of 1 °C min−1. The obtained catalysts were labelled as xW/SBA-15 where x represents WO3 content in weight percentage. The supported W catalysts with different supports such as SiO2, γ-Al2O3, and anatase-TiO2 were prepared by the same method. In addition, the unsupported WO3 used as a reference was prepared by calcination of AMT with same procedures for supported catalysts.2.2 Catalyst characterization
Brunauer–Emmett–Teller (BET) surface area, pore volume, pore size, and the adsorption–desorption isotherms were measured by using a Micromeritics ASAP 2020. The X-ray diffraction (XRD) patterns of samples were recorded on a Bruker AXS D8 Advance X-ray diffractometer using Cu (Kα) radiation with 0.02° step in the 2θ range from 10 to 80°. Diffuse-reflectance UV-vis spectra (UV-vis DRS) were obtained in the range of 200–700 nm with a Shimadzu UV-3600 Plus Spectrometer, using BaSO4 as reference. The Raman spectra of the samples were recorded on a Renishaw Micro Raman Spectrometer in the range from 200 to 1200 cm−1 (excitation line: 785 nm of diode solid-state laser). The XPS data were collected using X-ray photoelectron spectrometer Thermo SCIENTIFIC ESCALAB 250xi with an Al-Kα (1486.8 eV) X-ray source. The pyridine-absorption infrared spectra (pyridine-IR) were recorded on a Thermo Nicolet 5700 FT-IR Spectrometer. Temperature programmed reduction of hydrogen (H2-TPR) were recorded on a Xianquan TP5076 automated chemisorption analyzer equipped with a TCD detector.2.3 Catalytic evaluation
The catalytic experiments of W-based catalysts for metathesis of ethylene and 1-butene to propene were carried out in a fixed bed reactor. 0.5 g of shaped catalyst (40–60 mesh) was placed at the center of reactor. The W-based catalysts need to be pretreated at 550 °C and 1 bar for 4 h by pure N2 or other gases such as pure H2, 1% O2/N2, and water vapor in N2. It should be noted that the flow rate of pretreated gas was 35 mL min−1. Water vapor generated in a flask at 80 °C was introduced into reactor with a flowing N2.38 In the case of water pretreatment, the catalyst was further flushed with inert N2 at high temperature of 450 °C for 3 h in order to avoid the influence of the remaining presence of H2O prior to the reaction. After treatment and cooling to designed reaction temperature, for example 450 °C, the mixed feed gas (molar ratio ethylene/1-butene = 2/1) was fed into the catalyst bed. The gas weight hourly space velocity (WHSV), defined as the weight ratio of mixed gas to catalyst packed, was regulated at 1.8 h−1. The flow rates of ethylene and 1-butene were controlled by two mass flow controllers (MFC, Brooks, model 5800E). All the products were analyzed online by a gas chromatograph (7820A, Agilent) with a HP-plot/Al2O3 KCl column using a flame ionization detector (FID). The 1-butene conversion and products selectivity were calculated as the reaction reached steady state according to the following equations:
where (C
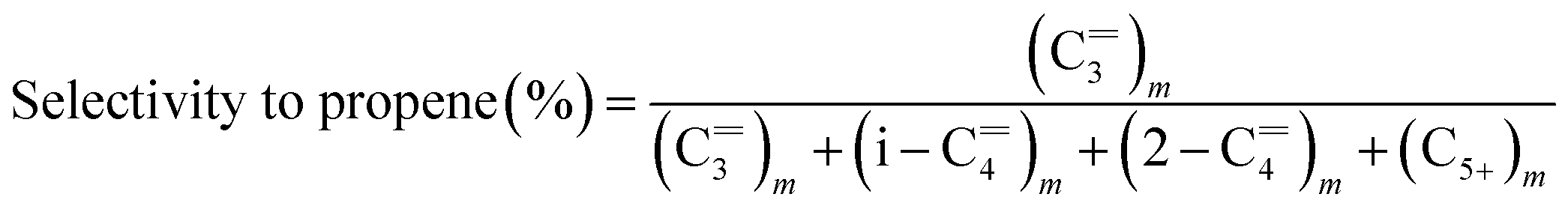 3)n is the molar number of propene, (C3)m is the weight percentage of propene in all hydrocarbon products. In addition, the specific activity is also calculated as the converted amount of 1-butene in molar base per gram of catalyst per hour.
3)n is the molar number of propene, (C3)m is the weight percentage of propene in all hydrocarbon products. In addition, the specific activity is also calculated as the converted amount of 1-butene in molar base per gram of catalyst per hour.
3. Results and discussion
3.1 Effect of support character
Table S1† summarizes the BET specific surface area, pore volume, pore size, W surface density, and acidity of as-prepared supports and W-contained catalysts. It is clear that SBA-15 exhibits the highest specific surface area and pore volume of 719 m2 g−1 and 1.09 cm3 g−1, respectively. In contrast, the corresponding values for TiO2 are the lowest at 13 m2 g−1 and 0.06 cm3 g−1. The structural properties of selected support directly determine the tungsten oxide surface density (W nm−2). As listed in Table S1,† the surface W density is linearly increased with a decrease in the surface area, which is calculated by using the initial BET surface area of the oxide support.39 For acidity, the 8W/SBA-15 and 8W/SiO2 display the low content in Brønsted acid which is not observed for 8W/γ-Al2O3 and 8W/TiO2. In spite of absence of Brønsted acid, 8W/γ-Al2O3 shows the significantly highest content in Lewis acid. The absence of Brønsted acid over 8W/γ-Al2O3 catalyst is similar to the result reported by Xu et al.17 In general, the content in Lewis acid is always much higher than that for Brønsted acid over as-prepared W-contained catalysts.The XRD patterns of supported W catalysts are shown in Fig. S1.† In the case of 8W/SiO2, the diffraction peaks assigned to monoclinic crystalline WO3 (PDF#72–1465) appear clearly, which are located at 23.1, 23.6, 24.3, 28.9, 33.3, and 34.1°. Those diffraction peaks could be observed but much weaker over 8W/TiO2 catalyst (Fig. S1a and b†). However, both 8W/SBA-15 and 8W/γ-Al2O3 catalysts do not show any diffraction peak for abovementioned WO3, which might be ascribed to the W highly dispersed on SBA-15 and its strong interaction with γ-Al2O3, respectively.40 The W loaded on TiO2 with small surface area leads to a smaller amount of the crystalline WO3 formed compared to that for SiO2 support, which might result from more favorable wetting of TiO2 by WO3 than that on SiO2. For comparison, the prepared unsupported WO3 reference exhibits the intensive diffraction peaks indicating the presence of WO3 (Fig. S1c†).
The UV-vis DRS spectra for various W-contained compounds and supported W catalysts are presented in Fig. 1. Comparison of the Na2WO4, AMT, and unsupported WO3 compounds with the supported W catalysts could provide reliable information about the local molecular coordination and bonding of supported tungsten oxide catalysts (Fig. 1a). In detail, Na2WO4 possesses a single ligand-to-metal charge transfer (LMCT) band maxima occurring at ∼208 nm; this represents the isolated tetrahedral structure of WOx. Ammonium metatungstate (AMT) precursor indicates characteristic spectrum with two maxima at 250 and 330 nm; this is related to the LMCT assigned to the oligomeric octahedral structure of WOx species. For as-prepared unsupported WO3, it exhibits a broad band at ∼400 nm attributed to d–d band of crystalline WO3.41 As exhibited in Fig. 1b, 8W/γ-Al2O3 gives rise to single band at ∼220 nm indicating the presence of isolated tetrahedral WOx.42 For W supported on TiO2, UV-vis spectra have no clear difference with that for TiO2 support except for a weak band at ∼400 nm which suggests that a small amount of crystalline WO3 is present.43 To be significantly different from above two catalysts, both 8W/SiO2 and 8W/SBA-15 display a typical band at ∼225 nm with a weaker band at ∼270 nm indicating the presence of the isolated tetrahedral structure and the oligomeric octahedral structure of WOx species, respectively.44 However, compared to the 8W/SBA-15, 8W/SiO2 exhibits one more band at ∼400 nm for crystalline WO3, which is well in agreement with the XRD results.
 | ||
| Fig. 1 UV-vis DRS spectra of (a) various W-containing compounds, and (b) 8 wt% W-containing catalysts and their corresponding support materials. | ||
Fig. S2† illustrates the Raman spectra of various supports and corresponding W-loaded catalysts compared at a similar net intensity. It can be seen that 8W/TiO2 exhibits a weak new band for crystalline WO3 phase at ∼805 cm−1 relative to that for TiO2 support. When W is loaded into γ-Al2O3, a single band at ∼975 cm−1 is assigned to the terminal νs (WO) of surface WOx.45 The Raman spectrum of 8W/SiO2 includes notable bands at 274, 715, 805, and 980 cm−1; these bands feature the deformation vibration mode of W–O–W, bending vibration mode of W–O, symmetric stretching vibration (νs) mode of W–O, and symmetric stretching vibration mode of terminal WO of surface WOx, respectively.42 8W/SBA-15 exhibits significantly weaker bands at 274, 715, and 805 cm−1 compared to that for 8W/SiO2, and similar intensity of band at 980 cm−1. To this end, we elucidated the structural differences of WOx over supported W catalysts through the XRD patterns, UV-vis spectra, and Raman spectra analysis.
The catalytic results of as-prepared W catalysts supported on different supports for metathesis of ethylene and 1-butene are shown in Fig. 2. The unsupported WO3 catalyst displays the lowest 1-butene conversion and nearly no detectable selectivity to propene. In spite of substantially higher 1-butene conversion with a gradual deactivation over supported 8W/TiO2, there is almost no propene formed via metathesis reaction. The 1-butene is dominantly transformed into trans-2-butene and cis-2-butene via double bond isomerization over both WO3 and 8W/TiO2 (Fig. 2c and Table S2†). According to the above results, it can be deduced that the crystalline WO3 did not provide active sites for the metathesis reaction to convert isomerized 2-butene and ethylene into propene. Over 8W/TiO2 catalyst, the catalytic results suggest that the metathesis reaction might be closely related to the terminal WO of surface WOx, since the Raman band of it in isolated tetrahedral WOx at ∼980 cm−1 is not present for 8W/TiO2 but clear for 8W/SiO2, 8W/SBA-15, and 8W/γ-Al2O3 (Fig. S2b†), and the latter three catalysts show much higher selectivity to propene except for 8W/γ-Al2O3. 8W/γ-Al2O3 exhibits relatively lower metathesis activity compared to that for 8W/SiO2 and 8W/SBA-15, which possibly results from their significant difference in acidity. It can be seen that 8W/γ-Al2O3 contains appreciable number of Lewis acid sites with no detectable Brønsted acid sites (Table S1†), which favors the isomerization of 1-butene to iso-butene via skeletal isomerization. The metathesis reaction is difficult to occur over iso-butene and ethylene.46 Besides, the presence of Brønsted acid sites might be favorable for promoting the metathesis reaction of 2-butene and ethylene for the formation of desired propene. Indeed, the higher selectivity to propene has been obtained over 8W/SiO2 and 8W/SBA-15 with the presence of suitable amount of Brønsted acid sites (Fig. 2c and Table S2†). As shown in Fig. 2, 8W/SBA-15 indicates the highest 1-butene conversion and selectivity to propene, which can be explained from two aspects. On the one hand, more Lewis and Brønsted acid sites are available for 8W/SBA-15 compared to that for 8W/SiO2; on the other hand, the W highly dispersed on SBA-15 with large surface area leads to more formation of the terminal WO in isolated tetrahedral structure of surface WOx (Fig. 1 and S2b†) due to more silanol group (Si–OH) interacted with the supported surface WOx species.30 Based on the abovementioned catalytic results and discussions, the metathesis reaction strongly depends on the terminal WO of isolated tetrahedral WOx and surface acidity.
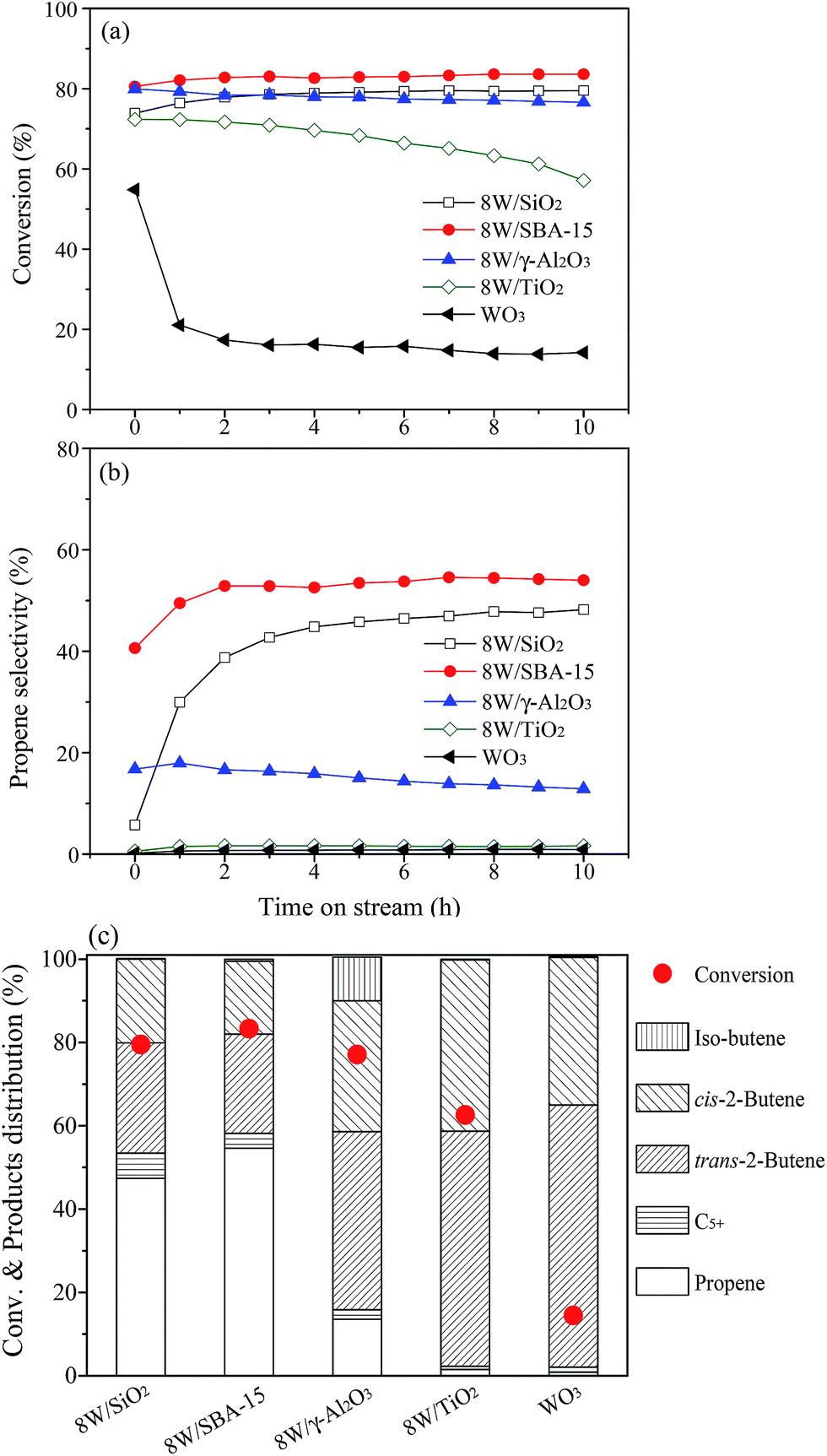 | ||
| Fig. 2 Time-dependence of 1-butene conversion (a), propene selectivity (b), and products distribution (c) obtained over WO3 and various 8 wt% W-containing catalysts at the reaction conditions of 450 °C, 0.1 MPa, 0.5 g of catalyst weight, 1.8 h−1 of WHSV, and an ethylene/1-butene molar ratio of 2. | ||
3.2 Effect of calcination temperature
Fig. S3† shows the XRD patterns of 8W/SBA-15 prepared with different calcination temperatures. It is clear that more crystalline WO3 phase is present on the surface of SBA-15 support with increasing calcination temperature from 400 to 550, and 700 °C as reflected with the notable increase in diffraction peaks at 23.1, 23.6, 24.3, and 34.1°; this is ascribed to the sintering of amorphous oligomeric octahedral WOx species to form the crystalline WO3 at higher temperature. Similarly, Praserthdam et al. investigated the effects of calcination ramp rate and calcination temperature on the amount of crystalline WO3. It was shown that a lower calcination ramp rate could enhance the catalyst activity in metathesis reaction from ethylene and 2-butene as compared to the ones calcined with a higher rate because they led to better dispersed catalysts.47 And the WO3 crystals were also clearly detected in XRD at a high calcination temperature.48 In our study, the speculated transformation of W phase is well confirmed by their UV-vis DRS spectra as shown in Fig. S4.† In detail, for 8W/SBA-15-700C catalyst, the relative intensity of band at ∼270 nm to that at ∼225 nm is substantially increased in contrast to that for 8W/SBA-15-400C and 8W/SBA-15-550C catalysts. Also, a new pronounced band of crystalline WO3 at ∼400 nm for 8W/SBA-15-700C is detectable, which means that the increased content in crystalline WO3 mainly comes from the evolution of oligomeric octahedral WOx species. Raman spectra for those prepared 8W/SBA-15 catalysts also demonstrate that the content of crystalline WO3 increases with an increase in the calcination temperature (Fig. S5†).It is somewhat curious that the abovementioned change of the state of W phase has negligible effect on the both isomerization of 1-butene and subsequent metathesis reaction as presented in Table S3.† It is worthy of note that, prior to catalytic reaction, the as-prepared 8W/SBA-15-400C is pretreated in a flow of N2 at 550 °C. The pretreatment procedure may lead to no significant discrepancies between 8W/SBA-15-400C and 8W/SBA-15-550C before the catalytic reaction.49 For 8W/SBA-15-550C and 8W/SBA-15-700C catalysts, the similar catalytic performance might be owing to their no distinct difference in the content for the isolated tetrahedral WOx (Fig. S4†). These reasons are responsible for almost unchanged catalytic results for 1-butene conversion and products selectivity.
3.3 Effect of W loading
Table 1 shows the BET surface area, pore volume, pore size, and acidity for the SBA-15 supported W catalyst with different W loadings. The BET surface area and pore volume continuously decrease with an increase in the W loading due to the additional mass introduced by the supported WOx phase. The reduced surface area possibly originates partially from bulky WOx species blocking the pores of SBA-15.50 It also can be seen from Fig. S6A† that all the samples exhibit the type IV isotherms with H1 hysteresis loops, which are typical for highly ordered hexagonal structure of mesoporous silica material.51 These results confirm the maintenance of ordered hexagonal arrangement of the SBA-15 frameworks upon W loading. Fig. S6B† shows the pure SBA-15 and its supported W samples have a narrow pore size distribution with an average size about ∼6 nm. As the W loading increases from 4 to 30 wt%, the surface density of W atoms is drastically enhanced from 0.18 to 2.84 W nm−2. Regarding the strength and types of acid sites, it can be determined from the pyridine adsorption measured by IR spectra (Fig. S7†). As presented in Table 1, the acidity for both Brønsted acid and Lewis acid firstly increases and then decreases with a higher W loading and the 15 wt% W loading leads to the maximum acidity. Note that the pure SBA-15 and a low W loading of 4 wt% do not indicate the presence of Brønsted acid. As shown in Fig. S7,† pure SBA-15 shows a band at ∼1445 cm−1 assigned to hydrogen-bonded pyridine (H), indicating that the H band seems to be come from the pyridine hydrogen bonded to the Si–OH.52 With a loading of WO3, a new band located at ∼1450 cm−1 formed is attributed to pyridine coordinately bonded to surface Lewis acid sites; these acid sites are ascribed to the isolated WOx in the tetrahedral coordination.53 When the WO3 is loaded in 8 wt%, a new band at ∼1540 cm−1 gradually appears, indicating the protonated pyridine bonded to surface Brønsted acid. The appearing surface Brønsted acid sites might be due to the protonation of terminal W sites in the tetrahedral coordination.54 With continuously increasing W loading, both Brønsted acid and Lewis acid are decreased after reaching the maximum acidity with a loading of 15 wt%; this is ascribed to the increased WO3 content leading to the formation of oligomeric species and further bulk WO3 species with lower content in the terminal W sites. To this end, we have clearly elucidated the type and content of acidity varied with the WOx content.| Catalyst sample | Surface areaa (m2 g−1) | Pore volumeb (cm3 g−1) | Pore sizeb (nm) | Surface density (W nm−2) | Acidity (μmol g−1) | |
|---|---|---|---|---|---|---|
| Brønsted acidc | Lewis acidc | |||||
| a Determined by BET method.b Evaluated by the BJH method.c Determined by pyridine-IR measurement. | ||||||
| SBA-15 | 719 | 1.09 | 6.1 | N/A | 0.0 | 0.0 |
| 4W/SBA-15 | 603 | 0.85 | 5.6 | 0.18 | 0.0 | 6.0 |
| 8W/SBA-15 | 551 | 0.83 | 5.8 | 0.41 | 2.4 | 12.0 |
| 15W/SBA-15 | 500 | 0.74 | 5.9 | 0.92 | 3.2 | 13.8 |
| 20W/SBA-15 | 470 | 0.74 | 5.9 | 1.39 | 1.6 | 13.2 |
| 30W/SBA-15 | 393 | 0.60 | 5.9 | 2.84 | 0.8 | 7.8 |
In order to further confirm the evolution of W phase and its molecular bonding with different W loadings, the as-prepared supported W catalysts are characterized by XRD, Raman, and XPS. The XRD patterns of SBA-15 and its supported W catalysts are given in Fig. 3. It is clearly illustrated that the crystalline WO3 (PDF#83-0948, triclinic) is present when the WO3 content is higher than 8 wt%, which is different from that (PDF# 72-1465, monoclinic) for 8W/SiO2 sample (Fig. S1†). The intensity of diffraction peaks located at 2θ of 23.1, 23.6, 24.3, and 33.6° becomes stronger with an increase in the WO3 content, which suggests that a certain amount of crystalline WO3 appears. As presented in Fig. 4, the Raman spectra for above samples imply that the aggregated WOx species including amorphous and crystalline W phase continuously increase with an increase in the WO3 content, as reflected with stronger bands at 274, 715, and 805 cm−1. The band at ∼980 cm−1 assigned to terminal WO bond firstly increases and then slightly decreases. The strongest band is present over 15W/SBA-15 catalyst. This can be inferred that the concentration of highly dispersed WOx species cannot be increased further, and the additional W loading contributes progressively to the growth of WO3 nanoparticles.26 XPS measurements are conducted to detect the oxidation state of surface W species. The XPS spectra of W 4f level are fitted according to the theory of Doniach and Sunjic.55 As shown in Fig. 5, the binding energy at 35.7/37.9 eV and 36.2/38.1 eV is ascribed to the 4f7/2 and 4f5/2 peaks of W atoms for aggregated W6+ oxide and dispersed W6+ hydroxide WO3(OH2)n, respectively.56,57 As illustrated in literature, the W6+ hydroxide could be transformed to surface tetrahedral WOx.58 The investigated catalysts of 4W/SBA-15, 15W/SBA-15, and 30W/SBA-15 exhibit the peaks of W6+ hydroxide, its peaks intensity displays maxima over 15W/SBA-15 catalyst, which can be explained that the isolated tetrahedral WOx is active to absorb H2O molecules in air for the formation of WO3(OH2)n. This result further confirms that 15W/SBA-15 has the highest content of isolated WOx.
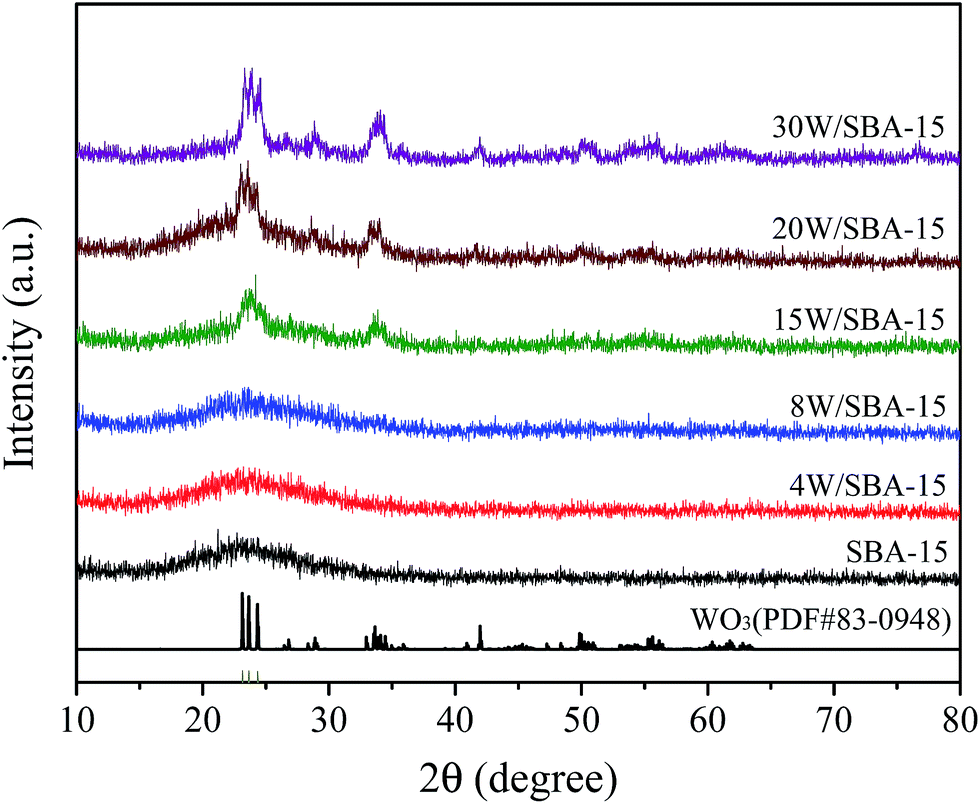 | ||
| Fig. 3 XRD patterns of fresh SBA-15 and W/SBA-15 catalysts with different W loadings. | ||
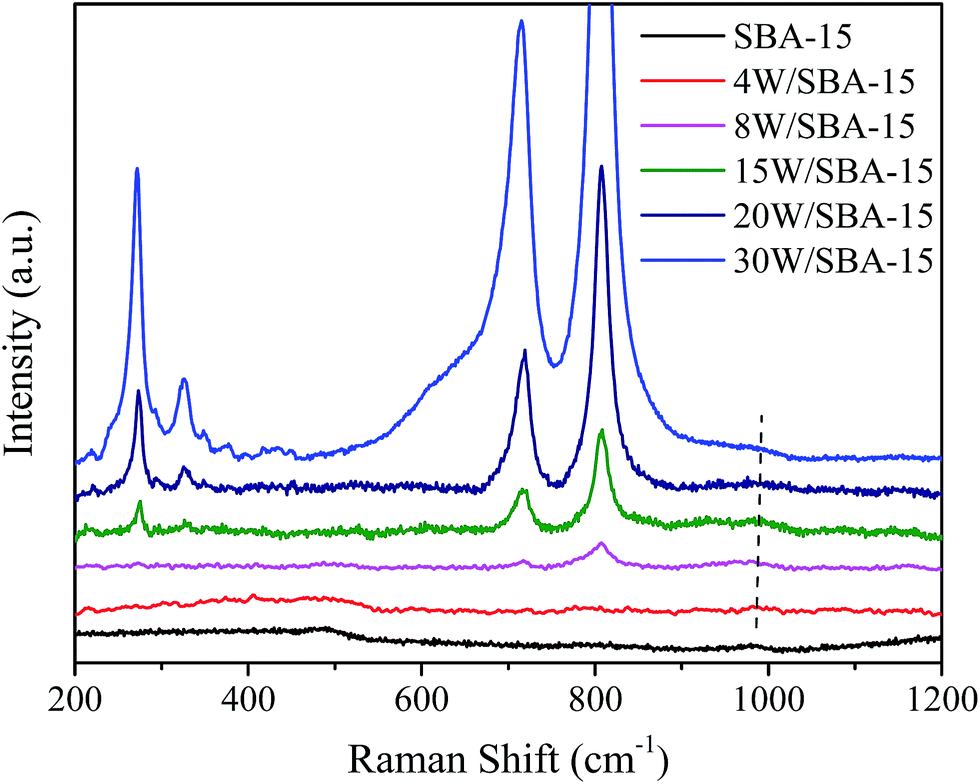 | ||
| Fig. 4 Raman spectra of fresh SBA-15 and W/SBA-15 catalysts with different W loadings. | ||
 | ||
| Fig. 5 XPS spectra of fresh 4W/SBA-15, 15W/SBA-15, and 30W/SBA-15 catalysts. | ||
The catalytic performance over SBA-15 supported W catalysts with different loadings is summarized in Fig. 6 and Table S4.† The pure SBA-15 exhibits a stable 1-butene conversion of 72.3% with a negligible selectivity to propene. The 1-butene is mainly converted into trans-2-butene and cis-2-butene via CC bond isomerization, which suggests that the Si–OH in SBA-15 has the isomerization activity.52 As W is introduced to SBA-15 with a 4 wt% loading, the selectivity to propene is notably enhanced from 2.9 to 37.4%, however, the 1-butene conversion increases slightly from 72.3 to 78.9% (Table S4†). This result reflects that the isolated WOx of tetrahedral coordination is responsible for the metathesis reaction to produce propene. With further increase in W loading from 4, 8 to 15 wt%, the 1-butene conversion continuously increases to a small extent from 78.9, 83.3, to 88.2%. Accordingly, the selectivity to propene increases remarkably from 37.4, 54.6, to 68.6% with marked increase in Brønsted acidity (Table 1). The above discussion demonstrates that both Lewis and Brønsted acid sites from the isolated tetrahedral WOx structure are favorable for the metathesis reaction. When W is introduced over a suitable amount, for example 15 wt%, both 1-butene conversion and selectivity to propene are decreased. In contrast, the metathesis activity is reduced to a larger extent. This is ascribed to the less amount of the formation of isolated tetrahedral WOx structure which results from its evolution into the inactive crystalline WO3.
 | ||
| Fig. 6 Time-dependence of 1-butene conversion (a), propene selectivity (b), and products distribution (c) obtained over SBA-15 and W/SBA-15 catalysts with different W loadings at the reaction conditions of 450 °C, 0.1 MPa, 0.5 g of catalyst weight, 1.8 h−1 of WHSV, and an ethylene/1-butene molar ratio of 2. | ||
In order to investigate the evolution of SBA-15 supported W catalysts during the catalytic reaction, the fresh and spent catalysts are characterized by UV-vis DRS spectra as presented in Fig. S8.† It is clear that the intensity of band at ∼225 nm gradually increases to a stable level when W loading is below 15 wt%, which is attributed to the increased content for the isolated tetrahedral WOx structure during the catalytic reaction. In addition, all spent catalysts exhibit gradually increased intensity of the band in the range of 400 to 700 nm. The higher W content supported on SBA-15, the more markedly elevated band intensity. This band is assigned to the W4+ and W5+ species.17 The lower oxidation state of W species is due to the reduction of W6+ during the reaction. To further confirm the presence of lower oxidation state of W species, the spent 15W/SBA-15 catalysts with different reaction time are measured by XRD as displayed in Fig. 7. One can see that the diffraction peak at 2θ of 33.6° assigned to crystalline WO3 is gradually shifted to lower value at 33.3° due to the appearance of nonstoichiometric phase WO2.92 and WO2.83, which suggests that the tungsten oxide species are gradually reduced during the metathesis reaction. It can be inferred that the reduction of W catalysts might be closely related to the induction period.59 As shown in Fig. S9,† the TPR profiles of fresh 8W/SBA-15, 15W/SBA-15, and 30W/SBA-15 catalysts display three typical reduction peaks for WO3 to WO2.90, WO2.90 to WO2, and WO2 to W, respectively.60 The temperature range for three different reduction stages is located at 400–500, 630–740, and 740–880 °C, respectively. This result further demonstrates that the tungsten oxide species could be reduced at the reaction temperature of 450 °C during metathesis reaction.
 | ||
| Fig. 7 XRD patterns of fresh 15W/SBA-15 and two spent catalysts recovered after 40 min and 10 h of reaction, respectively. | ||
3.4 Effect of pretreatment gas
The catalytic performance for 15W/SBA-15 pretreated with different gases is presented in Fig. 8 and Table 2. In contrast to N2-pretreated 15W/SBA-15 previously described as a reference, H2-pretreatment does not significantly affect the 1-butene conversion, however, the induction period becomes shorter to result in a higher initial selectivity to propene at 30% compared with that for N2-pretreatment at 20%. For 15W/SBA-15 pretreated by 1% O2/N2 gas, the 1-butene conversion is still kept unchanged. This indicates that the 1-butene isomerization is not closely related to the state of WOx. As expected, the initial selectivity to propene obtained in metathesis reaction is significantly lower at 5% and need a longer time to reach the stable stage. The abovementioned result analysis strongly demonstrates that the partially reduced WOx species is necessary precursor for the formation of active sites for metathesis reaction, but not for 1-butene isomerization. Liu et al. also confirmed that the partially reduced WO2.92 as active phase for metathesis reaction by adjusting the H2 concentration in prior reduction process.61 It is no doubt that the extent of reduction influences the metathesis activity as previously mentioned. However, it is hard to correlate the metathesis activity with quantifying the content of reduced WOx species. Indeed, without prior reduction process, the active sites for metathesis reaction are generated by the interaction of olefins with W/SBA-15 catalyst during the reaction, which takes more time for subsequent formation of propene. | ||
| Fig. 8 Time-dependence of 1-butene conversion (a), propene selectivity (b), and products distribution (c) over 15W/SBA-15 catalysts pretreated in different gases at the reaction conditions of 450 °C, 0.1 MPa, 0.5 g of catalyst weight, 1.8 h−1 of WHSV, and an ethylene/1-butene molar ratio of 2. a Spent 15W/SBA-15 catalyst pretreated in H2O followed by calcination, and the obtained catalyst further treated in N2 stream before reaction. | ||
| Pretreatment gas | Conversiona (%) | Selectivity (%) | Specific activity (mmol C4 per gcat per h) |
||||
|---|---|---|---|---|---|---|---|
| Propene | C5+ | trans-2-Butene | cis-2-Butene | Iso-butene | |||
| a Reaction conditions: T = 450 °C, P = 0.1 MPa, catalyst weight = 0.5 g, WHSV (E + B) = 1.8 h−1, n(E)/n(B) = 2.b Spent 15W/SBA-15 catalyst pretreated in H2O followed by calcination, and the obtained catalyst further treated in N2 stream before reaction. | |||||||
| N2 | 88.2 | 68.6 | 4.5 | 15.2 | 11.1 | 0.5 | 14.2 |
| H2 | 85.9 | 62.0 | 4.2 | 16.5 | 12.1 | 1.5 | 13.8 |
| O2 | 87.7 | 67.1 | 4.6 | 16.1 | 11.7 | 0.7 | 14.1 |
| H2O | 72.4 | 0.9 | 0.5 | 56.2 | 41.4 | 1.1 | 11.6 |
| H2Ob | 76.5 | 35.2 | 3.0 | 35.0 | 26.4 | 0.4 | 12.3 |
Interestingly, the pretreatment with water vapor introduced by a flowing N2 leads to the complete deactivation of metathesis reaction (Fig. 8b), however, the isomerization activity is similar to that for pure SBA-15 support (Fig. 6a). Furthermore, the spent H2O-pretreated 15W/SBA-15 catalyst is calcined in air at 550 °C like the preparation procedure for fresh catalyst. The obtained sample is re-evaluated using the same reaction conditions with the fresh N2-pretreated catalyst, which has not fully recovered the catalytic performance for 1-butene conversion and selectivity to propene. This result indicates that H2O-pretreatment seriously alters the existing structure of WOx species (Fig. 9a). For comparison, the XRD patterns for spent N2-, H2-, and O2-pretreated 15W/SBA-15 catalysts do not exhibit obvious evolution for tungsten oxide species (Fig. 9b). On the other hand, the diffraction peaks for H2O-pretreated 15W/SBA-15 catalyst illustrate the presence of large amount of crystalline WO3 phase. These intensive peaks are located at 23.1, 23.6, 24.3, and 33.3°, which represent the monoclinic WO3 similar to that for 8W/SiO2 (Fig. S1†). XPS spectra of fresh 15W/SBA-15 and H2O-pretreated 15W/SBA-15 catalysts are presented in Fig. S10.† In the case of H2O-pretreated 15W/SBA-15 catalyst, the peaks intensity of binding energy at 35.7 and 37.9 eV (black line) substantially increases, which suggests that the content for aggregated W6+ oxide increases. Correspondingly, the peaks intensity of binding energy at 36.2 and 38.1 eV (blue line) decrease to a remarkable extent with the decreased content for W6+ hydroxide WO3(OH2)n. Despite that WO3(OH2)n still exists, the H2O-pretreated 15W/SBA-15 catalyst does not display metathesis activity. This is ascribed to the remaining presence of H2O molecules at the surface of catalyst to be unfavorable for the transformation of WO3(OH2)n into active sites for metathesis reaction. In order to clarify the influence of H2O on the catalyst evolution, the H2-TPR experiments for fresh 15W/SBA-15 and calcined spent H2O-pretreated 15W/SBA-15 catalysts are carried out. As presented in Fig. S11,† the first reduction peak at the range of 400–500 °C shifts to higher temperature range and the peak intensity becomes much weaker. This result shows that the H2O pretreatment seriously affects the reducibility of tungsten oxide species. As previously demonstrated, the reduction behavior does really play an important role in forming the active sites for metathesis reaction. In our viewpoint, on the one hand, the catalyst pretreated by H2O leads to the irreversible deactivation due to the sintering of WOx species; on the other hand, the remaining presence of H2O may also poison the reduced W5+ sites to be interacted with olefins for further W-carbene formation. Although the catalyst was further flushed with inert N2 at relatively high temperature of 450 °C before the reaction, the metathesis reaction cannot proceed.
 | ||
| Fig. 9 XRD patterns of (a) fresh 15W/SBA-15, H2O-pretreated 15W/SBA-15, spent H2O-pretreated 15W/SBA-15, calcined spent H2O-pretreated 15W/SBA-15, and (b) spent 15W/SBA-15 catalysts pretreated in different gases (N2, H2, O2 and H2O). | ||
3.5 Effect of WHSV of ethylene and 1-butene
In order to obtain the optimal catalytic performance for the metathesis reaction of ethylene and 1-butene over 15W/SBA-15 catalyst, we have investigated the influence of WHSV of mixed feed gas in a fixed-bed reactor. The catalytic performances are evaluated by 1-butene conversion, specific activity and product selectivity. Note that the specific activity is defined as the converted amount of 1-butene in molar base per gram of catalyst per hour. In our study, the WHSV was regulated by changing the loaded amount of catalyst with a constant flow rate of feed gas at 9 mL min−1 (molar ratio ethylene/1-butene = 2/1). As presented in Fig. S12† and Table 3, the 0.25 g of 15W/SBA-15 (WHSV = 3.6 h−1) leads to the lowest 1-butene conversion and remarkably lower selectivity to propene but the highest specific activity at 25.7 mmol C4 per gcat per h. The 1-butene conversion and selectivity to propene reach the maximum value at 91.2% and 75.8% with a WHSV of 0.9 h−1, respectively. Accordingly, the specific activity is about 7.4 mmol C4 per gcat per h, which is higher than the value of 6.4 mmol C4 per gcat per h for W-FDU-12 combined with MgO catalysts under the similar reaction conditions.33 However, the overloaded catalyst (1.5 g, WHSV = 0.6 h−1) slightly decreases both 1-butene conversion and selectivity to propene with the lowest specific activity at 4.8 mmol C4 per gcat per h. On the basis of above catalytic results, the insufficient loaded catalyst exhibits much lower selectivity to propene, which suggests that the reaction rate of metathesis step is slower than that for isomerization step. In other words, the isomerized 2-butene could not be quickly converted into propene via metathesis reaction. This conclusion can also be obtained from the changing trend of 1-butene conversion, propene selectivity, and specific activity as listed in Table 3. As the WHSV is raised to a much higher value at 3.6 h−1, the 1-butene conversion decreases slightly while the selectivity to propene drops remarkably from 72.7 to 40.6%. Seen from the specific activity listed, the high WHSV of 3.6 h−1 exhibits about 5 times higher value than that for 0.6 h−1. This result also indicates that the conversion rate of 1-butene is quite fast. The metathesis reaction is rate-determining step for the enhancing selectivity to propene. In the case of overloaded catalyst, the selectivity to propene is decreased due to the reversible conversion between propene, 2-butene, and 1-butene.35 As the formed propene could not be run away from the catalyst surface, it will return back to 2-butene and ethylene. It is worth noting that a longer residence time (lower WHSV) gradually enhances the skeletal isomerization of 1-butene, despite that it is more difficult to proceed than that for CC bond isomerization.40 Therefore, a suitable WHSV is necessary to obtain the high selectivity to propene.
| WHSV (h−1) | Conversiona (%) | Selectivity (%) | Specific activity (mmol C4 per gcat per h) |
||||
|---|---|---|---|---|---|---|---|
| Propene | C5+ | trans-2-Butene | cis-2-Butene | Iso-butene | |||
| a Reaction conditions: T = 450 °C, P = 0.1 MPa, catalyst weight = 1.5, 1.0, 0.5, and 0.25 g, n(E)/n(B) = 2. | |||||||
| 0.6 | 90.6 | 72.7 | 4.3 | 12.3 | 8.9 | 1.7 | 4.8 |
| 0.9 | 91.2 | 75.8 | 4.1 | 10.9 | 7.8 | 1.2 | 7.3 |
| 1.8 | 88.2 | 68.6 | 4.5 | 15.2 | 11.1 | 0.5 | 14.2 |
| 3.6 | 79.8 | 40.6 | 4.2 | 31.5 | 23.4 | 0.3 | 25.7 |
3.6 Effect of reaction temperature
Finally, the reaction temperature is regulated to optimize the catalytic performance over 15W/SBA-15 catalyst under the above optimal WHSV of 0.9 h−1. As can be seen from Fig. S13† and Table 4, the lower reaction temperature gives slightly lower 1-butene conversion but significantly lower selectivity to propene. The decreased trend in 1-butene conversion is much milder than that for propene selectivity, which illustrates that the metathesis reaction is more difficult to occur relative to the isomerization reaction of 1-butene. The optimal selectivity to propene at 79.1% with 1-butene conversion of 92.1% is obtained over the reaction temperature of 500 °C. When the reaction temperature is further elevated to 550 °C, in spite of the stable 1-butene conversion, the selectivity to propene is obviously decreased due to the formation of C1–C3 alkane from thermal-cracking.62 Thereby, the suitable reaction temperature should be selected to obtain the highest selectivity to propene through efficient conversion of the formed intermediate 2-butene into propene via metathesis reaction, and avoid the side-reactions such as skeletal isomerization and thermal-cracking.| T (°C) | Conversiona (%) | Selectivity (%) | Specific activity (mmol C4 per gcat per h) |
||||
|---|---|---|---|---|---|---|---|
| Propene | C5+ | trans-2-Butene | cis-2-Butene | Iso-butene | |||
| a Reaction conditions: T = 350–550 °C, P = 0.1 MPa, catalyst weight = 1.0 g, WHSV (E + B) = 0.9 h−1, n(E)/n(B) = 2. | |||||||
| 350 | 79.6 | 12.5 | 1.8 | 49.6 | 34.3 | 1.8 | 6.4 |
| 400 | 84.6 | 49.5 | 4.2 | 26.0 | 18.6 | 1.7 | 6.8 |
| 450 | 91.2 | 75.8 | 4.1 | 10.9 | 7.8 | 1.2 | 7.3 |
| 500 | 92.1 | 79.1 | 3.8 | 8.7 | 6.5 | 1.9 | 7.4 |
| 550 | 92.3 | 72.8 | 5.9 | 7.8 | 6.0 | 3.1 | 7.4 |
3.7 Discussion on mechanism of metathesis reaction
Based on above experimental results combined with various characterizations such as XRD, UV-vis DRS, Raman, pyridine IR, and H2-TPR, it is well confirmed that the partially reduced isolated tetrahedral WOx species can be responsible for subsequent metathesis reaction of 1-butene and ethylene.When the 1-butene and ethylene are used as reactants for producing propene via metathesis reaction, there are three steps involved including (1) fast isomerization of 1-butene, (2) carbene formation, and (3) metathesis reaction as shown in Scheme 1. It is no doubt that SBA-15 exhibits significant isomerization activity, which is catalyzed by Si–OH (Scheme 1, 1). The Si–OH might be working as a weak Brønsted acid site for isomerization of CC bond, however, lack of ability of metathesis.63 Table S4† shows that the varied W loadings do not affect the level of 1-butene conversion in spite of the less amount of Si–OH on W/SBA-15 with well dispersed W over the support. As noted in the previous description, the isomerization rate of 1-butene is so fast that the decrease in the amount of Si–OH could not suppress the 1-butene isomerization. It is important to note that unsupported WO3 displays the negligible activity for 1-butene isomerization and inactive in metathesis reaction. The previously mentioned results suggest that the isolated tetrahedral WOx (W6+) species can be reduced by olefin or H2 to form the W5+ species, which might be regarded as the pre-requisite step for subsequent formation of W-carbene (Scheme 1, 2). Indeed, this viewpoint is strongly supported by following experimental facts: (1) shorter induction period for formation of propene after the catalyst pretreated in H2 stream; (2) pretreatment with O2 results in lower initial selectivity to propene (Fig. 8); (3) observed presence of W5+ in spent catalysts from UV-vis DRS spectra and XRD patterns (Fig. 7 and S8†) compared to fresh catalyst with only W6+ species (Fig. 5). It should be noted that the pretreatment with H2O directly deactivates the metathesis activity completely. The deactivation of catalyst might mainly result from the H2O-induced aggregation of isolated WOx species as the strong diffraction peaks for inactive WO3 phase are observed before and after reaction (Fig. 9a). However, the metathesis activity could be recovered to some extent due to the resulting re-dispersion of aggregated WO3 phase by calcination treatment.58 As reflected in Fig. 9a, the calcined spent H2O-pretreated 15W/SBA-15 exhibits much weaker diffraction peaks for crystalline WO3 phase compared to that before calcination treatment. In spite of the proposed route for formation of W-carbene, we cannot deny that the isolated tetrahedral W6+ species possibly play a similar role like W5+ species.
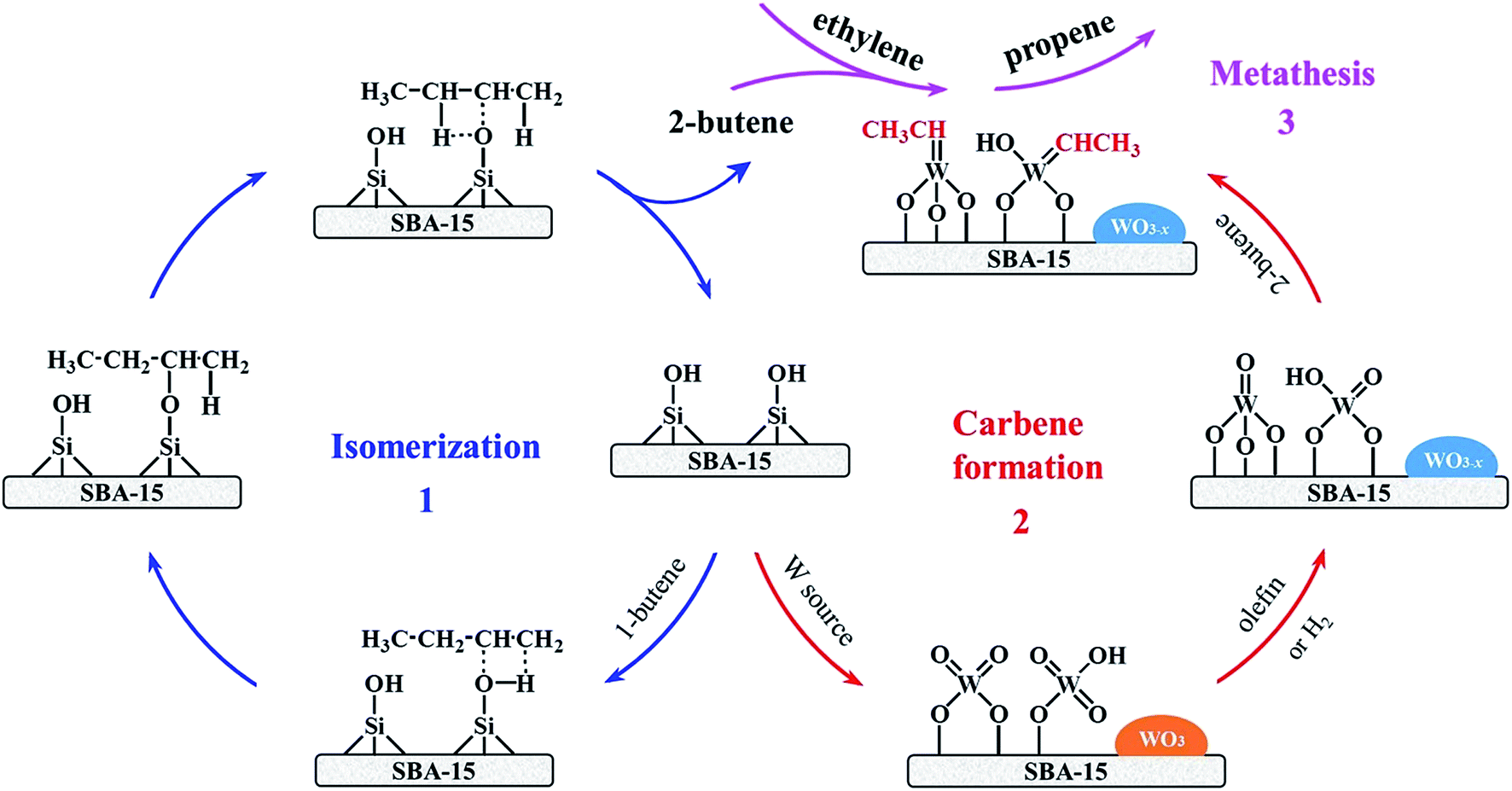 | ||
| Scheme 1 Proposed route for conversion of 1-butene and ethylene into propene via isomerization (1), carbene formation (2), and metathesis reaction (3) over SBA-15 supported W catalysts. | ||
The abovementioned isolated tetrahedral W5+ species consist of two types of structure, such as the terminal WO (Lewis acid sites) and W–OH (Brønsted acid sites). It should be noted that these acid sites are possible to catalyze CC bond isomerization.64 The widely accepted olefin metathesis mechanism involves metallocarbene intermediates, implying that the successful activation of supported-oxide based olefin metathesis catalysts requires the conversion of WO and W–OH species into W-carbene.65,66 The formation pathways of W-carbene over W/SBA-15 catalysts are illustrated in Scheme S1.† According to the DFT calculation, the 2-butene preferentially forms W-carbene active sites.67 In the case of terminal WO, the WO is firstly combined with 2-butene by [2+2] cycloaddition reaction to form a four-membered oxametallacycle intermediate. Subsequently, the decomposition of the four-membered oxametallacycle intermediate produces the acetaldehyde (H3C–CHO) and the final WCH–CH3 active sites (Scheme S1A†). For the terminal W–OH, the 2-butene is reacted with W–OH to form 2-butanoxide by protonation reaction. Then, β-H in 2-butanoxide is combined with WO to form the six-membered oxametallacycle intermediate which can decompose into the four-membered oxametallacycle intermediate and W–OH via migration of hydrogen from 2-butene to WO. Also, subsequent elimination of carbonyl compound (H3C–CHO) from the intermediate occurs to form WCH–CH3 active sites (Scheme S1B†). The above W-carbene formed via both the terminal WO and W–OH is regarded as the active sites for metathesis reaction.
The postulated metathesis mechanism for the formation of propene is presented in Scheme S2.† It is clear that the initially formed WCH–CH3 active sites can react with ethylene (Scheme S2a†) and 1-butene (Scheme S2b†) to give the metallocyclobutane intermediates by [2+2] cycloaddition reaction.68,69 Subsequently, these intermediates can be decomposed into propene with generation of new W-carbene, such as WCH2 and WCH–CH2CH3. The newly generated WCH2 and WCH–CH2CH3 further provide the active sites for cycloaddition with 2-butene to result in the metallocyclobutane intermediates. Similarly, the propene and 2-pentene are liberated from the metallocyclobutane intermediates with re-generation of initial WCH–CH3 active sites. To this end, the metathesis reaction process for conversion of 1-butene and ethylene into propene has been fully elucidated in this study.
4. Conclusions
The investigation of the structure of tungsten oxide supported on SBA-15 and its catalytic performance in the metathesis reaction of 1-butene and ethylene to propene has been carried out extensively. The main conclusions are summarized as following several aspects.For converting 1-butene and ethylene into propene, the 1-butene is firstly required to be isomerized to 2-butene. The results show the Si–OH serving as the primary active centers for 1-butene isomerization. And the terminal WO (Lewis acid sites) and W–OH (Brønsted acid sites) in the isolated tetrahedral WOx species might also play a role in the isomerization reaction. Over investigated W/SBA-15 catalysts, the isolated tetrahedral WOx (W6+) species are partially reduced by olefins or H2 to give rise to WOx (W5+) species as the precursor sites for the formation of W-carbene species which does contribute to olefin metathesis activity. In comparison to the isolated WOx species, the crystalline WO3 phase does not show the metathesis activity despite that they are partially reduced during the reaction.
The W/SBA-15 catalysts exhibit different initial selectivity to propene with tuning pretreatment atmosphere. The reducing gas H2 results in a shorter induction time compared to that for inert gas N2 and oxidizing gas O2, which provides strong evidence that the partially reduced isolated WOx (W5+) species is crucial for the subsequent generation of W-carbene. It is further confirmed by H2O pretreatment as the suppressed reduction of WOx species results in complete loss of metathesis activity. The sintering of isolated WOx species leads to a significant increase in the crystalline WO3 content, which is also possibly responsible for the deactivation. By optimizing the catalyst preparation methods and reaction conditions, the 1-butene conversion of 92.1% and propene selectivity of 79.1% have been achieved over 15W/SBA-15 catalyst. Accordingly, the specific activity is about 7.4 mmol C4 per gcat per h. The present study provides the molecular level structure–activity/selectivity relationship for the 1-butene/ethylene metathesis to propene. The comprehensive insights will guide us to develop advanced W-based catalysts for olefin metathesis to remarkably enhance the selectivity to desired olefins.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the financial support from the National Natural Science Foundation of China (21576119), the Natural Science Foundation of Jiangsu Province (BK20151125), the Fundamental Research Funds for the Central Universities (JUSRP51720B), the Jiangsu Provincial Six Talent Peaks Program (XNY-001), and the Programme of Introducing Talents of Discipline to Universities (111 Project B13025).References
- H. M. T. Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef.
- L. Yu, J. Yuan, Q. Zhang, Y. M. Liu, H. Y. He, K. N. Fan and Y. Cao, ChemSusChem, 2014, 7, 743–747 CrossRef CAS PubMed.
- Z. W. Xi, N. Zhou, Y. Sun and K. L. Li, Science, 2001, 292, 1139–1141 CrossRef CAS PubMed.
- A. Farshi, F. Shaiyegh, S. H. Burogerdi and A. Dehgan, Pet. Sci. Technol., 2011, 29, 875–885 CrossRef CAS.
- E. Epelde, M. Ibáñez, J. Valecillos, A. T. Aguayo, A. G. Gayubo, J. Bilbao and P. Castaño, Appl. Catal., A, 2017, 547, 176–182 CrossRef CAS.
- M. Sedighi, K. Keyvanloo and J. Towfighi, Fuel, 2013, 109, 432–438 CrossRef CAS.
- M. Bender, ChemBioEng Rev., 2014, 1, 136–147 CrossRef CAS.
- A. Corma, G. W. Huber, L. Sauvanaud and P. O'Connor, J. Catal., 2007, 247, 307–327 CrossRef CAS.
- Y. M. Liu, W. L. Feng, T. C. Li, H. Y. He, W. L. Dai, W. Huang, Y. Cao and K. N. Fan, J. Catal., 2006, 239, 125–136 CrossRef CAS.
- P. Tian, Y. X. Wei, M. Ye and Z. M. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- F. Jiang, M. Zhang, B. Liu, Y. B. Xu and X. H. Liu, Catal. Sci. Technol., 2017, 7, 1245–1265 CAS.
- F. Jiang, B. Liu, W. P. Li, M. Zhang, Z. J. Li and X. H. Liu, Catal. Sci. Technol., 2017, 7, 4609–4621 CAS.
- J. Zheng, J. Cai, F. Jiang, Y. B. Xu and X. H. Liu, Catal. Sci. Technol., 2017, 7, 4736–4755 CAS.
- W. G. Zhou, J. Y. Liu, X. Wu, J. F. Chen and Y. Zhang, Catal. Commun., 2015, 60, 76–81 CrossRef CAS.
- R. Westhoff and J. A. Moulijn, J. Catal., 1977, 46, 414–416 CrossRef CAS.
- E. Mazoyer, K. C. Szeto, N. Merle, S. Norsic, O. Boyron, J. M. Basset, M. Taoufik and C. P. Nicholas, J. Catal., 2013, 301, 1–7 CrossRef CAS.
- S. J. Huang, S. L. Liu, W. J. Xin, J. Bai, S. J. Xie, Q. X. Wang and L. Y. Xu, J. Mol. Catal. A: Chem., 2005, 226, 61–68 CrossRef CAS.
- T. Hahn, E. V. Kondratenko and D. Linke, Chem. Commun., 2014, 50, 9060–9063 RSC.
- X. J. Li, W. P. Zhang, S. L. Liu, S. J. Xie, X. X. Zhu, X. H. Bao and L. Y. Xu, J. Mol. Catal. A: Chem., 2009, 313, 38–43 CrossRef CAS.
- C. Meetings, Chemistry, 2003, 9, 971–975 CrossRef PubMed.
- S. Lwin and I. E. Wachs, ACS Catal., 2015, 6, 272–278 CrossRef.
- S. Lwin and I. E. Wachs, ACS Catal., 2014, 4, 2505–2520 CrossRef CAS.
- S. J. Huang, F. Chen, S. L. Liu, Q. Zhu, X. Zhu, W. J. Xin, Z. Feng, C. Li, Q. Wang and L. Y. Xu, J. Mol. Catal. A: Chem., 2007, 267, 224–233 CrossRef CAS.
- J. F. Wu, A. Ramanathan, W. K. Snavely, H. D. Zhu, A. Rokicki and B. Subramaniam, Appl. Catal., A, 2016, 528, 142–149 CrossRef CAS.
- V. Goelden, D. Linke and E. V. Kondratenko, ACS Catal., 2015, 5, 7437–7445 CrossRef CAS.
- S. Lwin, Y. Li, A. I. Frenkel and I. E. Wachs, ACS Catal., 2016, 6, 3061–3071 CrossRef CAS.
- S. Lwin and I. E. Wachs, ACS Catal., 2016, 7, 573–580 CrossRef.
- D. R. Hua, S. L. Chen, G. M. Yuan, Y. L. Wang, Q. F. Zhao, X. L. Wang and B. Fu, Microporous Mesoporous Mater., 2011, 143, 320–325 CrossRef CAS.
- E. Mazoyer, K. C. Szeto, S. Norsic, A. Garron, J. M. Basset, C. P. Nicholas and M. Taoufik, ACS Catal., 2011, 1, 1643–1646 CrossRef CAS.
- Y. D. Wang, Q. L. Chen, W. M. Yang, Z. K. Xie, W. Xu and D. Y. Huang, Appl. Catal., A, 2003, 250, 25–37 CrossRef CAS.
- E. Mazoyer, K. C. Szeto, J. M. Basset, C. P. Nicholas and M. Taoufik, Chem. Commun., 2012, 48, 3611–3613 RSC.
- J. L. Wang and H. R. Menapace, J. Catal., 1973, 28, 300–303 CrossRef CAS.
- W. Xu, C. Lin, H. Liu, H. B. Yu, K. Tao and S. H. Zhou, RSC Adv., 2015, 5, 23981–23989 RSC.
- K. Tao, Q. X. Ma, N. Tsubaki, S. H. Zhou and L. Han, J. Mol. Catal. A: Chem., 2016, 416, 39–46 CrossRef CAS.
- W. L. Jiang, R. L. Huang, P. D. Li, S. Feng, G. L. Zhou, C. C. Yu, H. J. Zhou, C. M. Xu and Q. Xu, Appl. Catal., A, 2016, 517, 227–235 CrossRef CAS.
- N. Gholampour, M. Yusubov and F. Verpoort, Catal. Rev., 2016, 58, 113–156 CAS.
- S. S. Geng, F. Jiang, Y. B. Xu and X. H. Liu, ChemCatChem, 2016, 8, 1303–1307 CrossRef CAS.
- J. Cai, F. Jiang and X. H. Liu, Appl. Catal., B, 2017, 210, 1–13 CrossRef CAS.
- T. Kim, A. Burrows, C. J. Kiely and I. Wachs, J. Catal., 2007, 246, 370–381 CrossRef CAS.
- V. M. Benitez and N. S. Fígoli, Catal. Commun., 2002, 3, 487–492 CrossRef CAS.
- E. Briot, J. Y. Piquemal, M. Vennat, J. M. Brégeault, G. Chottard and J. M. Manoli, J. Mater. Chem., 2000, 10, 953–958 RSC.
- E. I. Ross-Medgaarden and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 15089–15099 CAS.
- C. Martín, G. Solana, V. Rives, G. Marcì, L. Palmisano and A. Sclafani, Catal. Lett., 1997, 49, 235–243 CrossRef.
- B. Hu, H. Liu, K. Tao, C. R. Xiong and S. H. Zhou, J. Phys. Chem. C, 2013, 117, 26385–26395 CAS.
- C. Martín, G. Solana, P. Malet and V. Rives, Catal. Today, 2003, 78, 365–376 CrossRef.
- L. Harmse, C. van Schalkwyk and E. van Steen, Catal. Lett., 2010, 137, 123–131 CrossRef CAS.
- N. Poovarawan, K. Suriye, J. Panpranot, W. Limsangkass, F. J. Cadete Santos Aires and P. Praserthdam, Catal. Lett., 2015, 145, 1868–1875 CrossRef CAS.
- S. Maksasithorn, P. Praserthdam, K. Suriye and D. P. Debecker, Microporous Mesoporous Mater., 2015, 213, 125–133 CrossRef CAS.
- D. Hunyadi, I. Sajó and I. M. Szilágyi, J. Therm. Anal. Calorim., 2013, 116, 329–337 CrossRef.
- X. L. Yang, R. Gao, W. L. Dai and K. N. Fan, J. Phys. Chem. C, 2008, 112, 3819–3826 CAS.
- C. Lin, K. Tao, H. B. Yu, D. Y. Hua and S. H. Zhou, Catal. Sci. Technol., 2014, 4, 4010–4019 CAS.
- J. K. Jeon, H. Lee, J. H. Yim, Y. S. Kim, S. J. Lee, Y. K. Park, J. K. Shon and J. M. Kim, Catal. Lett., 2007, 119, 179–184 CrossRef CAS.
- X. L. Yang, W. L. Dai, R. H. Gao and K. N. Fan, J. Catal., 2007, 249, 278–288 CrossRef CAS.
- K. Amakawa, S. Wrabetz, J. Kröhnert, G. Tzolova-Müller, R. Schlögl and A. Trunschke, J. Am. Chem. Soc., 2012, 134, 11462–11473 CrossRef CAS PubMed.
- S. Doniach and M. Sunjic, J. Phys. Chem. C, 2001, 3, 285–291 Search PubMed.
- A. P. Shpak, A. M. Korduban, M. M. Medvedskij and V. O. Kandyba, J. Electron Spectrosc. Relat. Phenom., 2007, 156, 172–175 CrossRef.
- J. C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC.
- S. J. Huang, S. L. Liu, Q. J. Zhu, X. X. Zhu, W. J. Xin, H. J. Liu, Z. C. Feng, C. Li, S. J. Xie, Q. X. Wang and L. Y. Xu, Appl. Catal., A, 2007, 323, 94–103 CrossRef CAS.
- J. Howell, Y. P. Li and A. T. Bell, ACS Catal., 2016, 6, 7728–7738 CrossRef CAS.
- C. Martín, P. Malet, A. G. Solana and V. Rives, J. Phys. Chem. B, 1998, 102, 2759–2768 CrossRef.
- H. Liu, K. Tao, H. B. Yu, C. Zhou, Z. Ma, D. S. Mao and S. H. Zhou, C. R. Chim., 2015, 18, 644–653 CrossRef CAS.
- K. Ding, A. Gulec, A. M. Johnson, T. L. Drake, W. Wu, Y. Lin, E. Weitz, L. D. Marks and P. C. Stair, ACS Catal., 2016, 6, 5740–5746 CrossRef CAS.
- P. Arudra, T. I. Bhuiyan, M. N. Akhtar, A. M. Aitani, S. S. Al-Khattaf and H. Hattori, ACS Catal., 2014, 4, 4205–4214 CrossRef CAS.
- J. F. Wu, A. Ramanathan and B. Subramaniam, J. Catal., 2017, 350, 182–188 CrossRef CAS.
- A. G. Basrur, S. R. Patwardhan and S. N. Was, J. Catal., 1991, 127, 86–95 CrossRef CAS.
- G. Chen, M. Dong, J. F. Li, Z. W. Wu, G. F. Wang, Z. F. Qin, J. G. Wang and W. B. Fan, Catal. Sci. Technol., 2016, 6, 5515–5525 CAS.
- Z. Cheng and C. S. Lo, ACS Catal., 2012, 2, 341–349 CrossRef CAS.
- Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef PubMed.
- J. L. Hérisson and Y. Chauvin, Macromol. Chem. Phys., 1971, 141, 161–176 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13776a |
| This journal is © The Royal Society of Chemistry 2018 |