
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
UV-mediated hydrophosphinylation of unactivated alkenes with phosphinates under batch and flow conditions†
Fabien Gelat*,
Maxime Roger,
Christophe Penverne,
Ahmed Mazzad,
Christian Rolando and
Laëtitia Chausset-Boissarie*
and
Laëtitia Chausset-Boissarie*
USR 3290, MSAP, Miniaturisation pour la Synthèse l'Analyse et la Protéomique and FR 2638, Institut Eugène-Michel Chevreul, Université de Lille, F-59000 Lille, France. E-mail: Laetitia.chausset-boissarie@univ-lille1.fr; Fabien.gelat@univ-lille1.fr
First published on 23rd February 2018
Abstract
A UV-mediated hydrophosphinylation of unactivated alkenes with H-phosphinates and hypophosphorous acid under radical free conditions is presented. The reaction affords selectively a large number of structurally diverse organophosphorous compounds in moderate to good yields under mild reaction conditions in the presence of an organic sensitizer as catalyst irradiated by UV-A LEDs. Furthermore, the high yielding hydrophosphinylation in continuous flow is disclosed.
Introduction
Organophosphorus compounds have attracted much attention due to their wide range of applications in materials, catalysis, natural bioactive products and pharmaceuticals.1 In particular, phosphinates (P(O)(OR)R1R2, R1/R2 = hydrogen and/or carbon) have been the target of numerous synthetic efforts as they are versatile precursors to organophosphorus compounds and represent a sustainable alternative to the use of phosphorous trichloride.2 Indeed, phosphinates can be considered as the synthetic equivalents of phosphorous trichloride with several advantages in terms of stability and toxicity. In this regard, the development of efficient and selective methodologies for the preparation and functionalization of phosphinates (P(O)(OR)H2) and H-phosphinates (P(O)(OR)R1H) has been extensively studied, mostly by the group of Montchamp.2,3 Hydrophosphinylation of unactivated alkenes is one of these routes that provide a direct access to functionalized organophosphorous compounds.4 This atom economical process has been reported to proceed via transition metal catalysis,5 or radical activation (Scheme 1a and b).6 Despite the significant progress in this chemistry, these transformations suffer from some drawbacks like the relatively harsh conditions, narrow substrates scopes and costly catalytic system thus limiting their chemical and pharmaceutical applications. Recently, Lakhdar and co-workers reported a hydrophosphinylation of unactivated alkenes with ethyl and butyl phosphinates under photocatalytic conditions (Scheme 1c),7 even though diphenyl iodonium triflate which acted as sacrificial oxidant is not a heavy metal, the use of catalytic quantity is always preferred in term of sustainability and environmental benignity. Alternatively, pioneering work from the group of Dondoni8 demonstrated that P(O)-centered radical can be generated from H-phosphonates under UV-A irradiation, in the presence of 2,2-dimethoxy-2-phenylacetophenone (DMPA) as photoinitiator, and added to alkene functionalized carbohydrates. However, the reaction requires the use of a large excess of phosphonates (100 eq.). Mathé and coworkers9 then extended this free radical hydrophosphonylation to activated and unactivated alkenes, similar reaction with hypophosphorous acid and H-phosphinates derivatives10 have not been studied previously. | ||
| Scheme 1 Hydrophosphinylation of phosphinates with unactivated alkenes. | ||
In this context, we envisaged that continuous-flow systems in combination with a sensitizer irradiated by UV-A LEDs (λ = 365 nm) would result in a significant enhancement of the reaction. Over the last decade, interest has grown for flow chemistry based on microfluidic technology in particular towards large-scale application due to its significant improvement over traditional batch reactors concerning reduced consumption of chemicals, solvents and time together with enhanced yields, selectivity and control over reaction conditions. An easy scale up is also one of the characteristic of photo-reactions which are conducted in microreactors. In batch reactors, light penetration through the reaction media is limited which restrains the efficiency of photochemical processes. This drawback can be overcome with continuous microflow reactors since their small optical lengths improve sample irradiation and also enhance heat and mass transfer.11 Therefore, in view of the growing demand to develop efficient, mild and sustainable methods to access phosphinates and as a continuation of our interest in photocatalyzed processes,12 we herein report an original UV-mediated hydrophosphinylation of unactivated alkenes with hypophosphorous acid and less reactive H-phosphinates derivatives under batch and flow conditions.
Results and discussion
Hydrophosphinylation of unactivated alkenes with H-phosphinates under batch conditions
Initially, the hydrophosphinylation of relatively challenging H-phosphinate (1a) with octene (2a) in equimolar amounts in the presence of a photoinitiator under UV-A irradiation was selected as a model to investigate the reaction. The use of one equivalent of 4,4′-dimethoxybenzophenone (4,4′-DMBP) was found to be effective furnishing 3a after stirring in acetonitrile under an ambient inert atmosphere for 3 h (Table 1, entry 1, 42%). An investigation of solvents showed that the reaction efficiency was further enhanced when performed in DMSO (Table 1, entry 4, 69%) or acetic acid6b,13 (entry 5, 70%) as previously observed for radical process with H-phosphinates, whereas moderate yields were obtained with ethyl acetate (entry 2, 46%) or DMF (entry 3, 52%). Importantly a very low catalyst loading could promote the reaction yield up to 84% (entries 6 and 7). Moreover, the loading could be further decreased but a prolonged reaction time was required (Table 1, entry 8, 86%). Interestingly, the photoinitiator DMPA (2,2-dimethoxy-2-phenylacetophenone) was not efficient in our case since the phosphorylated product was formed in a moderate yield (Table 1, entry 15, 32%). Furthermore, none of the common photoinitiators (thioxantone, 4-methoxyacetophenone, benzophenone) exhibited a better catalytic effect (Table 1, entries 12–14). Control experiments revealed that the photoinitiator, inert conditions and light were essential for this reaction (Table 1, entries 9–11). It is noteworthy that using a 4,4′-DMBP derivative functionalized with an ionic liquid moiety14 (4,4′-(2-(1-methylimidazolium)ethoxy)benzophenone dibromide, DIBP) as photosensitizer led to the same conversions as previously observed furnishing an excellent way to simplify the purification procedure by simple washing of the reaction media thus improving the synthetic efficiency by avoiding chromatography (Table 1, entry 16).
|
|||||
|---|---|---|---|---|---|
| Entry | Photoinitiator | Equivalent | Solvent | Time (h) | Yieldb (%) |
| a Reaction condition: 1a (0.3 mmol, 1 equiv.), 2a (0.3 mmol, 1 equiv.), photoinitiator (x equiv.), solvent ([0.1 M]), under nitrogen and UV-A LED irradiation (λ = 365 ± 15 nm, 230 mW cm−2) at room temperature.b Derived from 31P crude NMR spectra on integration of all formed species.c 4,4′-DMBP = 4,4′-dimethoxybenzophenone.d Without irradiation.e Under ambient atmosphere.f 4-MAP = 4-methoxyacetophenone.g DMPA = 2,2-dimethoxy-2-phenylacetophenone.h DIBP = 4,4′-(2-(1-methylimidazolium)ethoxy)benzophenone dibromide.i The isolated yield is shown in parentheses. | |||||
| 1 | 4,4′-DMBPc | 1 | MeCN | 3 | 42 |
| 2 | 4,4′-DMBP | 1 | EtOAc | 3 | 46 |
| 3 | 4,4′-DMBP | 1 | DMF | 3 | 52 |
| 4 | 4,4′-DMBP | 1 | DMSO | 3 | 69 |
| 5 | 4,4′-DMBP | 1 | AcOH | 3 | 70 |
| 6 | 4,4′-DMBP | 0.5 | DMSO | 3 | 72 |
| 7 | 4,4′-DMBP | 0.1 | DMSO | 5 | 84 |
| 8 | 4,4′-DMBP | 0.05 | DMSO | 17 | 86 |
| 9 | — | — | DMSO | 5 | 10 |
| 10d | 4,4′-DMBP | 0.1 | DMSO | 5 | — |
| 11e | 4,4′-DMBP | 0.1 | DMSO | 5 | 5 |
| 12 | Thioxantone | 0.1 | DMSO | 5 | 76 |
| 13 | 4-MAPf | 0.1 | DMSO | 5 | 19 |
| 14 | Benzophenone | 0.1 | DMSO | 5 | 77 |
| 15 | DMPAg | 0.1 | DMSO | 5 | 32 |
| 16 | DIBPh | 0.1 | DMSO | 16 | 79 (76)i |
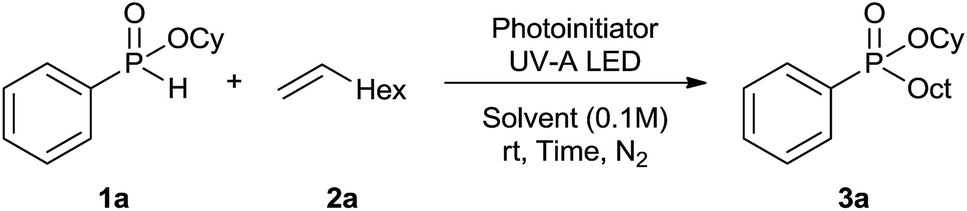
The generality of the method was investigated, with the scope of the reaction being explored with respect to the H-phosphinate component, and the results are summarized in Table 2. Various P(O)–H compounds were employed to react with 1-octene 2a giving the corresponding 3a–g with isolated yields ranging from 30 to 76% (Table 2, entries 1–7). As expected, in all of the cases the anti Markovnikov product was observed. The reaction is not limited to H-phosphinate esters as H-phosphinate acid 1b could also deliver the desired product with a good yield (entry 2, 64%) in contrast to H-phosphinate acid 1c whose corresponding phosphinate acid product 3c was difficult to obtain in high purity (entry 3, 30%). However, H-phosphinate including unactivated alkyl and benzyl derivatives reacted successfully regardless of the ester chosen (Table 2, entries 4–6). When using (hydroxymethyl)-H-phosphinate ester, which was reported to have several application,15 the branched product 3g was obtained with a significant yield of 75% (Table 2, entry 7). To further evaluate the substrate scope, a series of alkenes were tested with H-phosphinate 1a (Table 2, entries 8–12). The reaction with allyl benzene gave the corresponding product 3h with 48% of yield (Table 2, entry 8). Interestingly, 1a reacted with hindered alkene giving 3i with a modest yield of 27% (Table 2, entry 9). Note that halogens like Br and alcohol groups were well tolerated which indicates a potential for further functionalization (Table 2, entries 10–12).
|
||||
|---|---|---|---|---|
| Entry | H-Phosphinate 1 | Alkene 2 | Product 3 | Yield (%) |
| a Reaction condition: all reactions unless specified were carried out using 1 (0.3 mmol, 1 equiv.), 2 (0.3 mmol, 1 eq.), DIBP (0.1 equiv.) in DMSO ([0.1 M]) under nitrogen and UV-A LED irradiation (λ = 365 ± 15 nm, 230 mW cm−2) at room temperature for 16 h, isolated yield.b Derived from 31P crude NMR spectra on integration of all formed species.c (0.5 equiv.) of DIBP. | ||||
| 1 |  |
1-Octene 2a |  |
76 |
| 2 |  |
1-Octene 2a | 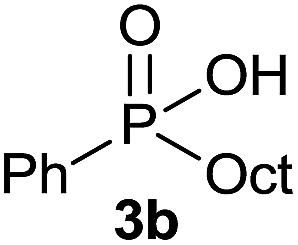 |
64 |
| 3 |  |
1-Octene 2a |  |
30b |
| 4 |  |
1-Octene 2a |  |
73 |
| 5 |  |
1-Octene 2a | 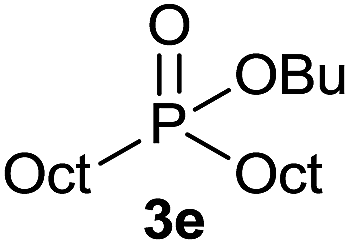 |
71 |
| 6c |  |
1-Octene 2a |  |
51 |
| 7 | 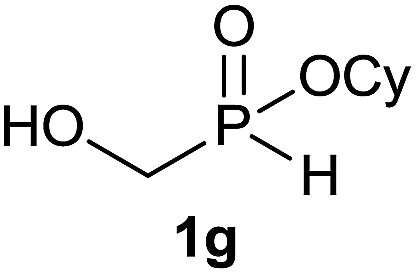 |
1-Octene 2a |  |
75 |
| 8 |  |
 |
 |
48 |
| 9c |  |
 |
 |
27 |
| 10 |  |
 |
 |
38 |
| 11c |  |
 |
 |
56 |
| 12c |  |
 |
 |
69 |

Although the detailed reaction mechanism is unclear, based on literature precedents6i,16 all these results could be well explained by a radical chain mechanism as depicted in Scheme 2. Upon irradiation, the excited state of the photoinitiator17 abstracts a proton from H-phosphinates to form the phosphoryl radical18 which adds to the terminal carbon of the alkenes. The carbon centered radical can then abstract a proton from H-phosphinate 1 to regenerate the phosphoryl radical.
 | ||
| Scheme 2 Proposed reaction mechanism for the photoinduced hydrophosphinylation of unactivated alkenes with H-phosphinates. | ||
Hydrophosphinylation of unactivated alkenes with H-phosphinates under flow conditions
With the successful results for the hydrophosphinylation under batch conditions, the reaction was then performed under continuous flow to further improve its efficiency in a shorter amount of time. The continuous flow hydrophosphinylation was performed using a continuous flow microfluidic system composed of a high pressure syringe pump delivering the homogeneous reaction mixture at specific flow rates to a commercially available microreactor (Mikroglas Dwell Device® microreactor from Invenios Europe, Langen, Germany) irradiated by HP UV-A lamp. This microreactor is made up of Foturan® glass that is transparent up to 300 nm allowing to work at a wide range of wavelengths (UV-A & visible).We were delighted to find that H-phoshinate derivatives (3a, b, k) could be obtained with 4,4′-DMBP as photoinitiator within a shorter reaction time than in batch (30 min vs. 5 h), indicating the importance of the short light path length provided by the microreactor, with slightly higher isolated yield, highlighting the potentials of this process (Scheme 3). This results offer the possibility to conduct the reaction on large scale without erosion of the yields.
 | ||
| Scheme 3 Hydrophosphinylation of alkenes with H-phosphinates 1 under batch and flow conditions. (a) Reactions conditions in batch: 1 (0.3 mmol, 1 equiv.), 2 (0.3 mmol, 1 eq.), 4,4′-DMPB (0.1 equiv.) in DMSO ([0.1 M]) under nitrogen and UV-A LED irradiation (λ = 365 ± 15 nm, 230 mW cm−2) at room temperature, 5 h, isolated yield. (b) Reaction conditions in continuous flow: 1 (0.3 mmol, 1 equiv.), 2 (0.3 mmol, 1 equiv.), 4,4′-DMPB (0.1 equiv.) in DMSO ([0.1 M]) under nitrogen and UV-A LED irradiation (λ = 365 ± 15 nm, 230 mW cm−2) with Dwell device manufactured by Mikroglass Chemtech Mainz, Germany with a rectangular shape of dimensions 115 mm × 2 mm × 0.5 mm as photo-microreactor, at room temperature for 30 min of residence time, isolated yield. | ||
Hydrophosphinylation of unactivated alkenes with hypophosphorous acid under batch conditions
To further demonstrate the utility of our protocol, we sought to test its potential for the hydrophosphinylation of various alkenes with hypophosphorous acid to form the previous precursor H-phosphinate acid (Scheme 4). | ||
| Scheme 4 Hydrophosphinylation of alkenes with hypophosphorous acid. All reactions (unless otherwise specified) were carried out using H3PO2 (0.3 mmol, 2 equiv.), 2 (0.15 mmol, 1 equiv.), DMPA (0.2 equiv.) in DMSO ([0.1 M]) under UV-A LED irradiation (λ = 365 ± 15 nm, 230 mW cm−2) at room temperature for 16 h. aDerived from 31P crude NMR spectra on integration of all formed species. bIsolated yield. | ||
A slight modification of the previous batch conditions (see Table 1, ESI†) allowed us to obtain the corresponding H-phosphinates 1 with 31P NMR yield ranging from 49 to 96%. A 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of hypophosphorous acid–alkenes was necessary to obtain a high conversion without the formation of the disubstituted by-product. However despite the observed high conversions, isolated yields were modest due to difficult purifications. In all of the cases, the reaction was chemoselective where only mono substituted phosphinates were observed. As for H-phosphinate, the reaction proceeded well with terminal (1c–h) and cyclic alkenes (1j). Hindered alkenes delivered the desired compounds like 1i with a moderate yield of 31%, which is still higher than that obtained with H-phosphinate ester. H-Phosphinate acid 1k however was difficult to obtain in high purity. Similar to the first reaction assessed, the reaction tolerates functional groups such as amines or alcohols (1l–n) but the obtained H-phosphinate acids were not isolated. Although the scope of this reaction seems to be limited, H-phosphinate acids could be easily esterified19 in situ and therefore the resulting procedure offers significant synthetic advantages.
:1 molar ratio of hypophosphorous acid–alkenes was necessary to obtain a high conversion without the formation of the disubstituted by-product. However despite the observed high conversions, isolated yields were modest due to difficult purifications. In all of the cases, the reaction was chemoselective where only mono substituted phosphinates were observed. As for H-phosphinate, the reaction proceeded well with terminal (1c–h) and cyclic alkenes (1j). Hindered alkenes delivered the desired compounds like 1i with a moderate yield of 31%, which is still higher than that obtained with H-phosphinate ester. H-Phosphinate acid 1k however was difficult to obtain in high purity. Similar to the first reaction assessed, the reaction tolerates functional groups such as amines or alcohols (1l–n) but the obtained H-phosphinate acids were not isolated. Although the scope of this reaction seems to be limited, H-phosphinate acids could be easily esterified19 in situ and therefore the resulting procedure offers significant synthetic advantages.
Experimental section
General information
All reagents were purchased from commercial suppliers (Strem Chemicals Inc., Sigma-Aldrich or Alfa Aesar) and were used without further purification. Thin-layer chromatography (TLC) was performed on Silica gel 60 F254 plates (Merck) and visualized under UV (254 nm) or by staining with potassium permanganate or phosphomolybdic acid. Column chromatography was performed with 63–200 mesh silica gel. NMR spectra were recorded on a Bruker AVANCE 300 spectrometer at 300 MHz (75 MHz). Chemical shifts are reported in parts per million relative to solvent signal and coupling constants are reported in hertz (Hz). High-resolution mass spectra (HRMS) were performed on a Thermo LTQ Orbitrap mass spectrometer using nanoESI ionization. H-Phosphinates derivatives 1 and 4,4′-(2-(1-methylimidazolium)ethoxy)benzophenone dibromide (DIBP)14 were prepared according to the reported procedure. The illumination was performed by UV-A LEDs (365 nm, irradiance = 230 mW cm−2) Omnicure® AC475 model from Lumen Dynamics (Excelitas Technologies, Waltham, MA, USA). Note that the irradiance was measured at the surface of the reactor using a radiometer.General procedure A for the hydrophosphinylation with H-phosphinate 1 derivatives under batch conditions
A solution of a selected H-phosphinate 1 (0.3 mmol, 1 equiv.), an alkene 2 (0.3 mmol, 1 equiv.) and DIPB (0.03–0.15 mmol, 10–50 mol%) in degazed DMSO (1.5 mL) was irradiated under 365 nm and N2 for 16 h. Ethyl acetate (30 mL) was added and the reaction mixture was washed with saturated solution of NaHCO3 (2 × 15 mL) and brine (2 × 15 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduce pressure. The residue was then purified by flash chromatography on silica gel if necessary to afford the corresponding organophosphorous compound.General procedure B for the hydrophosphinylation with hypophosphorous acid
To a 50% aqueous solution of hypophosphorous acid (79.2 mg, 0.6 mmol, 2 equiv.) in DMSO (3 mL) was added an alkene 2 (0.3 mmol, 1 equiv.), 4,4′-DMPA (15.5 mg, 0.06 mmol, 20 mol%). The reaction mixture was irradiated under 365 nm and ambient atmosphere for 16 h. Ethyl acetate (30 mL) was added and the organic layer was washed with brine (3 × 15 mL). To the organic layer a 0.03 M solution of NaHCO3 (30 mL, 1 mmol) was added. The aqueous layer was washed with ethyl acetate (2 × 15 mL), acidified with 1 M HCl (2 mL, 2 mmol), saturated with NaCl and extracted with ethyl acetate (30 mL). The last ethyl acetate layer was dried over Na2SO4, filtered and concentrated under reduce pressure. The residue was then purified by flash chromatography on silica gel using 9:1:0.5 DCM–MeOH–AcOH as the eluant to afford the desired pure H-phosphinate acid 1.
Hydrophosphinylation of cyclohexyl phenyl-H-phosphinate 1a under flow condition
A solution of a cyclohexyl phenyl-H-phosphinate20 1a (67.2 mg, 0.3 mmol, 1 equiv.), 1-octene 2a (33.6 mg, 0.3 mmol, 1 equiv.) and 10 mol% of 4,4′-dimethoxybenzophenone (7.2 mg) in degazed DMSO (1.5 mL) was pumped through the Mikroglas Dwell Device reactor (Vint = 1.15 mL) at 38.33 μL min−1, residence time of 30 min and irradiated by UV-A LEDs. Ethyl acetate (30 mL) was added to the collected reaction mixture and the reaction mixture was washed with saturated solution of NaHCO3 (2 × 15 mL) and brine (2 × 15 mL). To the organic layer was dried over MgSO4, filtered and the solvent was removed under reduce pressure to afford 3a (85.6 mg, 0.25 mmol, 85%) as a colorless oil.:1 of the both diastereoisomers). 31P NMR (121 MHz, CDCl3): δ = 42.7 (s), 46.3 (s); 1H NMR (300 MHz, CDCl3): δ = 7.76–7.66 (m, 2H), 7.49–7.34 (m, 3H), 4.26–4.10 (m, 1H), 2.31–1.48 (m, 7H), 1.42–1.03 (8H), 0.96–0.79 (m, 6H); 13C NMR (75 MHz, CDCl3): δ = 132.3 (d, JPCC = 5.1 Hz, 2 × 0.5C), 132.2 (d, JPCC = 5.2 Hz, 2 × 0.5C), 131.9 (d, JPCCCC = 2.2 Hz, 0.5C), 131.9 (d, JPCCCC = 2.2 Hz, 0.5C), 128.5 (d, JPCCC = 3.2 Hz, 2 × 0.5C), 128.3 (d, JPCCC = 3.1 Hz, 2 × 0.5C), 74.2 (d, JPOC = 7.2 Hz, 0.5C), 74.0 (d, JPOC = 7.1 Hz, 0.5C), 39.6 (d, JPC = 100 Hz 2 × 0.5C), 34.4 (d, JPOCC = 2.4 Hz, 0.5C), 34.4 (d, JPOCC = 2.3 Hz, 0.5C), 33.8 (d, JPOCC = 1.9 Hz, 0.5C), 33.8 (d, JPOCC = 1.9 Hz, 0.5C), 26.8 (d, JPCCC = 2.8 Hz, 0.5C), 26.1 (0.5C), 25.4 (2 × 0.5C), 23.8 (2 × 0.5C), 23.7 (2 × 0.5C), 22.6 (d, JPCC = 6.1 Hz, 0.5C), 22.4 (d, JPCC = 8.7 Hz, 0.5C), 18.3 (d, JPCC = 3.2 Hz, 0.5C), 17.7 (d, JPCCC = 2.2 Hz, 0.5C), 7.7 (0.5C), 6.9 (d, JPCC = 4.6 Hz, 0.5C) (1 signal missing); HRMS (ESI) m/z calcd for C17H28O2P ([M + H]+) 295.1821, found 295.1819.Conclusions
In conclusion, we developed an efficient UV-mediated hydrophosphinylation of unactivated alkenes with H-phosphinate under free radical conditions giving a straightforward access to a wide range of phosphorous compounds providing a metal free alternative for the preparation and functionalization of P(O)–H bonds. This reaction can be carried out using an ionic liquid soluble photosensitizer while maintaining good yield. Furthermore, the reaction with hypophosphorous acid is disclosed for the first time. Finally, a continuous flow hydrophosphinylation was also reported which allowed faster reaction time with high yields, highlighting the potential of our methodology for rapid scale up and in-line synthesis. Taking into account the simplicity of our reaction conditions we believe this procedure will be appealing for further chemical and pharmaceutical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the NMR and Mass Spectrometry facilities funded by the European Community (FEDER) and Région Haut de France (France), the CNRS, and the Université de Lille, Sciences et Technologies. The authors thank Dr Nassim El Achi for her help with editing this manuscript.Notes and references
- (a) L. D. Quin, A Guide to Organophosphorous Chemistry, Wiley Interscience, New York, 2000 Search PubMed; (b) New Aspects in Phosphorous Chemistry, ed. J.-P. Majoral, Springer, Berlin, 2002–2005, vol. 1–5 Search PubMed; (c) D. E. C. Corbridge, in Phosphorus: Chemistry, Biochemistry and Technology; CRC Press: London, edn 6, 2013 Search PubMed.
- J.-L. Montchamp, Acc. Chem. Res., 2014, 47, 77 CrossRef CAS PubMed.
- For selected reviews see: (a) O. Berger and J.-L. Montchamp, Beilstein J. Org. Chem., 2014, 10, 732 CrossRef PubMed; (b) N. Z. Kiss and G. Keglevich, Curr. Org. Chem., 2014, 18, 2673 CrossRef CAS; (c) J.-L. Montchamp, Top. Curr. Chem., 2015, 361, 217 CrossRef CAS PubMed.
- (a) L. Coudray and J.-L. Montchamp, Eur. J. Org. Chem., 2008, 3601 CrossRef CAS PubMed; (b) X.-Q. Pan, J.-P. Zou, W.-B. Yi and W. Zhang, Tetrahedron, 2015, 71, 7481 CrossRef CAS.
- (a) C. Petit, F. Fécourt and J.-L. Montchamp, Adv. Synth. Catal., 2011, 353, 1883 CrossRef CAS; (b) S. Deprele and J.-L. Montchamp, J. Am. Chem. Soc., 2002, 124, 9386 CrossRef CAS PubMed; (c) P. Ribiere, K. Bravo-Altamirano, M. I. Antczak, J. D. Hawkins and J.-L. Montchamp, J. Org. Chem., 2005, 70, 4064 CrossRef CAS PubMed; (d) S. Ortial, H. C. Fisher and J.-L. Montchamp, J. Org. Chem., 2013, 78, 6599 CrossRef CAS PubMed.
- (a) E. E. Nifant'ev, R. K. Magdeeva and N. P. Shchepet'eva, J. Gen. Chem., 1980, 50, 1416 Search PubMed; (b) S. Deprele and J.-L. Montchamp, J. Org. Chem., 2001, 66, 6745 CrossRef CAS PubMed; (c) O. Dubert, A. Gautier, E. Condamine and S. R. Piettre, Org. Lett., 2002, 4, 359 CrossRef CAS PubMed; (d) A. Gautier, G. Garipova, C. Salcedo, S. Balieu and S. R. Piettre, Angew. Chem., Int. Ed., 2004, 43, 5963 CrossRef CAS PubMed; (e) M. I. Antczak and J.-L. Montchamp, Synthesis, 2006, 3080 CAS; (f) Y. Feng and J. K. Coward, J. Med. Chem., 2006, 49, 770 CrossRef CAS PubMed; (g) T. Hirai and L.-B. Han, Org. Lett., 2007, 9, 53 CrossRef CAS PubMed; (h) H. C. Fisher, O. Berger, F. Gelat and J.-L. Montchamp, Adv. Synth. Catal., 2014, 356, 1199 CrossRef CAS; (i) P. Troupa, G. Katsiouleri and S. Vassiliou, Synlett, 2015, 26, 2714 CrossRef CAS; (j) Z. Li, F. Fan, Z. Zhang, Y. Xiao, D. Liu and Z.-Q. Liu, RSC Adv., 2015, 5, 27853 RSC.
- G. Fausti, F. Morlet-Savary, J. Lalevee, A.-C. Gaumont and S. Lakhdar, Chem.–Eur. J., 2017, 23, 2144 CrossRef CAS PubMed.
- A. Dondoni, S. Staderini and A. Marra, Eur. J. Org. Chem., 2013, 5370 CrossRef CAS.
- P.-Y. Geant, B. S. Mohamed, C. Perigaud, S. Peyrottes, J.-P. Uttaro and C. Mathe, New J. Chem., 2016, 40, 5318 RSC.
- During the preparation of the manuscript a similar approach was developed: P.-Y. Geant, J.-P. Uttaro, S. Peyrottes and C. Mathé, Eur. J. Org. Chem., 2017, 2850 Search PubMed.
- For recent reviews on flow photochemistry, see: (a) Z. J. Garlets, J. D. Nguyen and C. R. J. Stephenson, Isr. J. Chem., 2014, 54, 351 CrossRef CAS PubMed; (b) Y. H. Su, N. J. W. Straathof, V. Hessel and T. Noel, Chem.–Eur. J., 2014, 20, 10562 CrossRef CAS PubMed; (c) D. Cambie, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noel, Chem. Rev., 2016, 116, 1023 CrossRef PubMed.
- (a) M. Penhoat, T. Vanbésien, A. Cocud, A. Addad, H. Vezin and C. Rolando, New J. Chem., 2016, 40, 9460 RSC; (b) N. El Achi, M. Penhoat, Y. Bakkour, C. Rolando and L. Chausset-Boissarie, Eur. J. Org. Chem., 2016, 4284 CrossRef CAS; (c) N. El Achi, Y. Bakkour, L. Chausset-Boissarie, M. Penhoat and C. Rolando, RSC Adv., 2017, 7, 29815 RSC.
- (a) F. Effenberger and H. Kottmann, Tetrahedron, 1985, 41, 4171 CrossRef CAS; (b) O. Berger and J.-L. Montchamp, Chem.–Eur. J., 2014, 20, 12385 CrossRef CAS PubMed.
- S. C. Hubbard and P. B. Jones, Tetrahedron, 2005, 61, 7425 CrossRef CAS.
- O. Berger, L. Gavara and J.-L. Montchamp, Org. Lett., 2012, 14, 3404 CrossRef CAS PubMed.
- (a) O. Tayama, A. Nakano, T. Iwahama, S. Sakaguchi and I. Yasutaka, J. Org. Chem., 2004, 69, 5494 CrossRef CAS PubMed; (b) S.-I. Kawaguchi, A. Nomoto, M. Sonoda and A. Ogawa, Tetrahedron Lett., 2009, 50, 624 CrossRef CAS; (c) W.-J. Yoo and S. Kobayashi, Green Chem., 2013, 15, 1844 RSC.
- (a) A. Beckett and G. Porter, Faraday Trans., 1963, 59, 2038 RSC; (b) G. Porter and P. Suppan, Faraday Trans., 1965, 61, 1664 RSC; (c) J. Lalevee, F. Morlet-Savary, M. A. Tehfe, B. Graff and J. P. Fouassier, Macromolecules, 2012, 45, 5032 CrossRef CAS; (d) M. Marazzi, S. Mai, D. Roca-Sanjuán, M. l. G. Delcey, R. Lindh, L. González and A. Monari, J. Phys. Chem. Lett., 2016, 7, 622–626 CrossRef CAS PubMed; (e) G. R. Dormán, H. Nakamura, A. Pulsipher and G. D. Prestwich, Chem. Rev., 2016, 116, 15284 CrossRef PubMed.
- C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 11, 7981 CrossRef PubMed.
- S. Deprèle and J.-L. Montchamp, J. Organomet. Chem., 2002, 643, 154 CrossRef.
- O. Berger, C. Petit, E. L. Deal and J.-L. Montchamp, Adv. Synth. Catal., 2013, 355, 1361 CrossRef CAS.
- All attempts of purification by aqueous work-up or chromatography failed or conducted in a very low yield. Purity was determined by 31P NMR on a sample by mixing it with phenylphosphinic acid as a standard.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectral data. See DOI: 10.1039/c7ra12977g |
| This journal is © The Royal Society of Chemistry 2018 |