
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtective effects of a nanoemulsion adjuvant vaccine (2C-Staph/NE) administered intranasally against invasive Staphylococcus aureus pneumonia†
Liu-yang Yang,
Heng Zhou,
Yun Yang,
Ya-nan Tong,
Liu-sheng Peng,
Bao-hang Zhu,
Wei-bo Diao,
Hao Zeng,
Hong-wu Sun * and
Quan-ming Zou*
* and
Quan-ming Zou*
National Engineering Research Center of Immunological Products, Department of Microbiology and Biochemical Pharmacy, College of Pharmacy, Third Military Medical University, Chongqing 400038, PR China. E-mail: sunhongwu2001@163.com; qmzou2007@163.com
First published on 12th March 2018
Abstract
No licensed Staphylococcus aureus (S. aureus) vaccine is currently available. To develop an effective S. aureus vaccine, we selected the recombinant proteins staphylococcal enterotoxin B (rSEB) and manganese transport protein C (rMntC) as vaccine candidates and formulated a 2C-Staph vaccine. Based on the optimised formation of nanoemulsion (NE) technology, we constructed a novel NE adjuvant vaccine, 2C-Staph/NE. The 2C-Staph/NE particles showed a suitable diameter (24.9 ± 0.14 nm), a good protein structure of integrity and specificity, and high thermodynamic stability. 2C-Staph formulated with an NE adjuvant induced higher survival rates than a 2C-Staph/MF59 vaccine in sepsis and pneumonia models. Moreover, intramuscular vaccination with 2C-Staph/NE yielded protection efficacy in a sepsis model, and the intranasal vaccination route induced a potent protective effect in a pneumonia model. Intranasal vaccination with 2C-Staph/NE induced a strong mucosal response with high levels of IgA and IL-17A in bronchoalveolar lavage fluid (BALF), and the IgG levels in the BALF were comparable to those induced by the intramuscular vaccination route. Furthermore, the serum and BALF induced by intranasal administration showed potent opsonophagocytic activity against S. aureus. And, the IL-17A played a protective role in the pneumonia model demonstrated by a cytokine neutralization test. Taken together, our results showed that intranasal administration of 2C-Staph formulated with an NE adjuvant yielded ideal protection in a murine S. aureus pneumonia model.
1. Introduction
Staphylococcus aureus (S. aureus) is a Gram-positive bacterium that causes a variety of infections, including sepsis, pneumonia and skin soft-tissue infections.1 This bacterium acquires antibiotic resistance rapidly, and multi-drug resistant strains are emerging in medical care centres.2 In attempts to control the S. aureus infections, researchers tried many strategies to explore the effective antimicrobial agents, such as antimicrobial peptides (e.g. β-defensins, LL-37 and CAP18),3,4 metal nanoparticles (e.g. silver nanoparticles, titanium dioxide nanoparticles and gold nanoparticles)5–7 and metal nanoclusters (e.g. silver nanoclusters and gold nanoclusters).8,9 While these effective antimicrobial agents have improved patient outcomes, additional measures, such as prophylactic vaccine, are needed to prevent S. aureus infection.Although considerable research in recent years has focused on developing an effective S. aureus vaccine, a licensed S. aureus vaccine is not currently available. However, several important lessons for S. aureus vaccine development have been learned: (1) vaccines based on a single antigen are unlikely to be successful, and the use of multiple antigens is more promising;10 (2) novel technologies or adjuvants formulated with antigens may lead to more successful vaccine development;11 and (3) prophylactic vaccines should target several potential patient populations because S. aureus causes a broad range of diseases in different populations.10,12
To survive in different host niches, S. aureus expresses a wide array of virulence factors, including surface proteins and toxins.13 Vaccines based on these toxins or surface proteins have shown protective roles in different murine S. aureus infection models.14,15 Staphylococcal enterotoxin B (SEB), which is a critically conserved secreted toxin, plays a critical role in toxic shock syndrome induced by S. aureus.16 SEB has been regarded as a promising vaccine candidate for active or passive vaccination and has shown protective effects against S. aureus infection.17 Manganese transport protein C (MntC) is essentially a metal-binding protein and has been shown to confer protective immunity in an animal model.18 We previously found that the recombinant proteins rSEB and rMntC show protective effects in murine sepsis models.19,20
Adjuvants play an essential role in S. aureus vaccine components as shown by the failure of V710 (a non-adjuvant antigen vaccine (IsdB), Merck) in a phase III clinical trial.21 Because adjuvants can greatly improve antibody responses and cellular responses, many public and private initiatives have designed S. aureus vaccines containing adjuvants (such as aluminium, AS03 and SMIP.7–10) that are in pre-clinical or clinical trials.11,22 To date, only a few adjuvants have been approved for use in humans.23 Aluminium is a common adjuvant in humans that has been applied intramuscularly to elicit systemic immune responses.24 Emulsions, such as MF59 and AS-series adjuvants, are generally formulated with antigens to increase the efficacy of mucosal vaccines.25,26 However, certain disadvantages accompany the use of these adjuvants: (1) the aluminium adjuvant results in weak cellular immune responses and (2) the emulsion adjuvant is thermodynamically unstable because of its large size (>160 nm).27 Therefore, exploring a suitable adjuvant is critical in the successful vaccine development.
Historically, the development of S. aureus vaccines has primarily focused on pathogen and antigen selection and less on the host. S. aureus causes a broad spectrum of diseases, and immune responses vary among distinct populations.28 S. aureus is an important cause of pneumonia, and it has been implicated in hospitalized patients.29 Prophylactic vaccine maybe an effective method for combating S. aureus pneumonia, but there were no clinical trials related to S. aureus pneumonia yet.10,30 To date, most vaccines for combating S. aureus pneumonia were performed through intramuscular route, but some researchers reported that mucosal vaccines may also provide protective efficacy in S. aureus pneumonia model.31,32 Therefore, novel vaccine formulations and vaccination route may lead to more successful strategy for S. aureus pneumonia.
We previously reported that intramuscular immunisation of rSEB17,19 and rMntC20 in combination with an aluminium adjuvant induced protective effects in a murine S. aureus sepsis model. Consistent with the strategy of combining the toxin and surface proteins, we created the 2C-Staph vaccine by including the rSEB and rMntC proteins together in this study. Additionally, a novel nanoemulsion (NE) adjuvant with 2C-Staph was designed for the first time by low-energy methods. After evaluating the basic characteristics and stability, the intranasal and intramuscular administration of 2C-Staph/NE yielded differential immune protection in murine pneumonia and sepsis models. Moreover, we explored the different immune responses induced by different immunisation routes and evaluated the immune protection in murine pneumonia and sepsis models.
2. Materials and methods
2.1 Bacterial strains and mice
S. aureus strain MRSA 252 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The bacterial were cultured in tryptic soy broth, washed, and diluted with sterile phosphate-buffered saline (PBS) to an appropriate cell concentration determined spectrophotometrically at 600 nm (OD600).Balb/c mice (SPF grade, 6–8 weeks, female) used in this study were obtained from Beijing HFK Bioscience Co. Ltd. (Beijing, China). All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals after being approved by the Animal Ethical and Experimental Committee of the Third Military Medical University of PLA, Chongqing, China. All mice that underwent surgery were treated with sodium pentobarbital anesthesia to minimize suffering.
2.2 Expression and purification of recombinant proteins
The recombinant proteins rSEB and rMntC were prepared according to previously methods.19,20 Briefly, genes were ligated into the pGEX-6P-2 plasmid and expressed in the Escherichia coli BL21 (DE3) strain. The proteins were induced by adding the isopropyl-b-D-1-thiogalactopyranoside (IPTG) at 16 °C overnight, and rSEB and rMntC was expressed as a GST fusion protein. Finally, the proteins were subsequently purified using Capto™ MMC, and endotoxin was removed by the Triton X-114. The protein concentration was determined by the BCA method.2.3 Screening and preparation of the NE adjuvant vaccine
To determine the optimal Smix (mixture of surfactant and co-surfactant, oil), six mixing ratios of the surfactant (Cremophor EL-35, EL-35®) and co-surfactant (propylene glycol) (Smix, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3:1, 4:1, 5:1, 6:1 and 7:1) were evaluated. Combinations of oil (isopropyl myristate, IPM) and Smix (2:8), 200 μg mL−1 (rSEB and rMntC were 100 μg mL−1, respectively) were added drop-wise under gentle agitation to the aqueous phase based on previously described method.33 To determine the oil ratio (mixture of surfactant and co-surfactant) to optimise the Smix, nine different combinations of oil and Smix (1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2, and 9:1) were prepared. The optimal total protein concentrations (0, 100, 200, 300 and 400 μg mL−1) and order of addition were determined by previously described methods.34 Briefly, four orders of addition were evaluated: (1) PSCO-protein, surfactant, co-surfactant and oil; (2) SPCO-surfactant, protein, co-surfactant and oil; (3) SCPO-surfactant, co-surfactant, protein and oil; and (4) SCOP-surfactant, co-surfactant, oil and protein. Key factors, including the particle diameter, zeta potential and Poly dispersal index (PdI), of different NEs were measured by Nano ZS (Malvern, UK) at 25 °C.
:1, 3:1, 4:1, 5:1, 6:1 and 7:1) were evaluated. Combinations of oil (isopropyl myristate, IPM) and Smix (2:8), 200 μg mL−1 (rSEB and rMntC were 100 μg mL−1, respectively) were added drop-wise under gentle agitation to the aqueous phase based on previously described method.33 To determine the oil ratio (mixture of surfactant and co-surfactant) to optimise the Smix, nine different combinations of oil and Smix (1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2, and 9:1) were prepared. The optimal total protein concentrations (0, 100, 200, 300 and 400 μg mL−1) and order of addition were determined by previously described methods.34 Briefly, four orders of addition were evaluated: (1) PSCO-protein, surfactant, co-surfactant and oil; (2) SPCO-surfactant, protein, co-surfactant and oil; (3) SCPO-surfactant, co-surfactant, protein and oil; and (4) SCOP-surfactant, co-surfactant, oil and protein. Key factors, including the particle diameter, zeta potential and Poly dispersal index (PdI), of different NEs were measured by Nano ZS (Malvern, UK) at 25 °C.
According to the optimised Smix, oil ratio, protein concentration and order of addition results, an NE adjuvant vaccine was prepared by the addition of EL-35®, propylene glycol, IPM and antigen protein. This novel vaccine was considered complete when it achieved a low viscosity and clear appearance. The same protocol was used to prepare a blank NE (BNE), with distilled water replacing the protein as the aqueous phase as a control.
2.4 Characterisation and stability analysis of this novel vaccine
The morphology of this novel vaccine (diluted 50 fold with distilled water) was evaluated by transmission electron microscopy (TEM) as previously described.35 The average size, zeta potential and PdI of this novel system were determined with a Nano ZS (Malvern Instruments, Malvern, UK) instrument at 25 °C after a 100-fold dilution with water. Thermodynamic stability testing (13000 × g for 30 min) was subsequently performed as previously described.36 Next, the structural integrity and specificity of this novel NE vaccine were evaluated by SDS-PAGE and western blot analysis as previously described. Importantly, the protein stability was examined by matrix-assisted laser desorption/ionisation-time of flight mass spectrometry (MALDI-TOF MS) with a MALDI-7090 MS (Shimadzu, Japan). Briefly, the rSEB, rMntC, 2C-Staph, BNE, 2C-Staph/NE, and broken vaccine of 2C-Staph/NE were measured. The broken vaccine was generated as follows: two volumes of absolute ethanol were added to approximately 0.3 mL vaccine and shaken to ensure complete emulsification. Finally, long-term stability studies were performed. Samples were divided into three vials after production and stored at room temperature for 180 days. On days 0, 30, 90, and 180, all broken samples were measured by SDS-PAGE after the above treatment.
2.5 Immunisation procedure and S. aureus challenge
The immunisation procedure is consistent with the previous research.37 Briefly, mice were immunized with 10 μg protein by intramuscularly injected or intranasal administration on days 0, 14, and 21. BNE or PBS alone was used as the control. Serum samples were collected on day 28.For the sepsis infection model, BALB/c mice were intravenously infected with 1 × 109 CFUs MRSA 252 (lethal infection dose) on day 35 and monitored for survival for 10 days after infection. For the pneumonia infection model, BALB/c mice were inoculated with a MRSA 252 suspension (1 × 108 CFUs; 20 μL) in the naris on day 35 and monitored for survival for 10 days after infection. For detection the bacterial burdens, mice were inoculated with a MRSA 252 suspension (1 × 107 CFUs; 20 μL) in the naris on day 35. Lungs from corresponding animals were removed, weighed, and homogenised in 2 mL of PBS. Peripheral blood was collected in heparin anticoagulant tubes. All samples used for quantitative cultures were grown on tryptic soy broth for 24 hours at 37 °C.
2.6 Bronchoalveolar lavage fluid (BALF)
Lungs were lavaged five times with 1.0 mL saline supplemented with 3 mM EDTA and 1% fetal bovine serum as previously described.38 The lavage fluid was centrifuged at 3000 rpm for 5 min, then the supernatants were collected, and stored at −80 °C. The supernatants obtained from BALF were analysed for antibody and cytokine levels.2.7 Enzyme linked immunosorbent assay (ELISA)
Serum or BALF samples were used as primary antibodies to coat the wells of microtitre plates (Thermo Lab systems) overnight at 4 °C. Secondary antibodies included HRP-conjugated goat anti-mouse IgG, anti-IgG1, anti-IgG2a, anti-IgG2b, or anti-IgA (Sigma). The antibody responses were monitored at OD450.2.8 Opsonophagocytic killing assay (OPA)
The OPA was performed as previously described.37 Briefly, the HL60 cell line (ATCC, CCL-240) was used for the opsonophagocytic cells, and MRSA 252 was used as the target strain. HL60 cells were maintained in L-glutamine-containing 1640 medium (Corning) supplemented with 10% heat-inactivated FBS (HyClone), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (Gibco) at 37 °C in 5% CO2. In addition, 0.8% N,N-dimethylformamide (Sigma) was used to differentiate the HL60 cells (4 × 105 cells per mL) for 4 days. The viability results of differentiated HL60 cells ≥90% were considered acceptable for the OPA, and trypan blue assays were performed to verify the cell viability. First, 103 CFUs of S. aureus (10 μL) were incubated with the serum or BALF (20 μL) in 96-well round-bottom plates and then shaken on a mini-orbital shaker (700 rpm) at room temperature for 30 min. Then, 4 × 105 HL60 cells (40 μL) and complement (Pel Freeze, 10 μL) were added to 96-well round-bottom plates. After 1 hour of shaking on a mini-orbital shaker (700 rpm) in an incubator (37 °C with 5% CO2), the microtitre plates were placed on ice for 20 min to terminate the assay. Finally, 10 μL of the reaction mixture from each well was spotted onto tryptic soy agar, and the CFUs were calculated after overnight incubation. The control samples were incubated with opsonisation buffer B (OBB) instead of serum. These experiments were performed in duplicate for each sample, and the killing effect was defined as a reduction in CFUs compared with the OBB control.2.9 Splenocyte stimulation assays
Splenocytes were suspended in complete media (RPMI 1640 with 10% FBS) at a concentration of 2 × 106 cells per mL. The cells were stimulated with or without 10 μg mL−1 of protein at 37 °C for 5 days. The supernatants were collected, and the levels of IFN-γ and IL-17A were determined by ELISA using mouse IFN-γ ELISA and IL-17A ELISA kits (Biolegend), respectively.2.10 Inhibition of IL-17A immune pathways
Purified monoclonal anti-mouse IL-17A (αIL-17A; eBioscience) was prepared using endotoxin-free PBS. Vaccinated mice were injected with 100 μg (100 μL i.p.) of αIL-17A 2 days before infection. The vaccinated and nonvaccinated control groups were injected with the nonspecific isotype control IgG (100 μg i.p.).2.11 Statistical analysis
Data are presented as the mean ± standard deviation (SD). The survival rates were analysed using Kaplan–Meier survival curves. Antibody responses were analysed using Student's t-test. Bacterial burdens were analysed using the non-parametric Mann–Whitney test. GraphPad Prism 5.0 (GraphPad Software) was used for the data analyses, and significance was accepted at P < 0.05.3. Results
3.1 2C-Staph vaccination without adjuvant induced protective effects in S. aureus sepsis and pneumonia models
To select better antigen combinations against S. aureus-induced sepsis and pneumonia, we performed 3 vaccination and infection experiments. As shown in Fig. 1, vaccination with recombinant proteins yielded higher survival rates in the sepsis and pneumonia models than those in the PBS groups (6%; 3%). Furthermore, vaccination with the combination of rSEB and rMntC (2C-Staph) induced the highest survival rates in the sepsis model (53%) and pneumonia model (36%), and the rates were even higher than those in the rIsdB39 group (vaccine candidate V710 by Merck; P < 0.05). The results showed that the 2C-Staph group induced the high protective effects in sepsis and pneumonia models.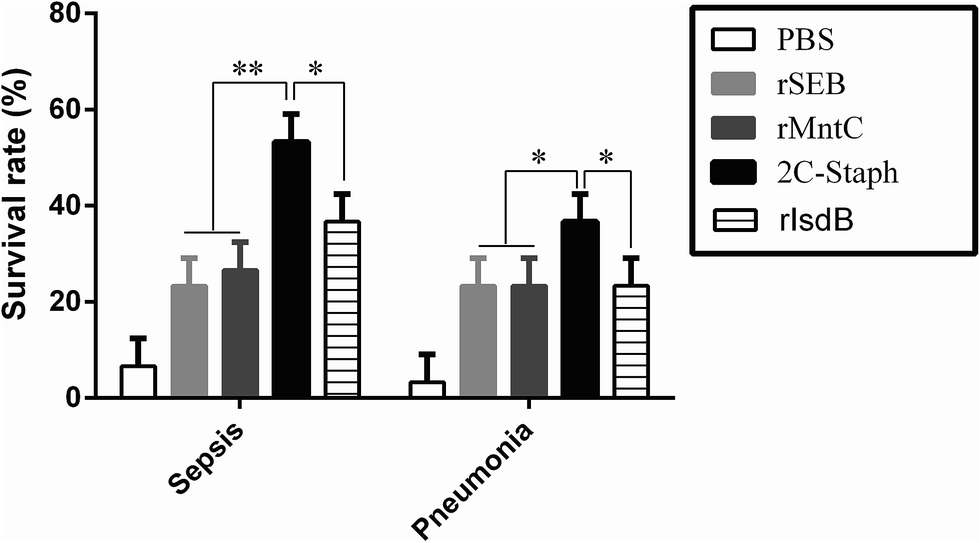 | ||
| Fig. 1 Survival rate of different antigens without adjuvant in the sepsis and pneumonia models. BALB/c mice (n = 10, per group) were immunised with PBS, rSEB, rMntC, rIsdB or the 2C-Staph vaccine. The mice were intravenously infected with MRSA 252 (1 × 109 CFUs) or intranasally infected with MRSA 252 (1 × 108 CFUs), and the survival rates were monitored for 10 days. The experiments were performed 3 times, and the results are shown as the mean ± SD. Asterisks indicate significant differences (*P < 0.05). | ||
Then, we further measured the effects of the mixture ratio of rSEB and rMntC (w/w) on the survival rates. As shown in Fig. S1,† a 1:1 mixture ratio showed better protective efficacy than the other two (2:1 and 1:2) mixtures in the sepsis and pneumonia models. Therefore, we chose the 1:1 mixture ratio for further study.
3.2 Screening and preparation of the novel NE vaccine
The optimised Smix, oil ratio, protein concentrations and order of addition results were obtained by measuring the diameter, zeta potential and PdI (Fig. 2 and S2†). The Smix affected the mean diameter and zeta potential. The average size was negatively correlated with the Smix (from 2:1 to 7:1), and the (5:1) Smix showed the smallest diameter (Fig. 2A). Importantly, the other key factor, the zeta potential of the NE, followed a pattern similar to that of the average size (Fig. 2E). Thus, a significant reduction in diameter and zeta potential was only evident in the (5:1) Smix. PdI is a key factor known to affect the spread of the particle size distribution, and smaller values reflect a narrower size range.33 The PdI value of the Smix (from 2:1 to 5:1) showed a narrow particle distribution (PdI value < 0.3; Fig. S2A†). Moreover, the appearance of different Smix NEs also showed that 5:1 was clearer than all the other ratios (Fig. S2F†). Therefore, the (5:1) Smix was chosen for the NE formulation.
 | ||
| Fig. 2 Influence of the different Smixes, oil ratios, addition orders and protein concentrations on the size and zeta potential. Average size (A–D) and zeta potential (ZP; E–H) of the NE vaccines were detected by Nano ZS. The results are shown as the mean ± SD. | ||
The oil ratio also plays a key role in NE formulation. The size, zeta potential, PdI and appearance of different oil ratios are shown in Fig. 2B, F, S2B and S2F.† The oil ratio (8:2) showed the smallest size and PdI values, a clearer appearance and more stable zeta potential, which demonstrated that it was optimal for the NE. Thus, we chose the oil ratio (8:2) for the NE formulation. We then found that a total protein concentration of 200 μg mL−1 NE showed a significant reduction in the particle diameter and zeta potential (Fig. 2C and G). In addition, the PdI values and appearance showed that a total protein concentration of 200 μg mL−1 was the optimal protein concentration for the NE formulation (Fig. S2C and S2G†). Finally, similar to the above evaluation standard, SCOP was the best addition order for 2C-Staph/NE (Fig. 2D, H, S2D and S2H†).
Based on the above results, we chose a Smix of 5:1 (w/w) of EL35® and propylene glycol, an oil ratio of 2:8, a total protein of 200 μg mL−1 and a SCOP addition order. This novel vaccine was ready for use when it achieved a low viscosity and clear appearance.
3.3 Characterisation and stability of the NE vaccine
As shown in the Fig. 3A, the NE vaccine showed a small particle size (<30 nm), and the NE droplet had good distribution without forming any aggregates. Based on the average size (24.90 ± 0.14 nm) and PdI value (0.218 ± 0.005, PdI < 0.3) in Fig. 3B, the NE vaccine was narrowly distributed. The zeta potential had a narrow distribution and was −6.86 mV for this NE (Fig. 3C). These results demonstrated that the 2C-Staph/NE vaccine displayed the basic characteristics of an NE. | ||
| Fig. 3 Characterisation and stability of 2C-Staph/NE. TEM (A), size distribution (B), zeta-potential distribution (C), structural integrity by SDS-PAGE (D), structural specificity by western blot (E) and long-term stability (F) for the NE adjuvant vaccine. (D and E): lane 1, supernatant of BNE; lane 2, supernatant BNE; lane 3, supernatant of BNE after being broken; lane 4, precipitate of BNE after being broken; lane 5, pre-stained protein ladder; lane 6, supernatant of 2C-Staph/NE; lane 7, precipitate of 2C-Staph/NE; lane 8, supernatant of 2C-Staph/NE after broken; lane 9, precipitate of 2C-Staph/NE after broken; and lane 10, 2C-Staph aqueous solution. | ||
The integrity and specificity of the protein structure results are shown in Fig. 3D and E. The precipitate in lane 9 (the NE vaccine after treatment) showed two clear and bright bands located at 28 kDa (rSEB) and 33 kDa (rMntC), which was similar to that of lane 10 (the antigen solution control). This result showed that the antigen mainly stayed in the precipitate when it was unbroken. The BNE had no obvious bands in lanes 1 to 4 (the supernatant and precipitate of BNE, the supernatant and precipitate of BNE after treatment). Importantly, obvious bands were not observed in lanes 6 to 8 (the supernatant and precipitate of the unbroken NE, the supernatant of the NE after breaking). These results confirmed that the protein had been loaded successfully (lane 6 and 7). However, significant degradation was not detected in the NE after breaking compared with its protein solution. Therefore, these results demonstrate the likely immunogenicity of this vaccine because of its good structural integrity and specificity.
Additionally, the stability of 2C-Staph in this NE system was measured by MALDI-TOF. Peaks were not observed in the BNE and 2C-Staph/NE groups (Fig. 4C and D). After the broken treatment, two obvious peaks were observed in the 2C-Staph/NE group (m/z = 28377, m/z = 32823), and the two peaks were similar to those of rSEB (28 kDa) and rMntC (33 kDa; Fig. 4A, B and E). These results demonstrate that the 2C-Staph was stable in this 2C-Staph/NE vaccine.
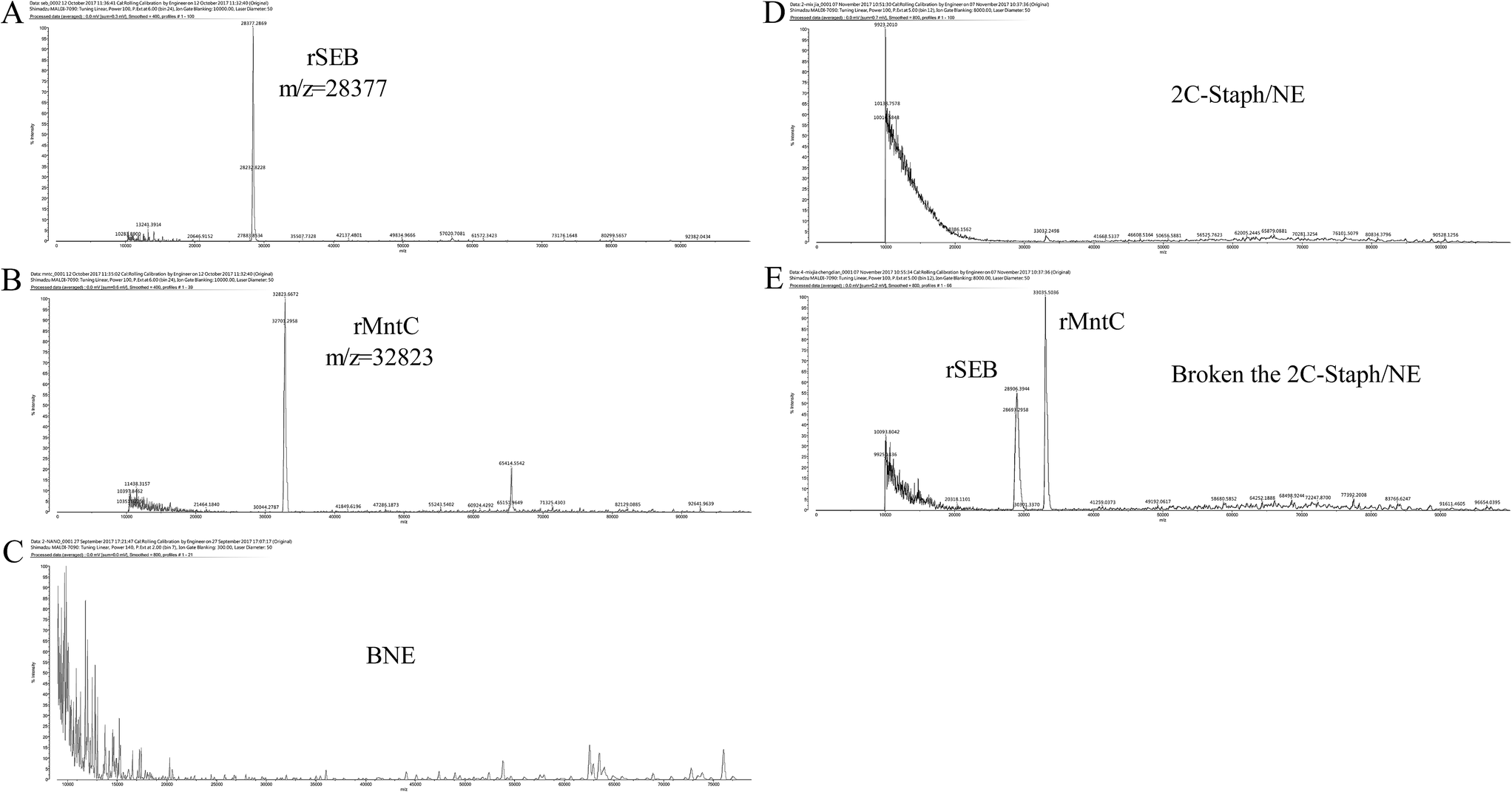 | ||
| Fig. 4 Mass spectrum of 2C-Staph in 2C-Staph/NE. rSEB (A), rMntC (B), BNE (C), 2C-Staph/NE (D) and 2C-Staph/NE after being broken (E) were detected by MALDI-TOF MS. The main peaks of rSEB (m/z = 28377) and rMntC (m/z = 32823) are indicated beside the peak. | ||
The NE vaccine did not exhibit turbidity, phase or drug separation, creaming, precipitation, and/or demulsification or any other form of instability after centrifugation (13000 × g for 30 min). The primary protein structure tests showed that the protein bands did not significantly differ between fresh samples and samples stored at room temperature for 6 months based on SDS-PAGE (Fig. 3F). These results show that 2C-Staph/NE is stable at room temperature.
3.4 Mice vaccinated intranasally with 2C-Staph/NE showed protective effects in an S. aureus pneumonia model but not in a sepsis model
Next, we analysed the survival rate in S. aureus sepsis and pneumonia models. As shown in Fig. 5A–D, mice vaccinated with 2C-Staph/NE all showed the highest survival rates in two different models with two immunisation routes, and 2C-Staph/NE showed higher protective efficacy than 2C-Staph/MF59, 2C-Staph, BNE and PBS. Therefore, 2C-Staph/NE greatly enhanced the immune responses.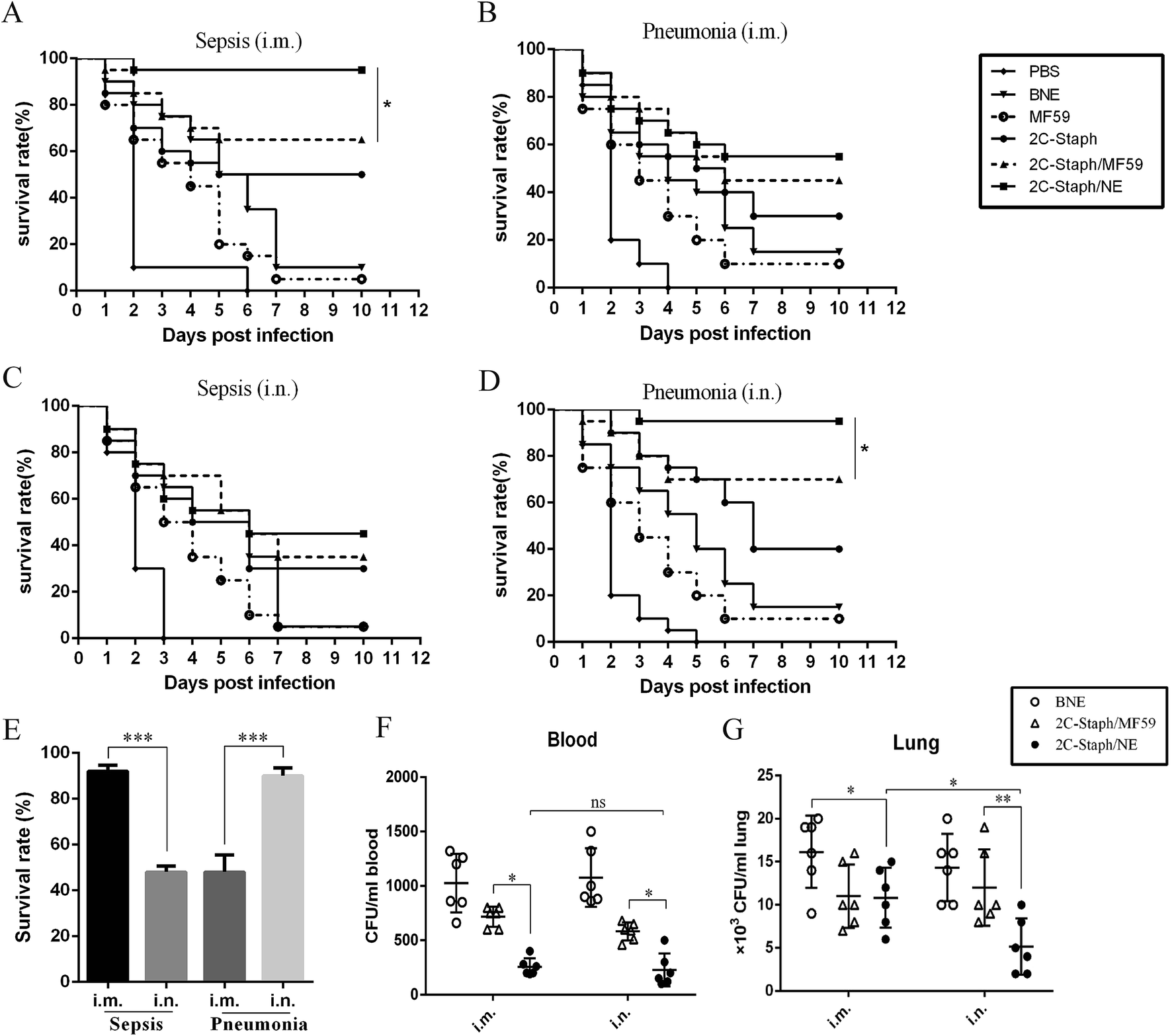 | ||
| Fig. 5 Protective effects of intramuscular or intranasal vaccination with 2C-Staph/NE vaccine in sepsis and pneumonia models. BALB/c mice (n = 10, per group) were intramuscularly (i.m.) or intranasally (i.n.) immunised with PBS, MF59, BNE, 2C-Staph, 2C-Staph/MF59 or 2C-Staph/NE. Mice were intravenously infected with MRSA 252 (1 × 109 CFUs; A and C) or intranasally infected with a lethal dose of MRSA 252 (1 × 108 CFUs; B and D), and the survival rates were monitored for 10 days. (E) Experiments were performed 3 times, and the results are shown as the mean ± SD. Asterisks indicate significant differences (*P < 0.05, **P < 0.01, ***P < 0.001). The bacterial burdens in the blood (F) and lungs (G) were assessed in pneumonia infection models by infectious doses of MRSA 252 (5 × 107 CFUs) at 24 hours post infection. | ||
In the sepsis model, mice vaccinated intramuscularly with 2C-Staph/NE showed a higher survival rate (95%) than the MF59 group (65%; P < 0.05) and 2C-Staph group (50%; Fig. 5A). However, the intramuscular vaccination route did not show a high protective effect in the pneumonia model (55% survival rate; Fig. 5B). Interestingly, vaccination by the intranasal route induced the highest survival rate (90%) among all the groups in the pneumonia models (Fig. 5D and E). In contrast, mice vaccinated intranasally with 2C-Staph/NE did not show protective effects in a sepsis model and presented a survival rate (45%) that was significantly lower than that achieved via the intramuscular route (95%; Fig. 5E). Moreover, the bacterial burdens in the pneumonia model were evaluated by vaccination with 2C-Staph/NE through two immune routes. As shown in Fig. 5F and G, intranasal vaccination with 2C-Staph/NE greatly reduced the bacterial burdens in the lung compared with the intramuscular route (P < 0.05), whereas the difference was not significant in the blood. Based on the data presented above, mice vaccinated by different routes showed different protective effects in different S. aureus infection models, and intranasally administered 2C-Staph/NE efficiently combated S. aureus infections in the lungs.
3.5 Vaccination with 2C-Staph/NE by intranasal or intramuscular routes both induced strong antibody responses in serum and BALF
Mice were vaccinated intramuscularly and intranasally at the same dose, and serum was harvested at 7 days after the last immunisation. As shown in Table 1, 2C-Staph without adjuvant induced moderate IgG responses by the intramuscular route and low IgG responses by the intranasal route. 2C-Staph combined with MF59 or the NE adjuvant both yielded higher IgG responses than 2C-Staph and BNE (P < 0.05). Furthermore, 2C-Staph/NE induced higher IgG and IgG subgroup levels than 2C-Staph/MF59 in mice vaccinated intranasally or intramuscularly (P < 0.05). Therefore, 2C-Staph formulated with the NE adjuvant induced higher levels of antibody responses than that formulated with MF59. Moreover, mice vaccinated intramuscularly with 2C-Staph/NE yielded higher IgG levels in the serum than those vaccinated by the intranasal route (P < 0.01). In conclusion, these results showed that mice vaccinated intramuscularly or intranasally with 2C-Staph/NE yielded strong IgG responses, and that vaccination by the intramuscular route induced higher IgG responses in serum than the intranasal route.| Anti-SEB antibodya | Anti-MntC antibodya | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IgG | IgG1 | IgG2a | IgG2b | IgG | IgG1 | IgG2a | IgG2b | ||
| a Antibody response is expressed as mean absorbance at 450 nm ± SD. All serum samples for detection IgG and IgG1 were diluted 1:4000. All serum samples for detection IgG2a and IgG2b were diluted 1:1000. Pa indicated the statistic difference between the 2C-Staph/NE and 2C-Staph/MF59 group. Pb indicated the statistic difference between the 2C-Staph/NE (i.m.) and 2C-Staph/NE (i.n.) group. |
|||||||||
| i.m. | BNE | 0.104 ± 0.002 | 0.098 ± 0.012 | 0.112 ± 0.013 | 0.103 ± 0.006 | 0.110 ± 0.006 | 0.094 ± 0.052 | 0.127 ± 0.004 | 0.090 ± 0.011 |
| 2C-Staph | 0.219 ± 0.067 | 0.248 ± 0.125 | 0.275 ± 0.009 | 0.379 ± 0.013 | 0.455 ± 0.137 | 0.387 ± 0.229 | 0.470 ± 0.012 | 0.291 ± 0.010 | |
| 2C-Staph/MF59 | 0.570 ± 0.118 | 0.409 ± 0.069 | 0.427 ± 0.048 | 0.629 ± 0.031 | 1.113 ± 0.086 | 0.977 ± 0.268 | 0.768 ± 0.136 | 0.507 ± 0.365 | |
| 2C-Staph/NE | 0.866 ± 0.231 | 0.714 ± 0.304 | 0.602 ± 0.125 | 0.821 ± 0.030 | 1.330 ± 0.086 | 1.231 ± 0.104 | 1.075 ± 0.493 | 0.838 ± 0.436 | |
| Pa < 0.05 | Pa < 0.01 | Pa < 0.05 | Pa < 0.05 | Pa < 0.05 | Pa < 0.01 | Pa < 0.001 | Pa < 0.01 | ||
| i.n. | BNE | 0.108 ± 0.002 | 0.103 ± 0.009 | 0.096 ± 0.011 | 0.107 ± 0.009 | 0.105 ± 0.005 | 0.092 ± 0.023 | 0.112 ± 0.011 | 0.092 ± 0.012 |
| 2C-Staph | 0.174 ± 0.013 | 0.168 ± 0.012 | 0.181 ± 0.014 | 0.184 ± 0.024 | 0.181 ± 0.015 | 0.175 ± 0.011 | 0.171 ± 0.013 | 0.192 ± 0.008 | |
| 2C-Staph/MF59 | 0.197 ± 0.016 | 0.205 ± 0.019 | 0.214 ± 0.011 | 0.243 ± 0.054 | 0.302 ± 0.043 | 0.273 ± 0.058 | 0.193 ± 0.011 | 0.221 ± 0.012 | |
| 2C-Staph/NE | 0.354 ± 0.073 | 0.303 ± 0.045 | 0.309 ± 0.013 | 0.413 ± 0.025 | 0.473 ± 0.106 | 0.368 ± 0.004 | 0.335 ± 0.009 | 0.452 ± 0.017 | |
| Pa < 0.05 | Pa < 0.05 | Pa < 0.05 | Pa < 0.01 | Pa < 0.05 | Pa < 0.05 | Pa < 0.01 | Pa < 0.001 | ||
| Pb < 0.001 | Pb < 0.01 | Pb < 0.01 | Pb < 0.001 | Pb < 0.001 | Pb < 0.001 | Pb < 0.001 | Pb < 0.001 | ||
Next, we detected the SEB-specific and MntC-specific antibodies in BALF. Among the four groups, the 2C-Staph/NE group induced the highest IgG responses by two immunisation routes (Fig. 6A). These results revealed that 2C-Staph formulated with the NE induced higher levels of antibody responses than that formulated with MF59. Furthermore, significant differences in IgG responses were not observed between the two immunisation routes (P > 0.05). These results showed that mice vaccinated intramuscularly and intranasally with 2C-Staph/NE yielded similar levels of IgG in BALF.
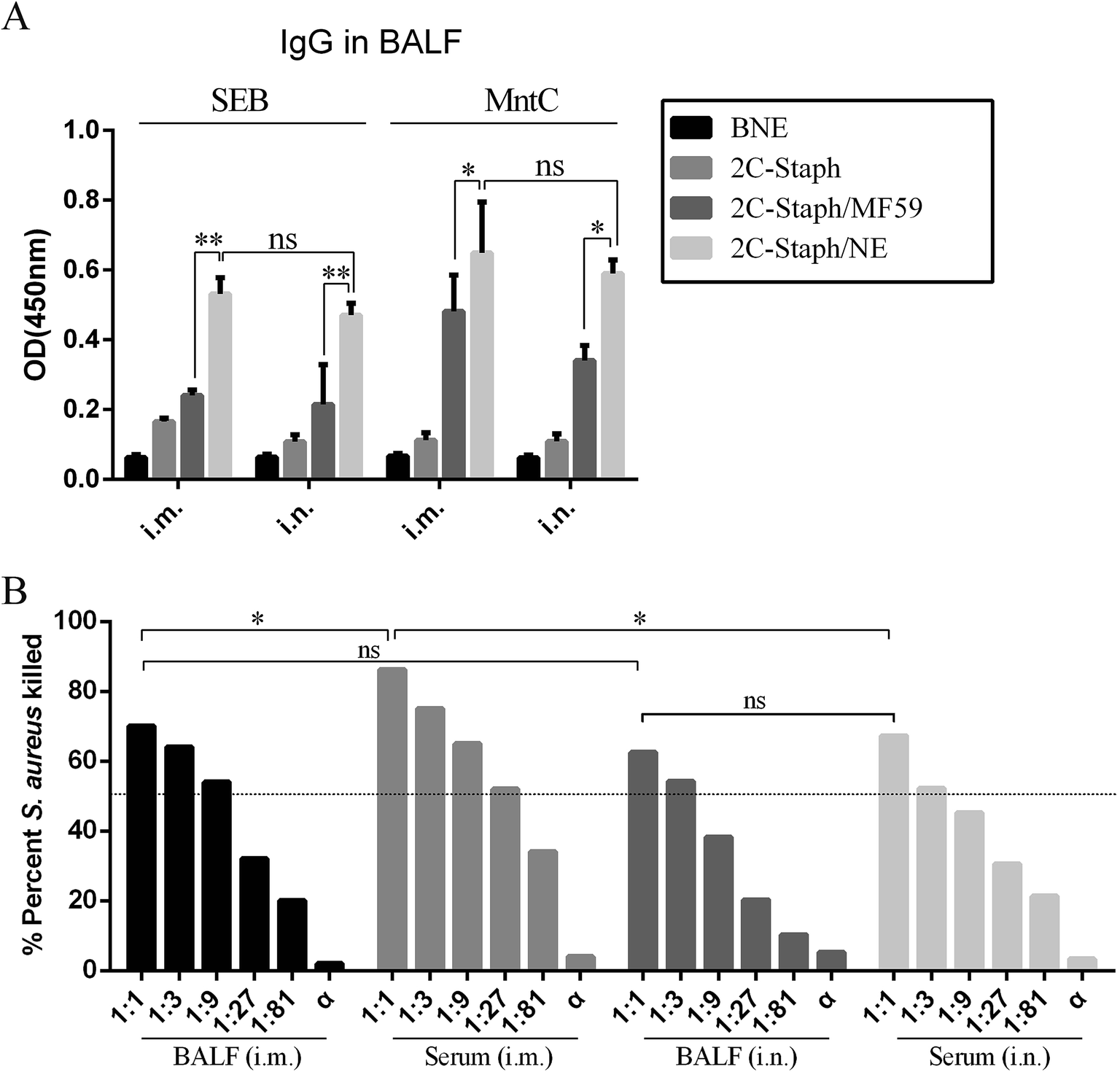 | ||
| Fig. 6 IgG responses in BALF and the OPA results. (A) BALB/c mice (n = 6, per group) were intramuscularly (i.m.) or intranasally (i.n.) immunised with BNE, 2C-Staph, 2C-Staph/MF59 or 2C-Staph/NE. Seven days after the last immunisation, the IgG levels in BALF were detected by ELISA. All serum samples were diluted 1:100. The results are shown as the mean absorbance at 450 nm ± SD. (B) Serum and BALF by intranasal or intramuscular vaccination with 2C-Staph/NE were used to perform OPA. The results are shown as the mean. Asterisks indicate significant differences (*P < 0.05, **P < 0.01, ***P < 0.001). | ||
We then performed OPA to detect the functional antibodies induced by the 2C-Staph/NE in serum and BALF (Fig. 6B). The serum and BALF of the mice vaccinated intramuscularly and intranasally with 2C-Staph/NE all showed high killing rates (>50%). The serum pooled from mice vaccinated by the intramuscular route showed the highest killing rate (83.3%), which was correlated with the IgG responses (Table 1). The OPA results in the BALF did not show significant differences in mice vaccinated intramuscularly and intranasally (P > 0.05). When the S. aureus specific antibodies were exhausted by incubating with S. aureus for 30 min, the serum and BALF did not show bacterial killing ability (<10%). Therefore, antigen-specific antibodies mediated OPA activity. These results showed that vaccination with 2C-Staph/NE by the intramuscular route induced higher IgG levels and more functional antibodies in serum, whereas the IgG levels and functional antibodies in BALF were similar between the two different vaccination routes.
3.6 Intranasal vaccination with 2C-Staph/NE induced high levels of IgA in serum and BALF
Several studies have revealed that intranasal vaccination induces high levels of IgA in serum and BALF.31,34 We then detected the IgA responses in serum and BALF by the two vaccination routes. From the results of the intranasal vaccination route (Fig. 7A), the 2C-Staph/NE group induced higher levels of IgA than the 2C-Staph/MF59 group (P < 0.001). However, vaccination through the intramuscular route induced similar IgA responses between the 2C-Staph/NE group and the 2C-Staph/MF59 group (P > 0.05). For the two vaccination routes, intranasal vaccination with 2C-Staph/NE induced higher IgA responses in the serum than the intramuscular route (P < 0.001). These results showed that intranasal vaccination with 2C-Staph/NE induced higher IgA levels in the serum than that with 2C-Staph/MF59 and that intranasal vaccination with 2C-Staph/NE induced higher IgA responses than vaccination through the intramuscular route.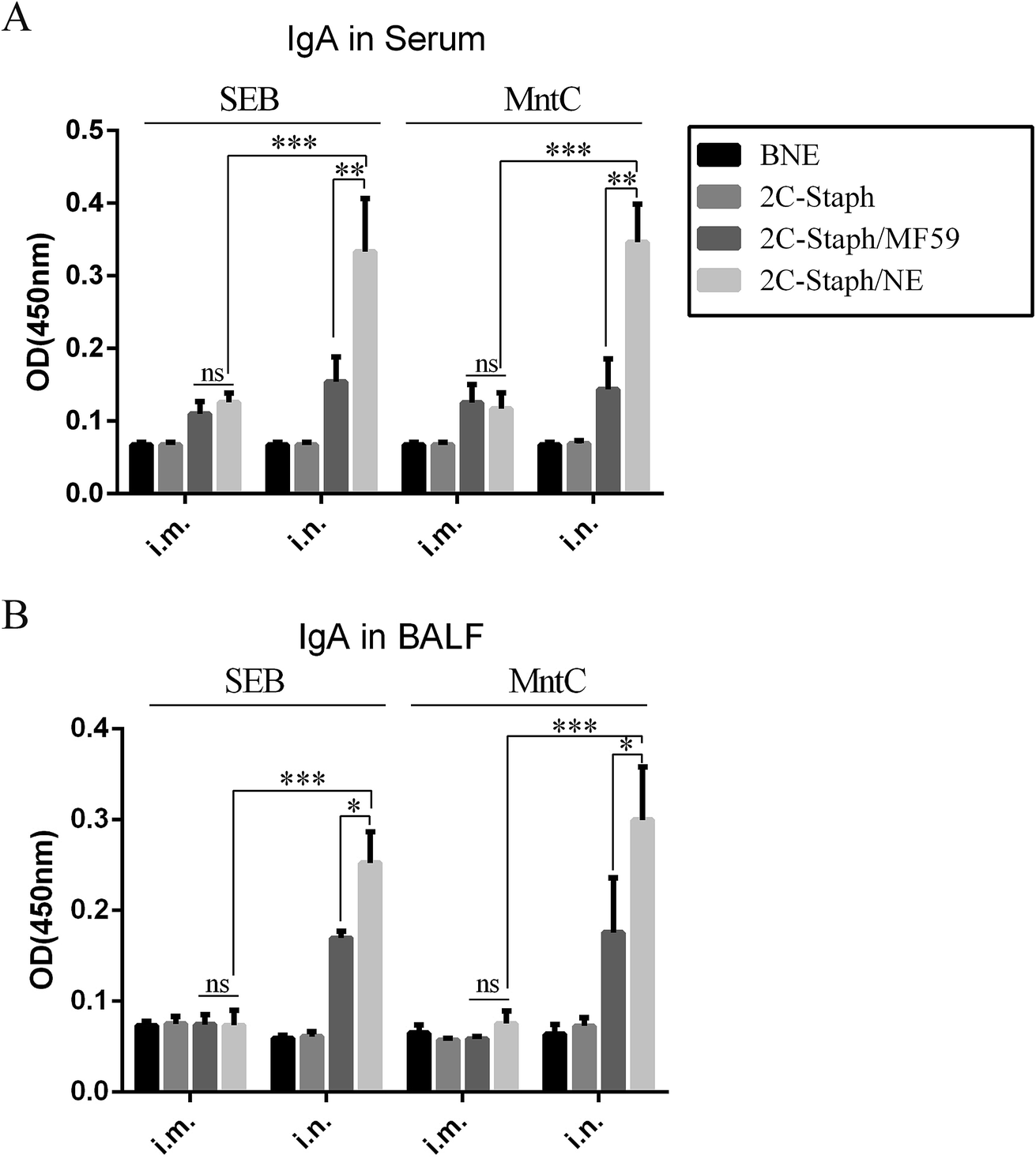 | ||
| Fig. 7 IgA responses in serum and BALF. BALB/c mice (n = 6 per group) were intramuscularly (i.m.) or intranasally (i.n.) vaccinated with BNE, 2C-Staph, 2C-Staph/MF59 or 2C-Staph/NE. Seven days after the last immunisation, IgA levels in serum (A) and BALF (B) were detected by ELISA. All serum samples were diluted 1:100, and all BALF samples were diluted 1:10. The results are shown as the mean absorbance at 450 nm ± SD. Asterisks indicate significant differences (*P < 0.05, **P < 0.01, ***P < 0.001). | ||
The IgA responses in BALF were similar to those in serum (Fig. 5B). Intranasal vaccination with 2C-Staph/NE induced higher levels of IgA in BALF than that with 2C-Staph/MF59 (P < 0.05). In addition, vaccination through the intranasal route induced higher levels of IgA in BALF than that through the intramuscular route (P < 0.05). These results showed that intranasal vaccination with 2C-Staph/NE induced higher IgA levels in BALF than that with 2C-Staph/MF59 and that intranasal vaccination with 2C-Staph/NE induced higher IgA responses than that via the intramuscular route.
3.7 Mucosal vaccination induced protective IL-17A responses in the lungs
Our research showed that the S. aureus antigen combined with the NE adjuvant induced strong cellular immune responses, especially for IFN-γ and IL-17A.34 The IFN-γ and IL-17A responses in the serum and BALF 7 days after the last vaccination were determined. As shown in Fig. 8A and B, vaccination with 2C-Staph/NE induced higher IL-17A responses intranasally or intramuscularly than that with 2C-Staph/MF59 (P < 0.05), whereas the IFN-γ responses between the two groups were similar (P > 0.05). Furthermore, intranasal vaccination with 2C-Staph/NE induced higher levels of IL-17A in the BALF and serum than the muscular route (Fig. 8A–C; P < 0.05). Therefore, intranasal vaccination with 2C-Staph/NE exhibited a significant increase in IL-17A in the serum and BALF, which may explain the high protective effect in the pneumonia model.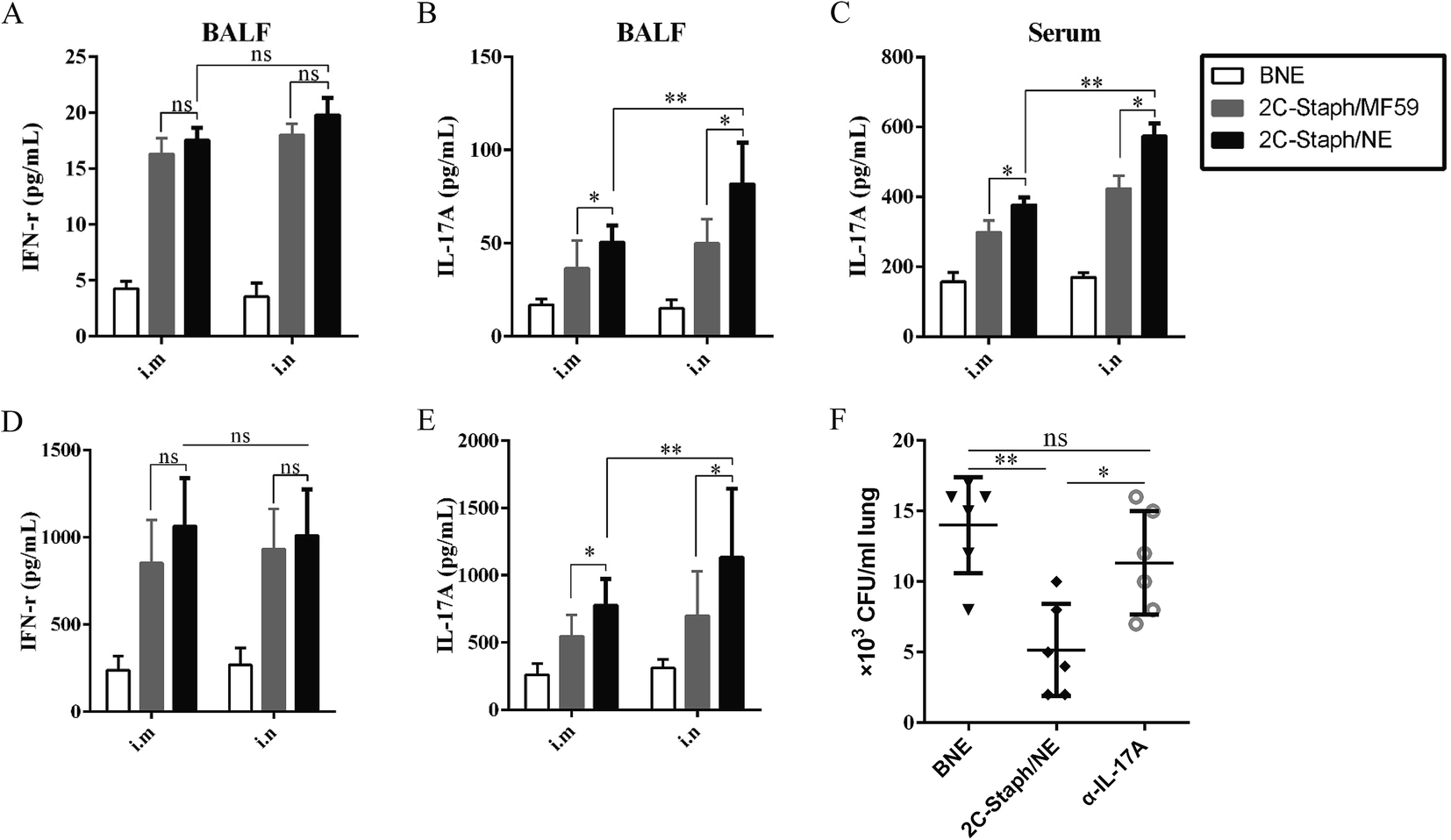 | ||
| Fig. 8 Cellular immune responses in serum and spleen. Seven days after the last immunisation, the IFN-γ and IL-17A in BALF (A, B) and serum (C) were detected by ELISA. (D, E) Supernatants from the splenocyte stimulation assays were harvested, and the IFN-γ and IL-17A levels were determined by ELISA (n = 6). (F) Vaccinated mice were injected with αIL-17A at 2 days before infection. The BNE unvaccinated mice and untreated vaccinated mice were regarded as the controls and vaccinated controls, respectively. Bacterial burdens were calculated in the lung at 24 hours post infection. Data are presented as scatter plots, and the medians are shown. Asterisks indicate significant differences (*P < 0.05, **P < 0.01, ***P < 0.001). | ||
And, we further measured the levels of IFN-γ and IL-17A produced by the splenocytes after stimulation with rSEB and rMntC. As shown in Fig. 8D, intranasal or intramuscular vaccination with 2C-Staph/NE induced similar levels of IFN-γ compared with that with 2C-Staph/MF59 (P > 0.05). However, the splenocytes from the 2C-Staph/NE group produced higher levels of IL-17A compared with those from the 2C-Staph/MF59 group (P < 0.05). Furthermore, intranasal vaccination with 2C-Staph/NE induced more IL-17A than the intramuscular route (Fig. 8E; P < 0.01). These results showed that intranasal vaccination with 2C-Staph/NE induced a strong Th17-based cellular response.
We then validated the IL-17A responses for immune protection. Blocking IL-17A caused significant increases in the mean organ bacterial burdens in the vaccinated mice compared with an untreated vaccinated group (Fig. 8F; P < 0.05). Because neutralisation of IL-17A notably reduced the 2C-Staph/NE immune protective effect, we demonstrated that the powerful IL-17A responses induced by 2C-Staph/NE played an important role in vaccine immune protection.
4. Discussion
Based on a multicomponent strategy, vaccine candidates must be conserved among the S. aureus strains and target the virulence determinants that mediate colonisation, nutrition acquisition or immune evasion.40 Both SEB and MntC are conserved among S. aureus strains; thus, they are suitable candidates for designing an S. aureus vaccine. Phase I clinical trials of vaccines containing the SEB (STEBVax, IBT/NIAID) or MntC (SA4Ag, Pfizer) have been completed, and these trials did not observe safety concerns and reported a good immunogenicity profile following vaccination of healthy subjects.41,42 Although 2C-Staph without adjuvant induced higher survival rates than single-antigen groups, the average survival rates in a sepsis model and pneumonia model were only 53% and 36% (Fig. 1). Vaccines formulated with adjuvants are increasingly needed to boost antigen effectiveness.21 We previously prepared an NE adjuvant that enhanced the systemic and mucosal immune response with recombination protein antigens.34 Thus, we attempted to design a novel NE adjuvant to enhance the immunogenicity of the 2C-Staph vaccine in our study.Smaller nanoparticles ranging from 1 to 100 nm can be easily transported to lymph nodes because they can be readily taken up by dendritic cells and retained for a longer period of time at the site of the injection.43 In this study, we evaluated the average size, zeta potential and PdI to characterise the Smix, oil ratio, addition order and total protein concentration. As shown in Fig. 2 and S2,† for the 2C-Staph/NE vaccine, the optimal average size of 2C-Staph/NE was 24.90 nm, optimal Smix was 5:1, oil ratio was 8:2, addition order was SCOP and total protein concentration was 200 μg mL−1 and an acceptable PdI was less than 0.3. The TEM results were correlated with those of Nano ZLS, which showed small particles ranging from 1 to 100 nm. The centrifugation test, SDS-PAGE, and western blotting analysis showed the stability of this novel NE vaccine. These short and long-term results indicated that the NE vaccine had good stability when stored at room temperature for 6 months.
Intramuscular or intranasal vaccination with the 2C-Staph/NE vaccine induced higher survival rates in both the sepsis and pneumonia models than that with the 2C-Staph and 2C-Staph/MF59 (P < 0.05). 2C-Staph formulated with the NE adjuvant enhanced the immune responses. As shown in Table 1, the NE adjuvant significantly improved the IgG and IgG subgroups responses of the 2C-Staph vaccine, and the values were even higher than those of the 2C-Staph/MF59 group (P < 0.05). For the splenocyte stimulation assays, intramuscular or intranasal immunisation with 2C-Staph/NE produced significantly higher levels of IL-17A than that with 2C-Staph/MF59 (P < 0.01), whereas the levels of IFN-γ were comparable (P > 0.05). Therefore, the 2C-Staph formulated with the NE adjuvant significantly improved the humoural and cellular responses.
In a sepsis model, intramuscular vaccination with 2C-Staph/NE induced the highest survival rate (95%). Interestingly, intranasal vaccination with 2C-Staph/NE significantly improved the survival rate in a pneumonia model compared with the intramuscular route (P < 0.05), suggesting that intramuscular vaccination induces the best protective effect in the sepsis model, whereas the intranasal route yielded the best protective efficacy in the pneumonia model (Fig. 5). Different vaccination routes of 2C-Staph/NE also induced different immune responses. First, compared with the intranasal route, the intramuscular vaccination route induced higher levels of IgG and IgG subgroups in the serum (Table 1; P < 0.05), whereas the IgG levels in the BALF were similar (Fig. 6A; P > 0.05). Second, the intramuscular vaccination route induced a higher level of functional antibodies in the serum than the intranasal route, whereas the OPA activity in BALF was comparable (Fig. 6B; P > 0.05). Third, intranasal vaccination with 2C-Staph/NE induced higher levels of IgA and IL-17A in the serum (P < 0.05) and BALF (P < 0.05) than the intramuscular route, although the level of IFN-γ in the BALF was similar. The IFN-γ and IL-17A responses in the splenocyte stimulation assays also showed similar results. Taken together, these data may explain why the intranasal vaccination route induced higher protective effects in the pneumonia model.
The formation of a strong mucosal barrier at the first point of contact with the host represents an effective strategy for preventing infection.44 Currently, few mucosal vaccines are on the market,43,45 and the NE adjuvant may supply a promising adjuvant for vaccine development. Intramuscular vaccination with 2C-Staph/NE induced strong systemic immune responses (e.g., high levels of IgG in serum) but only weak mucosal immunity (e.g., low levels of IgA and IL-17A in BALF). Mucosal vaccination routes could favour the production of mucosal antibodies and cellular responses.46 The IL-17A response showed a protective role in S. aureus infection pneumonia because IL-17A neutralization exhibited impaired clearance of S. aureus in vivo (Fig. 8F). Furthermore, suitable adjuvants are necessary for mucosal vaccines to enhance immune responses. The immune responses in the BALF indicated that NE adjuvant could be more efficient for delivery to the mucosal site and induce strong mucosal immune responses.
Our results showed that different immune routes might protect different populations. To date, pre-clinical prophylactic vaccines for S. aureus pneumonia were mainly administrated through intramuscular route.22,47 In our study, intranasal vaccination with 2C-Staph/NE induced higher protective effect in pneumonia model, compared with that by the intramuscular route (Fig. 5). Therefore, our results demonstrated that intranasal vaccination with 2C-Staph/NE may provide a more promising method of combatting S. aureus pneumonia.
5. Conclusions
In this study, we successfully constructed a 2C-Staph/NE vaccine. The NE adjuvant greatly increased the immune responses to 2C-Staph and corresponded to stronger humoural and cellular immune responses. Intramuscular or intranasal vaccination with 2C-Staph/NE induced distinct protective efficacy in sepsis and pneumonia models. The intramuscular vaccination route induced strong systemic immune responses and showed high protective effects in a sepsis model. Moreover, the intranasal vaccination route induced strong mucosal immune responses with higher levels of IgA and IL-17A in BALF and showed ideal protective effects in a pneumonia model. Furthermore, we demonstrated that IL-17A induced by intranasal administration of 2C-Staph/NE played a protective role in a pneumonia model. These results indicated that mucosal vaccination with 2C-Staph/NE may provide a new approach for treating S. aureus pneumonia.Conflicts of interest
There are no conflicts of interest associated with the present work.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 31670938 and No. 31600745), and the Natural Science Foundation Project Program of Chongqing CSTC (No. 2014jcyjA10107).References
- F. D. Lowy, Staphylococcus aureus infections, N. Engl. J. Med., 1998, 339, 520–532, DOI:10.1056/nejm199808203390806.
- T. J. Foster, The Staphylococcus aureus “superbug”, J. Clin. Invest., 2004, 114, 1693–1696, DOI:10.1172/jci23825.
- M. Zasloff, Antimicrobial peptides of multicellular organisms, Nature, 2002, 415, 389–395, DOI:10.1038/415389a.
- K. A. Brogden, Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria?, Nat. Rev. Microbiol., 2005, 3, 238–250, DOI:10.1038/nrmicro1098.
- L. Rizzello and P. P. Pompa, Nanosilver-based antibacterial drugs and devices: mechanisms, methodological drawbacks, and guidelines, Chem. Soc. Rev., 2014, 43, 1501–1518, 10.1039/c3cs60218d.
- K. P. Miller, L. Wang, B. C. Benicewicz and A. W. Decho, Inorganic nanoparticles engineered to attack bacteria, Chem. Soc. Rev., 2015, 44, 7787–7807, 10.1039/c5cs00041f.
- K. Zheng, M. I. Setyawati, D. T. Leong and J. Xie, Antimicrobial silver nanomaterials, Coord. Chem. Rev., 2018, 357, 1–17, DOI:10.1016/j.ccr.2017.11.019.
- K. Zheng, M. I. Setyawati, T. P. Lim, D. T. Leong and J. Xie, Antimicrobial Cluster Bombs: Silver Nanoclusters Packed with Daptomycin, ACS Nano, 2016, 10, 7934–7942, DOI:10.1021/acsnano.6b03862.
- K. Zheng, M. I. Setyawati, D. T. Leong and J. Xie, Antimicrobial Gold Nanoclusters, ACS Nano, 2017, 11, 6904–6910, DOI:10.1021/acsnano.7b02035.
- K. U. Jansen, D. Q. Girgenti, I. L. Scully and A. S. Anderson, Vaccine review: “Staphylococcus aureus vaccines: problems and prospects”, Vaccine, 2013, 31, 2723–2730, DOI:10.1016/j.vaccine.2013.04.002.
- F. Bagnoli, et al., Vaccine composition formulated with a novel TLR7-dependent adjuvant induces high and broad protection against Staphylococcus aureus, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3680–3685 CAS.
- R. A. Proctor, Challenges for a universal Staphylococcus aureus vaccine, Clin. Infect. Dis., 2012, 54, 1179–1186, DOI:10.1093/cid/cis033.
- L. Thomer, O. Schneewind and D. Missiakas, Pathogenesis of Staphylococcus aureus Bloodstream Infections, Annu. Rev. Phytopathol., 2016, 11, 343–364 CrossRef CAS PubMed.
- T. J. Foster, J. A. Geoghegan, V. K. Ganesh and M. Hook, Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus, Nat. Rev. Microbiol., 2013, 12, 49–62, DOI:10.1038/nrmicro3161.
- C. Kong, H. M. Neoh and S. Nathan, Targeting Staphylococcus aureus Toxins: A Potential form of Anti-Virulence Therapy, Toxins, 2016, 8, 72–93 CrossRef PubMed.
- M. Otto, Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus, Annu. Rev. Microbiol., 2010, 64, 143–162, DOI:10.1146/annurev.micro.112408.134309.
- Z. Zhao, et al., Fine-mapping of immunodominant linear B-cell epitopes of the Staphylococcus aureus SEB antigen using short overlapping peptides, PLoS One, 2014, 9, e90445, DOI:10.1371/journal.pone.0090445.
- J. E. Cassat and E. P. Skaar, Metal ion acquisition in Staphylococcus aureus: overcoming nutritional immunity, Semin. Immunopathol., 2012, 34, 215–235, DOI:10.1007/s00281-011-0294-4.
- Z. Zhao, et al., Multiple B-cell epitope vaccine induces a Staphylococcus enterotoxin B-specific IgG1 protective response against MRSA infection, Sci. Rep., 2015, 5, 12371, DOI:10.1038/srep12371.
- H. J. Yang, et al., Immunisation With Immunodominant Linear B Cell Epitopes Vaccine of Manganese Transport Protein C Confers Protection Against Staphylococcus aureus Infection, PLoS One, 2016, 11, e0149638, DOI:10.1371/journal.pone.0149638.
- V. G. Fowler, et al., Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: a randomized trial, Jama, 2013, 309, 1368–1378 CrossRef CAS PubMed.
- E. Monaci, et al., MF59- and Al(OH)3-Adjuvanted Staphylococcus aureus (4C-Staph) Vaccines Induce Sustained Protective Humoral and Cellular Immune Responses, with a Critical Role for Effector CD4 T Cells at Low Antibody Titers, Front. Immunol., 2015, 6, 439, DOI:10.3389/fimmu.2015.00439.
- H. Chen, Recent advances in mucosal vaccine development, J. Controlled Release, 2000, 67, 117–128 CrossRef CAS PubMed.
- P. He, Y. Zou and Z. Hu, Advances in aluminum hydroxide-based adjuvant research and its mechanism, Hum. Vaccines Immunother., 2015, 11, 477–488, DOI:10.1080/21645515.2014.1004026.
- D. T. O'Hagan, R. Rappuoli, E. De Gregorio, T. Tsai and G. Del Giudice, MF59 adjuvant: the best insurance against influenza strain diversity, Expert Rev. Vaccines, 2011, 10, 447–462, DOI:10.1586/erv.11.23.
- D. T. O'Hagan, A. Wack and A. Podda, MF59 is a safe and potent vaccine adjuvant for flu vaccines in humans: what did we learn during its development?, Clin. Pharmacol. Ther. Ser., 2007, 82, 740–744, DOI:10.1038/sj.clpt.6100402.
- Y. Perrie, A. R. Mohammed, D. J. Kirby, S. E. McNeil and V. W. Bramwell, Vaccine adjuvant systems: enhancing the efficacy of sub-unit protein antigens, Int. J. Pharm., 2008, 364, 272–280, DOI:10.1016/j.ijpharm.2008.04.036.
- C. Pozzi, et al., Vaccines for Staphylococcus aureus and Target Populations, Curr. Top. Microbiol. Immunol., 2017, 409, 491–528 Search PubMed.
- G. H. Dayan, et al., Staphylococcus aureus: the current state of disease, pathophysiology and strategies for prevention, Expert Rev. Vaccines, 2016, 15, 1373–1392, DOI:10.1080/14760584.2016.1179583.
- R. A. Proctor, Is there a future for a Staphylococcus aureus vaccine?, Vaccine, 2012, 30, 2921–2927, DOI:10.1016/j.vaccine.2011.11.006.
- B. G. Stiles, A. R. Garza, R. G. Ulrich and J. W. Boles, Mucosal vaccination with recombinantly attenuated staphylococcal enterotoxin B and protection in a murine model, Infect. Immun., 2001, 69, 2031–2036, DOI:10.1128/iai.69.4.2031-2036.2001.
- W. Xu, L. Chen, S. Guo, L. Wu and J. Zhang, Intranasal Administration of Recombinant Mycobacterium Smegmatis Inducing IL-17A Autoantibody Attenuates Airway Inflammation in a Murine Model of Allergic Asthma, PLoS One, 2016, 11, e0151581, DOI:10.1371/journal.pone.0151581.
- H. Sun, et al., Development and characterization of a novel nanoemulsion drug-delivery system for potential application in oral delivery of protein drugs, Int. J. Nanomed., 2012, 7, 5529–5543, DOI:10.2147/ijn.s36071.
- H. Sun, et al., Induction of systemic and mucosal immunity against methicillin-resistant Staphylococcus aureus infection by a novel nanoemulsion adjuvant vaccine, Int. J. Nanomed., 2015, 10, 7275–7290, DOI:10.2147/ijn.s91529.
- W. Cai, W. Deng, H. Yang, X. Chen and F. Jin, A propofol microemulsion with low free propofol in the aqueous phase: formulation, physicochemical characterization, stability and pharmacokinetics, Int. J. Pharm., 2012, 436, 536–544, DOI:10.1016/j.ijpharm.2012.07.008.
- A. Myc, et al., Development of immune response that protects mice from viral pneumonitis after a single intranasal immunization with influenza A virus and nanoemulsion, Vaccine, 2003, 21, 3801–3814 CrossRef CAS PubMed.
- L. Yang, et al., Protective efficacy of the chimeric Staphylococcus aureus vaccine candidate IC in sepsis and pneumonia models, Sci. Rep., 2016, 6, 20929, DOI:10.1038/srep20929.
- G. Harris, et al., A mouse model of Acinetobacter baumannii-associated pneumonia using a clinically isolated hypervirulent strain, Antimicrob. Agents Chemother., 2013, 57, 3601–3613, DOI:10.1128/aac.00944-13.
- A. Joshi, et al., Immunization with Staphylococcus aureus iron regulated surface determinant B (IsdB) confers protection via Th17/IL17 pathway in a murine sepsis model, Hum. Vaccines Immunother., 2012, 8, 336–346, DOI:10.4161/hv.18946.
- D. Parker and A. Prince, Immunopathogenesis of Staphylococcus aureus pulmonary infection, Semin. Immunopathol., 2012, 34, 281–297, DOI:10.1007/s00281-011-0291-7.
- R. W. Frenck Jr, et al., Safety, tolerability, and immunogenicity of a 4-antigen Staphylococcus aureus vaccine (SA4Ag): Results from a first-in-human randomised, placebo-controlled phase 1/2 study, Vaccine, 2017, 35, 375–384, DOI:10.1016/j.vaccine.2016.11.010.
- W. H. Chen, et al., Safety and Immunogenicity of a Parenterally Administered, Structure-Based Rationally Modified Recombinant Staphylococcal Enterotoxin B Protein Vaccine, STEBVax, Clin. Vaccine Immunol., 2016, 23, 918–925, DOI:10.1128/cvi.00399-16.
- S. G. Reed, M. T. Orr and C. B. Fox, Key roles of adjuvants in modern vaccines, Nat. Med., 2013, 19, 1597–1608, DOI:10.1038/nm.3409.
- T. van der Poll and S. M. Opal, Pathogenesis, treatment, and prevention of pneumococcal pneumonia, Lancet, 2009, 374, 1543–1556, DOI:10.1016/S0140-6736(09)61114-4.
- K. Eriksson and J. Holmgren, Recent advances in mucosal vaccines and adjuvants, Curr. Opin. Immunol., 2002, 14, 666–672 CrossRef CAS PubMed.
- S. Chadwick, C. Kriegel and M. Amiji, Nanotechnology solutions for mucosal immunization, Adv. Drug Delivery Rev., 2010, 62, 394–407, DOI:10.1016/j.addr.2009.11.012.
- A. R. Spaulding, et al., Vaccination against Staphylococcus aureus pneumonia, J. Infect. Dis., 2014, 209, 1955–1962 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13630g |
| This journal is © The Royal Society of Chemistry 2018 |