DOI:
10.1039/C8RA01119B
(Paper)
RSC Adv., 2018,
8, 10009-10016
Acidic ionic liquid based UiO-67 type MOFs: a stable and efficient heterogeneous catalyst for esterification†
Received
5th February 2018
, Accepted 3rd March 2018
First published on 13th March 2018
Abstract
A facile strategy for the synthesis of acidic ionic liquid based UiO-67 type MOFs was developed in this study. Brønsted acids (H2SO4, CF3SO3H and hifpOSO3H (hexafluoroisopropyl sulfuric acid)) were introduced into UiO-67–bpy (bpy = 2,2′-bipyridine-5,5′-dicarboxylic acid) frameworks by reacting with bipyridyl nitrogen to introduce the properties of an acidic ionic liquid into the frameworks. The prepared catalysts, denoted as UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3, were characterized by XRD, SEM, FT-IR, EA, TGA and N2 adsorption–desorption studies. The relatively high surface area was still maintained and acidic active groups were uniformly dispersed in the frameworks. The catalytic performance of UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3 was evaluated by the esterification of acetic acid with isooctyl alcohol. The prepared catalysts showed good catalytic activities in the esterification, of which UiO-67–CF3SO3 gave the maximum isooctyl alcohol conversion of 98.6% under optimized conditions. The catalyst could be reused five times without a significant decrease in the conversion of isooctyl alcohol, and almost no active species were leached, indicating the excellent stability and reusability of the catalyst. Our study provides one effective way to synthesize heterogeneous acidic ionic liquid catalysts consisting of isolated, well defined acidic groups that will probably attract interest in acid catalyst chemistry.
1. Introduction
In the recent past, ionic liquids (ILs) have attracted much attention due to their unique properties such as eco-friendly nature, negligible vapour pressure, high thermal stability and low melting points.1–4 Specially, acidic ILs have shown efficient catalytic activities in various kinds of reactions including esterification,5 alkylation,6 dehydration7 and Diels–Alder reactions.8 However, their actual applications are greatly soured by shortcomings such as high viscosity and being difficult to recycle.9 In order to overcome these drawbacks, attempts have been made to introduce ILs onto various supports, such as mesoporous silica,10 activated carbon11 and zeolites,12 which will supress the cost of ILs and improve the reusability of ILs.
Metal–organic frameworks (MOFs) as a novel group of hybrid materials which have generated considerable research interest and have been applied widely in the areas of catalysis,13 adsorption,14 drug delivery,15 sensor,16 and catalyst carriers.17 Compared to conventional porous materials, MOFs have unique advantages including large surface area, high porosity, tunable topologies and low structure density which make MOFs as ideal catalyst carriers.18–21 Moreover, MOFs are highly designable, tuneable host–guest interactions can be created between MOFs and guest molecules. During the past decade, the introduction of ILs into MOFs has been reported, which are usually carried out by post-synthetic modification (PSM) strategy. Up to now, several approaches have been reported for the PSM of MOFs with ILs such as impregnating MOFs into ILs solutions22 and adopting the ship-in-bottle method for the introduction of ILs.23 By adopting PSM, ILs will be effectively introduced into MOFs and the basic frameworks remain unchanged,24 and the resulting materials had the properties of both ILs and MOFs which greatly extends the applications.25 Therefore, using PSM strategy in the areas of MOFs and ILs has become a promising research field.
Since its discovery, zirconium-based MOFs have drawn much attention due to its high thermo-chemical stability and acids resistive properties.26–28 The robust nature of Zr-MOFs endows them as excellent platforms for the applications in various fields.29 Among the reported Zr-based MOFs, UiO-67–bpy (bpy = 2,2′-bipyridine-5,5′-dicarboxylic acid) has been studied widely due to the rich coordination chemistry of the 2,2′-bipyridine moieties. The structure of UiO-67–bpy is octahedral and it crystalizes in cubic space group Fm3 with the fcu topology as shown in Fig. 1. Studies highlight the incorporation of noble metals/complexes or organometallic species on/in UiO-67–bpy by coordinating with the N atoms of the 2,2′-bipyridine moieties, and the resulting materials exhibit outstanding performance in catalysis and ratiometric thermometer.30–32 However, to the best of our knowledge, only Lei Xu et al. has reported the introduction of ILs into UiO-67–bpy by N-quaternization of bipyridine sites.29 In the delight of the idea of Kavitha who prepared 2,2-bipyridinium hydrogensulfate by treating 2,2′-bipyridine with sulfuric acid,33 it is feasible to introduce the properties of acidic ILs unto UiO-67–bpy by reacting bipyridyl groups with Brønsted acids. Due to the acidic nature, the resulting materials could be applied as heterogeneous catalysts in various chemical reactions.
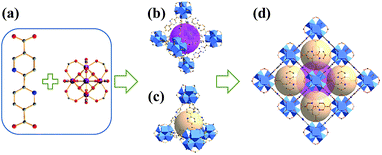 |
| | Fig. 1 (a) Linker of bpydc and Zr6 cluster (gray: C; red: O; blue: N; dark pink: Zr), (b) the octahedral cage, (c) the tetrahedral cage, (d) unit cell of UiO-67–bpy. Hydrogen atoms are omitted for clarity. | |
Herein, we report a facile PSM strategy to synthesize acidic ionic liquid based UiO-67 type MOF catalysts, by treating the bipyridyl groups of UiO-67–bpy with different Brønsted acids (H2SO4, CF3SO3H and hifpOSO3H (hexafluoroisopropyl sulfuric acid)). The newly prepared materials were characterized by XRD, SEM, FT-IR, EA, TGA and N2 adsorption–desorption studies. The catalytic performance of synthesized catalysts was then evaluated by esterification of isooctyl alcohol with acetic acid. The reaction conditions were further optimized and the reusability of the catalysts was also carried out.
2. Experimental
2.1 Materials
N,N-Dimethylformamide (DMF, 99.5%), isooctyl alcohol (≥99%) and anhydrous methanol were obtained from Xilong Chemical Industry Incorporated Co., Ltd. Benzoic acid was purchased from Sinopharm Chemical Reagents Co., Ltd. 2,2′-Bipyridine-5,5′-dicarboxylicacid (bpydc, 98%) was provided by Zhengzhou Alpha Chemical Co., Ltd. Zirconium tetrachloride (ZrCl4, 98%), hexafluoroisopropanol (99.5%) and trifluoromethanesulfonic acid (CF3SO3H, 98%) was supplied by Aladdin Chemical Co., Ltd. Chlorosulfonic acid (ClHSO3, 97%) was purchased from Beijing Ouhe Technology Co., Ltd. Acetic acid (99.5%), anhydrous dichloromethane (CH2Cl2) and sulfuric acid (H2SO4) were obtained from Beijing Chemical Works. De-ionized water (99.5%) was obtained from home-built distillation assembly. All chemicals were used without further purification.
2.2 Characterization techniques
NMR data (1H, 13C and 19F) were collected on the AVANCE III HD-600 MHz NMR spectrometer. Powder X-ray diffraction (PXRD) patterns were performed on the Rigaku smartlab X-ray diffractometer equipped with a Cu-Kα1 radiation source (λ = 0.15406 nm) and the XRD patterns were collected in the steps of 0.02° between the 2θ range of 5–50°. The simulated powder pattern was carried out based on crystal structure data of UiO-67–bpy as reported.34 The morphologies and structures were investigated by the Hitachi SU8020 scanning electron microscopy (SEM) equipped with Energy Dispersive Spectrometry (EDS) at an accelerating voltage of 5 kV and 20 kV, respectively. All the samples were mounted on a carbon tape and coated with gold prior to measurement. Fourier transformed infrared (FT-IR) was conducted on the Nicolet 380 spectroscopy of Thermo Electron Corporation, using the KBr pellet methodology, in the range of 4000–400 cm−1 with a resolution of 4 cm−1. Element Analysis (EA) was performed with the Elementar Vario Macro Cube to determine the N and S content of the prepared catalysts. The N2 adsorption–desorption isotherms were collected using the Micromeritics ASAP 2020 HD88 surface area and porosity analyzer at 77 K. Prior to the measurements, the samples were activated under vacuum at 120 °C for 12 h. Thermogravimetric analysis (TGA) was performed on the Shimadzu TA-60WS Thermal analyzer in the range of 40–700 °C at a heating rate of 5 °C min−1 under air flow. Inductively coupled plasma-optical emission spectrometry (ICP-OES) experiments were conducted on the Shimadzu Plasma Atomic Emission Spectrometer. Gas Chromatography Mass Spectrometry (GC-MS) analysis was carried out on the Shimadzu GCMS-QP2020 equipped with a HP-5 capillary column (30 m × 0.25 mm, using N2 as carrier gas) and GC analysis were performed on the Shimadzu GC-2014 equipped with a HP-5 capillary column (30 m × 0.25 mm, using N2 as carrier gas).
2.3 Preparation of UiO-67–bpy
The synthesis method of UiO-67–bpy was derived from Andreas Schaate35 and Sigurd Øien36 with slight modification. Typically, ZrCl4 (2.58 mmol, 602 mg) and DMF (100 mL, 500 eq. based on Zr salt) were placed in a 100 mL Teflon-capped glass flask. Benzoic acid (9.46 g, 30 eq. based on Zr salt) was also added to the flask which acted as a modulator. The precursors were sonicated for 30 minutes for dissolution. Then bpydc (2.58 mmol, 626 mg) and H2O (3.4 mmol, 62 μL) was added to the solution, dispersed by ultrasound until no suspended particles were visible. The tightly capped flask was kept in a pre-heated oven at 95 °C for 4 days. After the flask cooled down to room temperature naturally, the white precipitate was collected by filtration and washed 3 times in anhydrous DMF (15 mL), followed by washed 3 times in anhydrous methanol (15 mL). Finally, the exchanged sample was dried under reduced pressure at 120 °C overnight.
2.4 Preparation of hexafluoroisopropyl sulfuric acid (hifpOSO3H)
The preparation method of hifpOSO3H was following a modified procedure reported by Witali Beichel.37 Typically, ClHSO3 (0.1 mol, 11.652 g) was placed in a three-necked flask at −78 °C to precooling. Following by slowly adding hexafluoroisopropanol (0.1 mol, 16.804 g) to the flask using a constant pressure dropping funnel and then stirring at 140 °C under reflux until the gas evolution had ceased. Finally, the product was distilled at reduced pressure at 140 °C gave the pure acid as a colorless liquid. The structure was confirmed by NMR analysis and NMR data was available in ESI.†
2.5 Preparation of acidic ionic liquid based UiO-67 type MOF
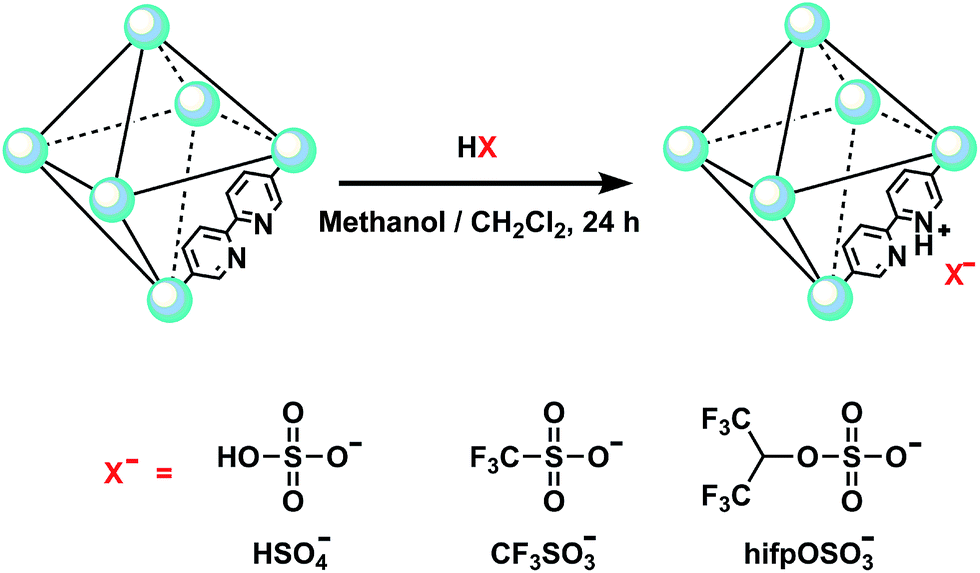
The simplified preparation processes of acidic ionic liquid based UiO-67 type MOF (denoted as UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3) were shown in Scheme 1. Before preparation, the as-synthesized UiO-67–bpy powders were evacuated at 120 °C for 6 h to active powders. For the preparation of UiO-67–HSO4, the activated powders (0.2 g) were immersed in H2SO4 (200 mL, 0.01 M) using anhydrous methanol as solvent for 24 h, during which time the mixture was stirred one minute every two hours. After treatment, the solution was decanted off and the remained powders were collected by filtration and washed several times with anhydrous methanol (15 mL). Finally, the powders were dried under dynamic vacuum at 120 °C for 12 h.
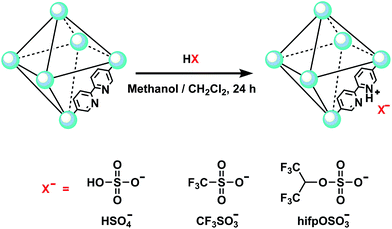 |
| | Scheme 1 Scheme for the post-synthesis of acidic ionic liquid based UiO-67 type MOF and the structures of the Brønsted acid anions. The topology is shown in a simplified form as an octahedral cage. | |
The preparation of UiO-67–CF3SO3 and UiO-67–hifpOSO3 were similar to the preparation of UiO-67–HSO4, except that CF3SO3H (2 mmol, 0.3002 g) and hfipOSO3H (2 mmol, 0.4962 g) were added dropwise to anhydrous CH2Cl2 (200 mL) in an ice bath, respectively. Then activated powders (0.2 g) were immersed in the resulting solution for 24 h, during which time the mixture was stirred one minute every two hours. After treatment, the solution was decanted and the remaining powders were collected by filtration and washed several times with anhydrous CH2Cl2 (15 mL). Finally, the powders were dried under dynamic vacuum at 120 °C for 12 h.
2.6 Catalytic reaction
The catalytic activity of the prepared catalysts was evaluated by the esterification of acetic acid with isooctyl alcohol. The reaction was carried out in the round bottom flask equipped with a magnetic stirrer and connected to a reflux condenser. In a typical reaction, a mixture of isooctyl alcohol and acetic acid with molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 were charged into the flask containing 0.2 g the prepared catalysts. Then the reaction mixture was heated to 80 °C with continuously stirring for 18 h. After the reaction, the reaction mixture was withdrawn, the catalyst was separated by centrifugation and the liquid phase was analyzed by GC-MS to define the structure of products, and GC was applied to analyse the conversion of isooctyl alcohol and the yield of isooctyl acetate.
:6 were charged into the flask containing 0.2 g the prepared catalysts. Then the reaction mixture was heated to 80 °C with continuously stirring for 18 h. After the reaction, the reaction mixture was withdrawn, the catalyst was separated by centrifugation and the liquid phase was analyzed by GC-MS to define the structure of products, and GC was applied to analyse the conversion of isooctyl alcohol and the yield of isooctyl acetate.
3. Results and discussion
3.1 Catalysts characterization
The powder XRD patterns of the simulated UiOy-67–bpy, the pristine UiO-67–bpy, and the synthesized catalysts that treated with different Brønsted acids were shown in Fig. 2. The diffraction peaks for the pristine UiO-67–bpy agreed with the simulated one which based on the single crystal data of UiO-67–bpy as reported.34 All the main diffraction peak positions of UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3 were matched well with the simulated pattern and the pristine one, apart from some slight variations in the Bragg intensities. Furthermore, from the inserted picture in Fig. 1, the three sharp peaks at 5.76° (1 1 1), 6.66° (2 0 0) and 9.46° (2 2 0) for the synthesized catalysts indicated that they were highly crystalline. These results indicated that the integrity of framework was preserved for the prepared catalysts through the PSM processes under acidic conditions.
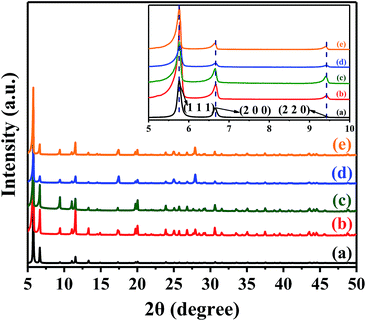 |
| | Fig. 2 Powder XRD patterns of (a) the simulated UiO-67–bpy, (b) the pristine UiO-67–bpy, (c) UiO-67–HSO4, (d) UiO-67–CF3SO3, (e) UiO-67–hifpOSO3. | |
To investigate the surface morphologies and crystalloid structures of the synthesized catalysts, scanning electron microscopy (SEM) was carried out and the results were given in Fig. 3. The pristine UiO-67–bpy crystals had well defined octahedral microcrystals with a smooth surface, and the average crystals size was around 1.3 μm (Fig. S1(a)†). The well-defined octahedral morphology was still maintained for the synthesized catalysts and the average particles size almost remain the same, about 1.2 μm (Fig. S1(b), (c), (d)†), after treated by Brønsted acids solutions of H2SO4, CF3SO3H and hifpOSO3H, respectively. However, the surface of UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3 tended to be rough. Meanwhile, it could be also observed that a small number of UiO-67–bpy crystallites were disrupted or slightly changed to irregular ones which could be attributed to the collapse of UiO-67–bpy frameworks.
 |
| | Fig. 3 SEM images of (a) the pristine UiO-67–bpy, (b) UiO-67–HSO4, (c) UiO-67–CF3SO3, (d) UiO-67–hifpOSO3. | |
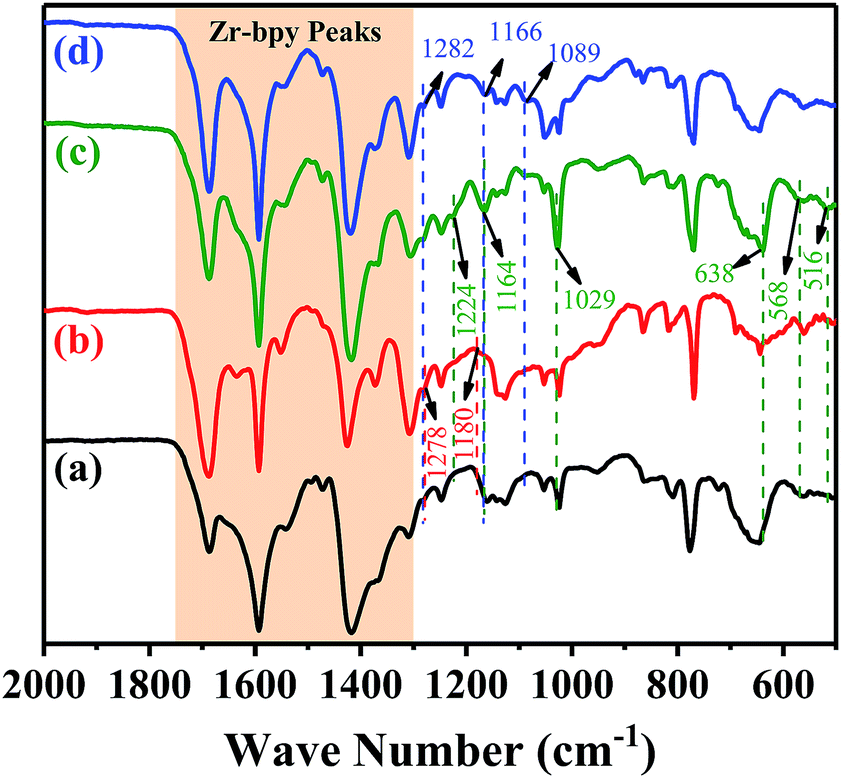
FT-IR studies were used to confirm whether the acidic characteristic groups had been successfully introduced into the frameworks. The enlarged partial FT-IR spectra of the pristine UiO-67–bpy and the prepared catalysts were shown in Fig. 4. For all samples, the observed vibration bands between 1300–1750 cm−1, which corresponding to carboxylate linkers38 of the pristine UiO-67–bpy were well maintained in the all resulting materials. For the UiO-67–HSO4, two peaks were observed at 1278 cm−1 and 1180 cm−1 corresponding the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO symmetric and asymmetric stretching vibrations, respectively. In the case of UiO-67–CF3SO3, two bands at 1224 cm−1 and 1164 cm−1 could be ascribed to the OSO symmetric and asymmetric stretching vibrations, respectively, and the peak at 1029 cm−1 corresponding to the C–F stretching. Furthermore, the bands at 638 cm−1, 568 cm−1, and 516 cm−1 could be attributed to the C–S stretching. The peaks at 1282 cm−1 and 1166 cm−1 observed over UiO-67–hifpOSO3 catalyst were the OSO symmetric and asymmetric stretching vibrations, respectively, and the peak at 1089 cm−1 corresponding to the C–F stretching. These results indicated that the acidic characteristic groups of the synthesized catalysts were introduced into the UiO-67–bpy matrix successfully.
SO symmetric and asymmetric stretching vibrations, respectively. In the case of UiO-67–CF3SO3, two bands at 1224 cm−1 and 1164 cm−1 could be ascribed to the OSO symmetric and asymmetric stretching vibrations, respectively, and the peak at 1029 cm−1 corresponding to the C–F stretching. Furthermore, the bands at 638 cm−1, 568 cm−1, and 516 cm−1 could be attributed to the C–S stretching. The peaks at 1282 cm−1 and 1166 cm−1 observed over UiO-67–hifpOSO3 catalyst were the OSO symmetric and asymmetric stretching vibrations, respectively, and the peak at 1089 cm−1 corresponding to the C–F stretching. These results indicated that the acidic characteristic groups of the synthesized catalysts were introduced into the UiO-67–bpy matrix successfully.
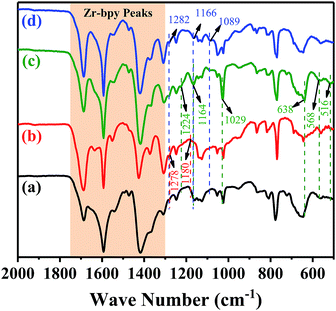 |
| | Fig. 4 FT-IR spectra for (a) the pristine UiO-67–bpy, (b) UiO-67–HSO4, (c) UiO-67–CF3SO3, (d) UiO-67–hifpOSO3. | |
To confirmed the N and S content in UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3, element analysis (EA) was carried out and the results were summarized in Table 1. It could be observed that, for the prepared catalysts, the molar ratio of N to S was all around 2:1, which indicated that each linker of UiO-67–bpy was reacted with one acids molecule. Furthermore, as indicated by EDS elemental mapping (Fig. S2†) the acidic characteristic groups for the synthesized catalysts were uniformly dispersed in the frameworks.
Table 1 N and S content for the synthesized catalysts
| Entry |
Sample |
N (wt%) |
S (wt%) |
N:Sa |
| Molar ratio. |
| 1 |
UiO-67–HSO4 |
6.50 |
7.28 |
2.04:1 |
| 2 |
UiO-67–CF3SO3 |
5.57 |
6.65 |
1.91: 1 |
| 3 |
UiO-67–hifpOSO3 |
4.65 |
5.08 |
2.09:1 |
N2 adsorption–desorption isotherms of the pristine UiO-67–bpy UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3 were shown in Fig. 5 and the pore size distributions for the all samples were depicted in Fig. S3.† Table 2 summarized the textural properties of the all samples including BET surface area, pore volume and average pore size. The pristine UiO-67–bpy exhibited type-I isotherm, which revealed its microporous nature. The pristine UiO-67–bpy showed a BET surface area and pore volume of 1802.5 m2 g−1 and 0.6926 cm3 g−1, which were comparable with the reported results.39 The isotherms for UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3 also exhibited type-I isotherms which was consistent with the pristine one and showed no sign of bulging indicating the robust nature of UiO-67–bpy and the pores were not damaged during the preparation conditions. Compared with the pristine UiO-67–bpy, both surface area and pore volume for all of the synthesized catalysts had been decreased from 1802.5 m2 g−1 to 1014.7 m2 g−1 and from 0.6926 cm3 g−1 to 0.3880 cm3 g−1, respectively. The decrease in BET surface area and pore volume could be attributed to the introduction of acidic characteristic groups into the cages of UiO-67–bpy and might partially occupied the channels of the frameworks. Furthermore, the optimized geometric structures of acetic acid, isooctyl alcohol and isooctyl acetate were shown in Fig. S4.† Comparing their sizes with the prepared catalysts, it could be concluded that the open cavities for the prepared catalysts could still benefit the diffusion of the reactants and products.
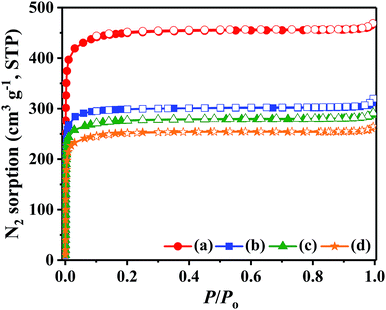 |
| | Fig. 5 N2 adsorption–desorption isotherms for (a) the pristine UiO-67–bpy, (b) UiO-67–HSO4, (c) UiO-67–CF3SO3, (d) UiO-67–hifpOSO3. | |
Table 2 The textural properties of the pristine UiO-67–bpy and the synthesized catalysts
| Entry |
Sample |
BET surface area (m2 g−1) |
Pore volumea (cm3 g−1) |
Average pore size (nm) |
| Calculated by using Horvath–Kawazoe (HK) method. |
| 1 |
Pristine UiO-67–bpy |
1802.5 |
0.6926 |
1.4257 |
| 2 |
UiO-67–HSO4 |
1232.9 |
0.4610 |
1.1795 |
| 3 |
UiO-67–CF3SO3 |
1124.4 |
0.4254 |
1.0701 |
| 4 |
UiO-67–hifpOSO3 |
1014.7 |
0.3880 |
1.0201 |
Since the synthesized catalysts were achieved by treating UiO-67–bpy powders with strong Brønsted acids, it was necessary to substantiate the thermal and structural stability. Thermogravimetric analysis (TGA) was conducted and TGA curves were depicted in Fig. 6. The two-stepped thermal degradation was observed for the pristine UiO-67–bpy which was consistent with the reported result.35 The initial weight loss before 150 °C was correspond to the vaporization of absorbed water and solvent inner/outer the surfaces of the framework. The second degradation step was emerged about 450 °C which was attributed to the decomposition of the frameworks. The first degradation step of the synthesized catalysts were occurred before 200 °C which was comparable to the pristine one. The rapid weight loss in the temperature range of 300–380 °C for UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3 may be attributed to decomposition of HSO4−, CF3SO3− and hifpOSO3−, respectively. When the temperature increased around 400 °C, the frameworks of UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3 tended to degrade and collapse. The results indicated the decomposition temperature for the synthesized catalysts were slightly lower than the pristine one. But the thermostability of the prepared catalysts was much better than commercial catalyst Amberlyst-15 of which usually used in esterification reactions with the thermostability of 120 °C.40 Thus the synthesized UiO-67–HSO4, UiO-67–CF3SO3 and UiO-67–hifpOSO3 catalysts can be employed as heterogeneous catalysts for various organic reactions.
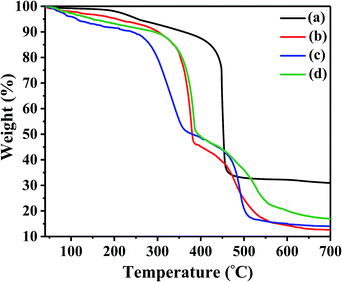 |
| | Fig. 6 TGA curves of (a) the pristine UiO-67–bpy, (b) UiO-67–HSO4, (c) UiO-67–CF3SO3, (d) UiO-67–hifpOSO3. | |
3.2 Catalytic assessment
Esterification is one of the fundamental reactions in the field of organic synthesis. The product organic esters are considered as high production-volume (HPV) chemicals. They are important intermediates in pharmaceutical, cosmetic and polymer industries for the production of fragrances, polymers, polyesters, paints and plasticizers.41–43 Thus, we chose this reaction to probe the catalytic performance of prepared catalysts.
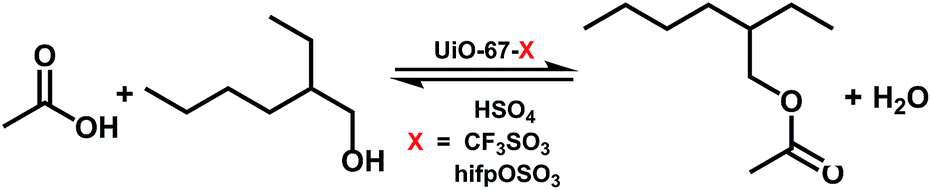
3.2.1 Catalytic activities of the prepared catalysts. The catalytic performance of the synthesized catalysts was evaluated by choosing esterification of acetic acid and isooctyl alcohol as the probe reaction as shown in Scheme 2. The results under different reaction time by using the prepared catalysts were shown in Fig. 7. The conversion of isooctyl alcohol was increased with increasing the reaction time and became steady after 18 h. Without catalyst, the conversion of isooctyl alcohol was only 66.4% and the yield of isooctyl acetate was 56.7%. With the pristine UiO-67–bpy, the conversion of isooctyl alcohol was 77.5% after 21 h with 70.5% yield of isooctyl acetate due to the unsaturated site at Zr ion clusters with could provide Lewis acidity.48 It was uplifting that the synthesized catalysts showed increased catalytic performance. By employing these three prepared catalysts, the conversion of isooctyl alcohol was over 85.0% because of the strong acidity provided by the introduction of the acidic active sites that promote the esterification effectively. Obviously, the catalytic performance of UiO-67–CF3SO3 was the best amongst all the catalysts employed and gave 98.0% isooctyl alcohol conversion which may attribute to the stronger acidity as compared to UiO-67–HSO4 and UiO-67–hifpOSO3. Thus, the reaction conditions were further optimized by employing the UiO-67–CF3SO3 catalyst.
 |
| | Scheme 2 Esterification of acetic acid with isooctyl alcohol using the prepared catalysts. | |
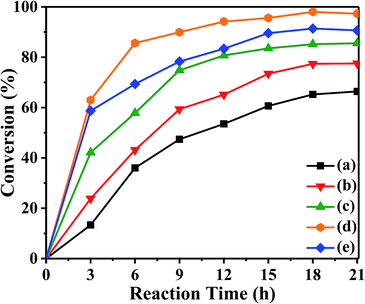 |
| | Fig. 7 The catalytic activities of prepared catalysts (a) blank, (b) the pristine UiO-67–bpy, (c) UiO-67–HSO4, (d) UiO-67–CF3SO3, (e) UiO-67–hifpOSO3. Reaction conditions: catalyst amount 0.2 g, isooctyl alcohol to acetic acid molar ratio 1:6, at 80 °C. | |
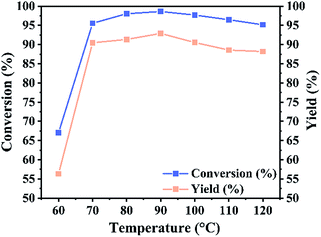
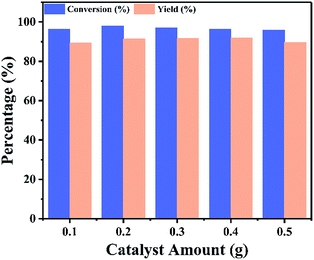
3.2.2 Optimization of reaction variables. The effect of catalyst amount on the conversion of isooctyl alcohol was investigated at 18 h, 80 °C and molar ratio of isooctyl alcohol to acetic acid 1:6, the reaction results were displayed in Fig. 8. The conversion of isooctyl alcohol was increased with the increasing of catalyst amount. However, the conversion of isooctyl alcohol was not affected by further increasing the catalyst amount, due to the reaction reached equilibrium. Since reaction temperature is the dominant factor in every chemical reaction, the effect of temperature in our system was subsequently investigated from 60 to 120 °C. As shown in Fig. 9, it could be observed that the optimum temperature for the esterification was 90 °C. Further increased reaction temperature did not show any significant change on the conversion of isooctyl alcohol, however when decreased the reaction temperature to 60 °C, the reaction was greatly affected, only 67.0% conversion and 56.3% yield obtained as compared to 98.6% conversion and 92.9% yield at 90 °C.
 |
| | Fig. 8 Catalyst amount effect upon the catalytic performance of UiO-67–CF3SO3. Reaction conditions: isooctyl alcohol to acetic acid molar ratio 1:6, for 18 h, at 80 °C. | |
 |
| | Fig. 9 Reaction temperature effect upon the catalytic performance of UiO-67–CF3SO3. Reaction conditions: catalyst amount 0.2 g, isooctyl alcohol to acetic acid molar ratio 1:6, for 18 h. | |
3.2.3 Catalytic activities of different catalysts. In order to investigate the catalytic performance of target catalyst UiO-67–CF3SO3, the catalytic performance of different catalysts in this work and reported by others for the esterification of acetic acid and isooctyl alcohol were summarized in Table 3. It should be noted that the reaction conditions performed for the investigation were catalyst amount 0.2 g, molar ratio of isooctyl alcohol to acetic acid 1:6, reaction temperature 90 °C and reaction time 18 h. As shown in Table 2, in this work, under optimized conditions, the pristine UiO-67–bpy could achieve 80.7% conversion of isooctyl alcohol. While, the UiO-67–CF3SO3 obtained much better catalytic performance and the conversion of isooctyl alcohol could reach 98.6%. Moreover, we used commercial H-Beta zeolite for the reaction and could reach 92.2% conversion of isooctyl alcohol. In Table 3, some reported catalysts were listed for comparison (entries 4–7), and the results showed that the catalytic activity of UiO-67–CF3SO3 was comparable to or even better than the reported catalysts indicating that UiO-67–CF3SO3 would be an excellent catalyst for esterification.
Table 3 Catalytic performance of different catalysts for the esterification of isooctyl alcohol
| Entry |
Catalyst |
Ioaa:Aab |
Conversion (%) |
Yield (%) |
Reference |
| Ioa = isooctyl alcohol. Aa = acetic acid. Mesoporous zirconium oxophosphate. |
| 1 |
Pristine |
1:6 |
80.7 |
73.7 |
This work |
| 2 |
UiO-67–CF3SO3 |
1:6 |
98.6 |
92.9 |
This work |
| 3 |
H-Beta |
1:6 |
92.2 |
90.1 |
This work |
| 4 |
H-Beta |
1:17.5 |
93.0 |
— |
44 |
| 5 |
MZrPc |
1:3 |
84.5 |
— |
45 |
| 6 |
(NH4)8[CeW10O36]·20H2O |
1:52 |
— |
96.0 |
46 |
| 7 |
[ChCl][CrCl3·6H2O]2 |
1:5 |
— |
64.9 |
47 |
In order to explore the catalytic application of UiO-67–CF3SO3 as a catalyst for other esterification, different acids and alcohols were chosen as the substrates for investigation. As shown in Table 4, the UiO-67–CF3SO3 showed excellent catalytic performance in these reactions which indicated that UiO-67–CF3SO3 could be used as an efficient catalyst for esterification.
Table 4 Results of esterification for different acids and alcohols catalysed by UiO-67–CF3SO3a
| Entry |
Acids |
Alcohols |
Conversion of alcohols (%) |
| Reaction conditions: catalyst amount 0.2 g, alcohols to acids molar ratio 1:6, for 18 h, at 90 °C. |
| 1 |
Acetic acid |
Methanol |
99.2 |
| 2 |
Acetic acid |
Ethanol |
97.8 |
| 3 |
Acetic acid |
Cyclohexanol |
85.1 |
| 4 |
Formic acid |
Isopropanol |
90.7 |
| 5 |
Formic acid |
1-Pentanol |
94.6 |
| 6 |
Formic acid |
n-Octanol |
93.5 |
3.3 Reusability of the catalyst
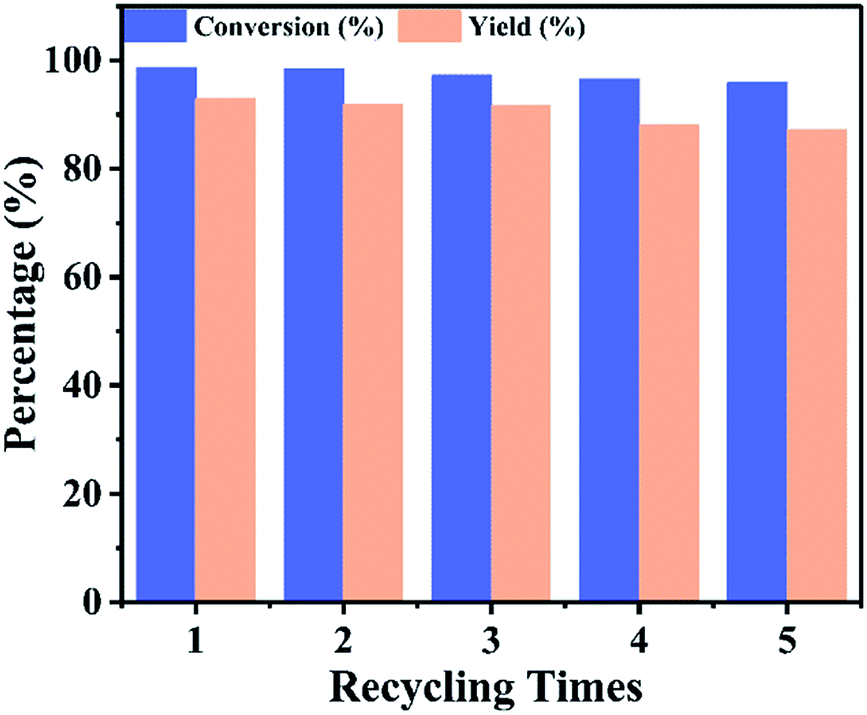
The reusability of a catalyst is prime important, consequently the reusability of UiO-67–CF3SO3 was investigated, the esterification reaction was repeated several times under the optimized experimental conditions, the results were shown in Fig. 10. After each reaction, the catalyst was separated from the reaction mixture by using simple centrifugation (8000 rpm for 10 min), washed with anhydrous dichloromethane and dried under vacuum overnight at 80 °C, then reused in the next run. The conversion of isooctyl alcohol decreased from 98.6% to 95.9% and the yield of isooctyl acetate decreased from 92.9% to 87.1%. These results indicated high reusability of the UiO-67–CF3SO3 catalyst. The decrease in conversion and yield for the UiO-67–CF3SO3 catalyst might be attributed to the blockage of MOFs channels and partially decomposition of catalyst which could be also observed in Fig. S5(a)† of the recovered catalyst, however, the size still maintained around 1.2 μm (Fig. S5(b)†). Next, the leaching of the active species of UiO-67–CF3SO3 was investigated. After the reaction, the solid catalyst was separated from the reaction mixture, the liquid phase was diluted and then tested by ICP-OES, the result showed that almost no S content was detected. According to these results, it could be confirmed the active species were effectively introduced into UiO-67–bpy and UiO-67–CF3SO3 possessed excellent catalytic activity.
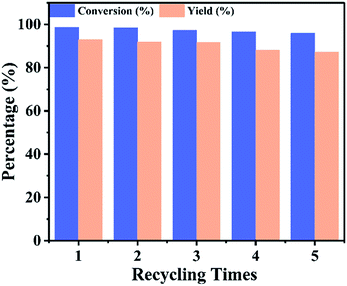 |
| | Fig. 10 Catalyst reusability of UiO-67–CF3SO3. Reaction conditions: catalyst amount 0.2 g, isooctyl alcohol to acetic acid molar ratio 1:6, for 18 h, at 90 °C. | |
4. Conclusion
Brønsted acids (H2SO4, CF3SO3H and hifpOSO3H) were introduced into the frameworks of UiO-67–bpy effectively by a facile PSM strategy for the synthesis of acidic ionic liquid based UiO-67 type MOF. The relative high surface area for all prepared catalysts were maintained and acidic active groups were dispersed in the frameworks uniformly. The prepared catalysts showed better catalytic activities in the esterification of acetic acid with isooctyl alcohol and UiO-67–CF3SO3 catalyst showed best catalytic performance. Under optimized conditions, catalyst amount 0.2 g, isooctyl alcohol to acetic acid molar ratio 1:6, for 18 h, at 90 °C, the UiO-67–CF3SO3 catalyst could give maximum isooctyl alcohol conversion 98.6% and isooctyl acetate yield 92.9%. The UiO-67–CF3SO3 catalyst could be reused five times with no obvious reduction in catalytic performance and no obvious leaching was detected, which indicated the excellent stability and reusability of UiO-67–CF3SO3.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We gratefully acknowledge the National Key Research and Development Program of China (No. 2016YFB0601303), the National Natural Science Foundation of China (No. 21676278), the Instrument Developing Project of the Chinese Academy of Sciences (No. YZ201624) and Science and Technology Open Cooperation Project of Henan Province (No. 172106000066) for financial support.
References
- N. Luo, Y. Lv, D. Wang, J. Zhang, J. Wu, J. He and J. Zhang, Chem. Commun., 2012, 48, 6283–6285 RSC.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- K. Dong, X. Liu, H. Dong, X. Zhang and S. Zhang, Chem. Rev., 2017, 117, 6636–6695 CrossRef CAS PubMed.
- Z. Ouyang, D. Bao, X. Zhang, H. Dong, R. Yan, X. Zhang and S. Zhang, Sci. China: Chem., 2015, 58, 1918–1928 CrossRef CAS.
- F. Su and Y. H. Guo, Green Chem., 2014, 16, 2934–2957 RSC.
- K. S. Yoo, V. V. Namboodiri, R. S. Varma and P. G. Smirniotis, J. Catal., 2004, 222, 511–519 CrossRef CAS.
- Y. Zhang, E. A. Pidko and E. E. Hensen, Chem.–Eur. J., 2011, 17, 5281–5288 CrossRef CAS PubMed.
- K. Matuszek, S. Coffie, A. Chrobok and M. Swadźbakwaśny, Catal. Sci. Technol., 2017, 7, 1045–1049 CAS.
- H. Wan, C. Chen, Z. Wu, Y. Que, Y. Feng, W. Wang, L. Wang, G. Guan and X. Liu, Chemcatchem, 2015, 7, 441–449 CrossRef CAS.
- C. Paun, J. Barklie, P. Goodrich, H. Q. N. Gunaratne, A. Mckeown, V. I. Pârvulescu and C. Hardacre, J. Mol. Catal. A: Chem., 2007, 269, 64–71 CrossRef CAS.
- M. J. Schneider, M. Haumann, M. Stricker, J. Sundermeyer and P. Wasserscheid, J. Catal., 2014, 309, 71–78 CrossRef CAS.
- B. Karimi and M. Vafaeezadeh, Chem. Commun., 2012, 48, 3327–3329 RSC.
- B. An, L. Zeng, M. Jia, Z. Li, Z. Lin, Y. Song, Y. Zhou, J. Cheng, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 17747–17750 CrossRef CAS PubMed.
- N. A. Khan, Z. Hasan and S. H. Jhung, Chem. Commun., 2016, 52, 2561–2564 RSC.
- S. Hashemipour and H. A. Panahi, J. Mol. Liq., 2017, 243, 102–107 CrossRef CAS.
- B. Yuan, J. Zhang, R. Zhang, H. Shi, N. Wang, J. Li, F. Ma and D. Zhang, Sens. Actuators, B, 2016, 222, 632–637 CrossRef CAS.
- Q. X. Luo, B. W. An, M. Ji, S. E. Park, C. Hao and Y. Q. Li, J. Porous Mater., 2015, 22, 247–259 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. Okeeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276–288 RSC.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS.
- H. Y. Min, K. L. Fow and G. Z. Chen, Green Energy & Environment, 2017, 2, 218–245 Search PubMed.
- X. L. Sun, W. H. Deng, H. Chen, H. L. Han, J. M. Taylor, C.
Q. Wan and G. Xu, Chem.–Eur. J., 2017, 23, 1248–1252 CrossRef CAS PubMed.
- M. Han, Z. Gu, C. Chen, Z. Wu, Y. Que, Q. Wang, H. Wan and G. Guan, RSC Adv., 2016, 6, 37110–37117 RSC.
- C. Chen, Z. Wu, Y. Que, B. Li, Q. Guo, Z. Li, L. Wang, H. Wan and G. Guan, RSC Adv., 2016, 6, 54119–54128 RSC.
- F. P. Kinik, A. Uzun and S. Keskin, ChemSusChem, 2017, 10, 2842–2863 CrossRef CAS PubMed.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- H. Fei, S. Pullen, A. Wagner, S. Ott and S. M. Cohen, Chem. Commun., 2015, 51, 66–69 RSC.
- Z. Hu and D. Zhao, Dalton Trans., 2015, 44, 19018–19040 RSC.
- L. Xu, Y. Luo, L. Sun, S. Pu, M. Fang, R. Yuan and H. Du, Dalton Trans., 2016, 45, 8614–8621 RSC.
- M. I. Gonzalez, E. D. Bloch, J. A. Mason, S. J. Teat and J. R. Long, Inorg. Chem., 2015, 54, 2995–3005 CrossRef CAS PubMed.
- Y. Zhou and B. Yan, J. Mater. Chem. C, 2015, 3, 9353–9358 RSC.
- K. Manna, T. Zhang and W. Lin, J. Am. Chem. Soc., 2014, 136, 6566–6569 CrossRef CAS PubMed.
- S. J. Kavitha, K. Panchanatheswaran, J. N. Low, G. Ferguson and C. Glidewell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o165–o169 Search PubMed.
- L. Li, S. Tang, C. Wang, X. Lv, M. Jiang, H. Wu and X. Zhao, Chem. Commun., 2014, 50, 2304–2307 RSC.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- S. Øien, G. Agostini, S. Svelle, E. Borfecchia, K. A. Lomachenko, L. Mino, E. Gallo, S. Bordiga, U. Olsbye and K. P. Lillerud, Chem. Mater., 2015, 27, 1042–1056 CrossRef.
- W. Beichel, J. M. U. Panzer, J. Hatty, X. Ye, D. Himmel and I. Krossing, Angew. Chem., Int. Ed., 2014, 53, 6637–6640 CrossRef CAS PubMed.
- W. Salomon, C. Rochmarchal, P. Mialane, P. Rouschmeyer, C. Serre, M. Haouas, F. Taulelle, S. Yang, L. Ruhlmann and A. Dolbecq, Chem. Commun., 2015, 51, 2972–2975 RSC.
- H. Fei and S. M. Cohen, Chem. Commun., 2014, 50, 4810–4812 RSC.
- S. Suzuki, Y. Ono, S. Nakata and S. Asaoka, J. Phys. Chem., 1987, 91, 1659–1663 CrossRef CAS.
- V. C. Gyani, B. Reddy, R. Bhat and S. Mahajani, Ind. Eng. Chem. Res., 2014, 53, 15811–15823 CrossRef CAS.
- Y. Leng, J. Wang, D. Zhu, X. Ren, H. Ge and L. Shen, Angew. Chem., Int. Ed., 2009, 48, 168–171 CrossRef CAS PubMed.
- A. K. Chakraborti, B. Singh, S. V. Chankeshwara and A. R. Patel, J. Org. Chem., 2009, 74, 5967–5974 CrossRef CAS PubMed.
- K. V. V. K. Mohan, N. Narender and S. J. Kulkarni, Green Chem., 2006, 8, 368–372 RSC.
- S. K. Das, M. K. Bhunia, A. K. Sinha and A. Bhaumik, ACS Catal., 2011, 1, 493–501 CrossRef CAS.
- V. Mirkhani, S. Tangestaninejad, M. Moghadam, B. Yadollahi and L. Alipanah, Monatsh. Chem., 2004, 135, 1257–1263 CrossRef CAS.
- J. Cao, B. Qi, J. Liu, Y. Shang, H. Liu, W. Wang, J. Lv, Z. Chen, H. Zhang and X. Zhou, RSC Adv., 2016, 6, 21612–21616 RSC.
- U. S. F. Arrozi, H. W. Wijaya, A. Patah and Y. Permana, Appl. Catal., A, 2015, 506, 77–84 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01119b |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Guoying Zhao*a,
Latif Ullaha,
Meng Wanga,
Aoyun Wanga,
Yanqiang Zhang
ab,
Guoying Zhao*a,
Latif Ullaha,
Meng Wanga,
Aoyun Wanga,
Yanqiang Zhang