
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVEGF heparinized-decellularized adipose tissue scaffolds enhance tissue engineering vascularization in vitro
Mei Songa,
Yu Zhou*b and
Yi Liua
aBurns and Plastic Surgery Center, Lanzhou General Hospital of the People's Liberation Army, Lanzhou, Gansu, 730050 P. R. China
bBeijing Lu Daopei Institute of Hematology, Beijing, 100176 P. R. China. E-mail: liuyi196402@126.com; yu.zhou@ludaopei.com
First published on 1st October 2018
Abstract
Scaffolds based on decellularized adipose tissue (DAT) are gaining popularity in the adipose tissue engineering field due to their high biocompatibility and vascularizing properties. Previous studies involving decellularized adipose tissue (DAT) scaffolds have not fully demonstrated their ability to induce in vitro vascularization of engineered tissue. With the aim of developing a more effective adipose tissue engineering vascularization technique based on DAT, we investigated the vascularizing potential of a vascular endothelial growth factor (VEGF) delivery system utilizing a heparinized DAT (Hep-DAT) scaffold in vitro. To generate this system, heparins were cross-linked to DATs with 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide and N-hydroxysuccinimide. Encapsulated Hep-DATs were able to control the release of a significantly higher amount of VEGF in vitro than non-heparinized DATs. Human bone marrow stromal cells (hBMSCs), when seeded on these VEGF–Hep-DATs, differentiated into endothelial cells which expressed vascular endothelial markers CD34 and VWF, thus resulting in accelerated vascularization of transplanted tissue as compared to the control DAT-only scaffold. In conclusion, these studies demonstrate that VEGF–Hep-DATs promoted greater tissue vascularization as compared to the DAT control scaffold and that VEGF–Hep-DATs are an effective and biocompatible angiogenesis system.
1. Introduction
Since the National Science Foundation put forward the concept of “tissue engineering” in 1987, research in this field has developed rapidly.1 However, to date, the number of tissue engineering products that can be used clinically is limited to just a few skins and cartilages.2 One of the main reasons for these limitations is that a good technique for vascularization of engineered tissue has been elusive thus far.3 In order to construct an organized three-dimensional engineered tissue of any volume, the key issue of early vascularization must be considered. If internal blood circulation can not be established, tissue engineering can only be limited to building thin tissue. Therefore, in recent years, much attention has been invested in developing improved techniques for the vascularization of engineered tissue, though, not much progress has been made.Currently, the main strategy for vascularizing engineered tissue involves three steps: (1) choosing a reasonable scaffold material, (2) maintaining sustained release of growth factors within the material and (3) induction of differentiation of plant cells.4 Ideally, the scaffold material should be biocompatible and degradable, and have a three-dimensional porous structure. While many useful attempts have been made to create the ideal scaffolding material including the use of polyglycolide acid (PGA), polyethylene terephthalate (PET), poly(lactic-co-glycolic acid) (PLGA), polytetrafluoroetylene (PTFE) and collagen, hyaluronic acid, silk protein and other artificial polymer materials for tissue engineering research,5,6 these scaffolds still have many issues with histocompatibility, absorption rate and immune rejection, thus lacking clinical utility. With a recent emphasis on the utilization of extracellular matrix (ECM) as a biologically available source for scaffolding, some researchers have begun to shift their attention to the extraction of scaffolds from the organism rather than engineering them with artificial polymers due to the enhanced biocompatibility and biometrics of ECM. ECM is a naturally occurring bio-derived material, which is prepared by removing the cellular components from the organism's prototype tissue using special physical and chemical methods. Studies have shown that ECM's three-dimensional structure is not only the ideal scaffold for supporting tissue organization and cell growth, but also plays an imperative role in the induction and regulation of cell adhesion, growth, proliferation and differentiation, as well as promoting tissue repair and regeneration.7,8 These biologically advantageous properties of ECM are a necessary component in tissue repair and reconstruction. Recently, a variety of adipose tissue ECM-based scaffolds from decellularized adipose tissues (DATs) have been generated for applications in adipose tissue engineering.9,10 After decellularization, DAT scaffolds effectively retain the three-dimensional structure of the original tissues and organs, have a beneficial biomechanical properties, good biocompatibility, slow degradation process, providing a good micro-environment for cell migration, proliferation, and differentiation.9,11
Incorporation of growth factors in the scaffolding material is essential due to their ability to induce vascularization, with the most effective vascularizing growth factor being vascular endothelial growth factor (VEGF). Biologically, VEGF promotes endothelial cell proliferation, increases microvascular permeability and induces angiogenesis. VEGF specifically promotes mitosis in vascular endothelial cells, regulates angiogenesis and induces neovascularization, and is therefore, the most important cytokine for endothelial cell development and the further differentiation and development of endothelial cells to a vascular endothelial cells fate.12,13 The ability of VEGF in promoting angiogenesis is important for normal embryonic development as well as pathologic tumorigenesis,14 and has a potential value for applications in clinical practice. A recent study incorporated VEGF into tissue-engineered scaffolds by chemical binding to form a VEGF-modified scaffold. When this VEGF-modified scaffold was implanted into chicken chorioallantoic membrane, newly generated blood vessels were increased throughout the scaffolding. When the VEGF-modified scaffold was implanted in the subcutaneous tissue of the rat, the scaffold was remodeled into subcutaneous tissue and promoted angiogenesis.15 The affinity for heparin to the DAT scaffold surface may protect the biological activity of VEGF in the microenvironment.16–19
Another key issue that affects the vascularization of DAT is the ability of the seeded cells to differentiate. Bone Marrow Stromal Cell (BMSC), which originate from mesoderm, have been shown to differentiate into vascular structures such as vascular endothelial cells20 and smooth muscle cells.21 In addition, BMSCs are capable of secreting some angiogenic growth factors such as VEGF.22 Previous reports have demonstrated that BMSCs can increase the level of local VEGF concentration in vitro and in vivo, and thereby promoting localized blood vessels.23 Therefore, BMSCs could in fact be the ideal seed cells for the promotion of tissue engineering vascularization, the prospects of which would result in extremely broad applications.
In this study, we prepared the decellularized adipose tissue (DAT) by a modified freeze-thawing method and modified the surface of the scaffold with heparin cross-linked to VEGF in order to control the release of VEGF. We hypothesized that these VEGF–Hep-DATs not only promotes differentiation of BMSCs into vascular endothelial cells, but also enhance angiogenesis, thus forming a vascular network.
2. Materials and methods
2.1 Materials
Unless otherwise stated, all chemicals used in the studies were purchased from Sigma-Aldrich (St. Louis, Missouri, USA).2.2 Procurement of adipose tissue
Human adipose tissue was obtained from 8 healthy female donors (20–40 years old patients) who had undergone routine abdominoplasty at the Burns and Plastic Surgery Center of General Hospital of Lanzhou Military Command of the People's Liberation Army (Lanzhou, China) between October 2013 and January 2014. Written informed consent was obtained from all patients, and experiments were performed according to the Human Research Guidelines by the Institutional Review Board of General Hospital of Lanzhou Military Command of China. The samples were transported to the Clinical Laboratories of General Hospital of Lanzhou Military Command at room temperature (RT) within 30 min.2.3 Preparation of decellularized adipose tissue (DAT)
Preparation of the decellularized adipose tissue (DAT) by modified freeze-thaw method: the abdominal skin was removed from the subcutaneous adipose tissue and the adipose tissue was then cut into 0.3 to 0.5 cm tissue blocks. The adipose tissue was then rinsed thoroughly with sterile PBS to remove blood impurities, and snap frozen in liquid nitrogen for 20 min. Immediately following liquid nitrogen treatment, the adipose tissue was transferred to a 37 °C water bath for temperature recovery and this freeze/thaw cycle was then repeated 5 more times. Adipose tissue was centrifuged at 1500 rpm for 10 min in order to remove the upper layer of lipid droplets, followed by 3 washes with 70% ethanol and sterile PBS. The adipose tissue was then treated with 0.05% trypsin, 0.05% EDTA, 20 ng ml−1 DNAse, and 20 ng ml−1 RNAse for 4 hours with mild oscillation, followed by three-30 min washes with sterile PBS, and then treated for 8 hours with isopropyl alcohol, followed by 3 more 30 min washes with sterile PBS. Finally, the adipose tissue was incubated overnight at 4 °C in a 1% dose of streptomycin. All the procedures are completed at 37 °C constant temperature in an oscillation chamber of the gas bath and DAT were freeze dried for 24 hours in a vacuum freeze dryer followed by disinfection with ethylene oxide at a low temperature.For evaluation of biological properties of acellular matrix, H&E staining, oil red O staining, and scanning electron microscopy (SEM) were used to observe the effect of cell removal and alterations to the matrix structure.
The DAT scaffolds were fixed with 4% paraformaldehyde for 24 hours followed by dehydration with ethanol and then embedded in Leica JUNG tissue-freezing medium. The embedded tissue were then cut into 8 μm sections with the Leicacm1850 frozen slicer. These sections were then utilized for immunohistochemical and tissue staining procedures.
The expression of type IV collagen and LAMIN β1 protein in DAT scaffolds were analyzed by immunofluorescence (IF) staining. The sections were pre-incubated at room temperature for 1 hour in a 1% BSA blocking solution, and then the sections were incubated overnight with the corresponding primary antibody at 4 °C and washed three times with PBS. Then the sections were incubated with fluorescently labeled secondary antibody at room temperature for 1 hour. After washing with PBS, the sections were stained with DAPI.
For oil red O staining, frozen DAT sections were allowed to dry, followed by treatment with the oil red O solution (SIGMA LOT: O1391) and allowed to react for 7 minutes at room temperature. Dye solution was removed from the sections and they were treated with 1% hydrochloric acid to activate the oil red O solution for 3 seconds. Sections were then rinsed with distilled water for 5 seconds to quench the reaction, counterstained with hematoxylin for 1 minute, rinsed in tap water for 2 minutes, sealed with gum arabic and observed under a microscope. Red color indicated the presence of lipids.
The expression of related proteins was observed under microscope. The staining intensity of the samples was calculated and classified into weak expression (less than 50% of cells showing intense staining) and strong expression (at least 50% of cells showing intense staining). To avoid possible bias caused by the selection of the fields to be examined, the samples were evaluated by three independent blind-observers.
2.4 Heparin cross-linking and Hep-DAT characterization
Heparin cross-linking procedures were modified from the previous methods.24 Heparin sodium salt (200 mg) was dissolved in 20 ml 0.1 M MES buffer (ThermoFisher Scientific, MA, USA) and reacted with 3 M M1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 5 M MN-hydroxysuccinimide (NHS) for 20 min at room temperature. Two pieces of hydrated DATs (180–200 mg each) were then transferred to the reaction solution to initiate cross-linking. After reacting for 16 hours at room temperature, heparinized DATs were washed with 0.1 M Na2HPO4 for 1 hour, followed by 3 washes with 4 M NaCl (30 min each) and a final 3 washes in distilled H2O (30 min each). Cross-linked heparins were visualized by staining DAT in 0.1% toluidine blue solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 mixture of 1% toluidine blue and 1% NaCl, pH 2–2.5) for 5 min.
:9 mixture of 1% toluidine blue and 1% NaCl, pH 2–2.5) for 5 min.
The contents of the primary amine groups in the DATs were assessed by incubation with 2,4,6-trinitrobenzenesulfonic acid.25 The amount of cross-linked heparins within Hep-DATs were quantified by toluidine blue assay as previously described,26 the basal staining absorbance of control DAT was subtracted from the absorbance of Hep-DAT to remove the effect of background staining. For both experiments, a series of diluted standards (glycine for amino group detection and heparin for toluidine blue assay) were used to generate standard curves for quantification. Absorbance measured with a fluorescence microplate reader (Tecan Infinite M200, Tecan Group Ltd, AG, Switzerland).
2.5 Determination of VEGF loading and release on Hep-DAT scaffolds
For VEGF binding, 100 mg (wet weight) Hep-DATs were immersed in 500 μl 0.09% NaCl containing 2 μg ml−1 VEGF for overnight binding at 4 °C. Following VEGF binding, DATs were washed 3 times (5 min each) in endothelial cell medium (EC medium) containing basal medium and 5% fetal bovine serum to remove the unbound VEGF. The amount of VEGF left in the binding tube (VEGF(L)) and the total amount of VEGF washed away (VEGF(W)) were assessed by ELISA. VEGF encapsulated by DAT was quantified by the following equation: encapsulated VEGF (ng) = 1000 − VEGF(L) − VEGF(W). For the in vitro release assay, VEGF-binding Hep-DATs (n = 3) were transferred to 24 wells plates (each well containing 1 ml EC medium) and incubated at 37 °C. EC medium containing released VEGF was collected at different time points during the incubation. DATs were transferred to new wells containing 1 ml fresh medium after medium collection at each time point. Released VEGF was quantified by ELISA (Peprotech, NJ, USA).2.6 Identification and labeling of hBMSC
Human BMSC (hBMSC) were purchased from Cyagen Biosciences Inc. (Guangzhou, China), and cell morphology was assessed. hBMSC cell surface markers, such as CD29, CD34 and CD44, were detected by flow cytometry, and the cell types were determined. hBMSCs were then differentiated into adipogenic and osteogenic lineages in vitro. Oil red O staining and alizarin red staining were used for identification. The second and third generation hBMSCs were used for subsequent experiments. CellTracker Green-5-chloromethylfluorescein diacetate (CMFDA) powder (100 μg, ThermoFisher Scientific, MA, USA) was thawed in 100 μl of dimethylsulfoxide (DMSO) solution. 1 μl of the CMFDA solution was then diluted in 300 μl PBS. Third generation hBMSCs were collected and centrifuged at 1200 rpm for 5 min. The hBMSCs were then resuspended in 200 μl of DiI working solution, allowed to stand at room temperature for 5 to 10 min, mixed with 7 ml PBS, and centrifuged at 1300 rpm for 5 min. The hBMSCs were resuspended in culture medium and seeded on the scaffold. Scaffolds were analyzed by immunofluorescence on the second day.2.7 Growth of stem cells on scaffold
Hep-DAT scaffolds were placed in a 24-well plate with complete medium, and incubated overnight at 37 °C, with 5% CO2. After discarding the medium, 100 μl of CMFDA-labeled hBMSCs were uniformly inoculated into the scaffold at 1 × 106 cells per mL. After incubation for 48 hours, hBMSCs were seeded in VEGF–Hep-DAT in the same manner. Cell growth was observed by fluorescence uptake on day 7 and day 14 following inoculation. Frozen sections of the cell-seeded scaffolds were collected and cell adhesion was observed under electron microscope.The CCK-8 method was used to detect the cytoxicity of the Hep-DAT and VEGF–Hep-DAT. The scaffolds were seeded with hBMSCs and placed in a 24-well culture plate. DMEM medium was added followed by incubation at 37 °C with 5% CO2 saturated humidity incubator. Each following day, 10 μl of the CCK-8 solution was added to each well, and after 4 hours of incubation the OD value at 450 nm was measured.
2.8 Differentiation of stem cells on scaffolds
CMFDA-labeled hBMSCs (100 μl of 1 × 106 cells per mL) were inoculated on either Hep-DAT or VEGF–Hep-DAT scaffolds for 2 days, followed by incubation with serum-free culture medium for 21 days. DATs were then embedded in Leica JUNG tissue-freezing medium. Embedded tissue were cut into 8 μm sections with the Leicacm1850 frozen slicer (Leicacm, Berlin, GER). Sections were then blocked in a 1% BSA solution at room temperature for 1 hour, followed by overnight incubation with primary antibody against Von Willebrand Factor (VWF) (anti-VWF, Abcam, Cambridge, USA). Sections were then rinsed three times with PBS, incubated with Cy3 fluorescently labeled secondary antibody (Abcam), for 1 hour at room temperature, washed again in PBS, and finally stained with DAPI and sealed with anti-quencher sealer. The staining intensity of the samples was calculated and classified as weakly expressed (less than 50% of the cells showing strong staining) or strongly expressed (at least 50% of the cells showing strong staining). To avoid possible biases caused by the selection of the fields to be examined, the samples were evaluated by three independent blind-observers.2.9 In vivo DATs transplantation and histological analysis
CMFDA-labeled hBMSCs (1 × 106 cells) were inoculated on either Hep-DAT or VEGF–Hep-DAT scaffolds (0.3 × 0.5 cm2) for 3 days. Scaffolds were washed gently with PBS before transplantation. Sprague Dawley rattus norregicus were purchased from (SJA Laboratory Animal Co, Changsha, China) and surgical protocol was approved by the Institutional Animal Care and Use Subcommittee of the Lanzhou Army General Hospital. Hep-DAT or VEGF–Hep-DAT scaffolds were implanted bilaterally into the subcutaneous tissue of the back of the Sprague Dawley rattus norregicus. After 2 or 4 weeks of transplantation, scaffolds and surrounding tissue were collected from Sprague Dawley rattus norregicus from each experimental group (n = 5). Collected samples were fixed in 10% formalin for 24 hours and then processed in Shandon Citadel 2000 automatic tissue processor (ThermoScientific), followed by embedding in paraffin and sectioning at 5 μm thickness. Hematoxylin and eosin (H&E) staining was performed. For immunofluorescence staining, sections were incubated in 0.05% Trypsin–EDTA at 37 °C for 30 min for antigen retrieval. Immunohistochemistry was performed to recognize VWF stained vascular network.2.10 RNA isolation and real time PCR
Recellularized Hep-DATs or VEGF–Hep-DATs (wet weight 50 mg, n = 3) were collected and the total RNA was extracted using TRIzol reagent (ThermoFisher Scientific, MA, USA). Reverse transcription of 1 mg total RNA into cDNA was carried out with PrimeScript RTreagentkit (Takara, Tokyo, JPN). Quantitative RT-PCR assay was performed on a 7900HT PCR system (Applied Biosystems, ThermoFisher Scientific, MA, USA) according to the SYBR Premix Ex Taq TM II kit protocol (Takara, Tokyo, JPN). Each reaction was performed in triplicate, and the relative expression levels of mRNA were calculated using the 2−ΔΔCT method.27 The primer sequences are as follows: VWF (forward: CCTTGACCTCGGACCCTTATG; reverse: GATGCCCGTTCACACCACT);GAPDH(forward: GGAGCGAGATCCCTCCAAAAT; reverse: GGCTGTTGTCATACTTCTCATGG).2.11 Statistical analyses
The experimental results were analyzed with SPSS 16 software (IBM, NY, USA). Comparison between groups was performed using a two-tailed paired t-test, variance analysis test, and Mann Whitney U test. GraphPad Prism software was used for scientific mapping. A p-value of < 0.05 was considered statistically significant.3. Results
3.1 Characterizations of DAT
After decellularization, highly hydrated DAT scaffolds were generated (Fig. 1A). SEM analysis of the scaffolds revealed a crisp cross-sectional network structure (Fig. 1B). H&E staining on frozen sections demonstrated that the hydrated DAT was mainly composed of loosely stacked acellular ECM networks (Fig. 1C). No significant quantities of nuclei or lipid droplets were detected in the H&E or oil red O stained DAT samples (Fig. 1C and D), indicating that residual nucleic acids and lipids had been effectively removed. Both collagen IV (Fig. 1E) and laminin β1 (Fig. 1F), immunostaining performed for the detection of ECM proteins in the DAT, were both extensively present on the scaffold, indicating that the main ECM components of the basement membrane were well preserved during the decellularization process.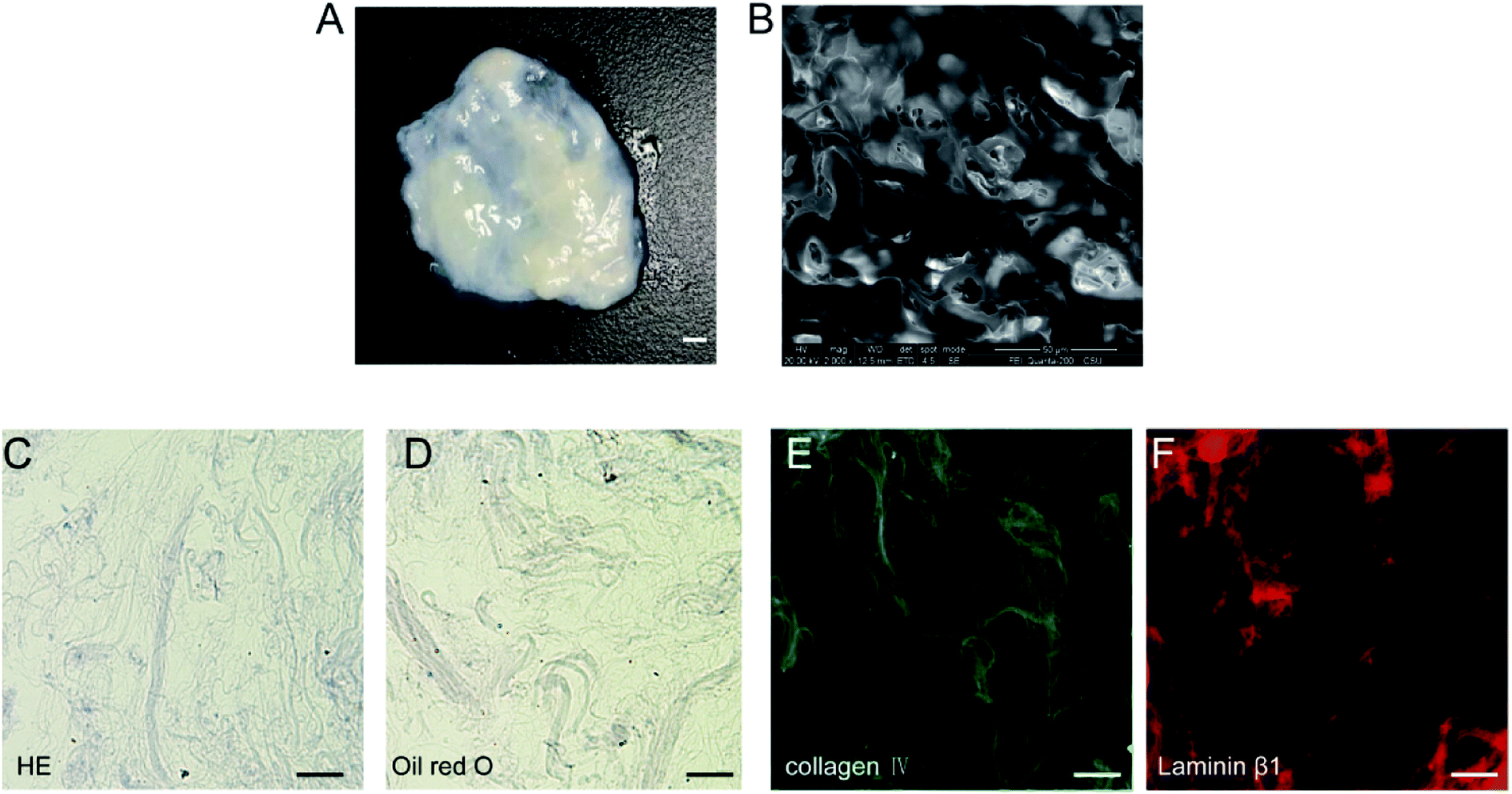 | ||
| Fig. 1 Hydrated DAT morphology (A) and DAT SEM analysis (B). H&E stained DAT frozen sections (C) and oil red O stained DAT frozen sections (D). Immunofluorescent staining of fir collagen IV (green, E) and laminin β1 (red, F). | ||
3.2 Detection of cross-linked heparin
After cross-linking, the presence of heparin was visualized by toluidine blue staining. The darker staining color of the Hep-DAT indicated that a greater quantity of heparin was attached (Fig. 2A). As quantified by the toluidine blue assay, the amount of heparin linked to the DAT was nearly 43.8 ± 3.7 μg per 100 mg wet weight (Table 1). The infrared spectra of the Hep-DAT showed that the transmittance of the Hep-DAT at each wavelength was higher than that of the DAT alone (Fig. 2B), indicating that the Hep-DAT can encapsulate a greater quantity of heparin than the DAT. To further investigate the extent of heparin cross-linking, the availability of primary free amine group in different types of DATs was performed using a 2,4,6-trinitrobenzenesulfonic acid assay. For the non-heparinized DAT, the average free amine groups were 261.5 ± 14.2 nmol per 100 mg wet weight. After heparin cross-linking, this number changed to 150.8 ± 25.9 nmol for the Hep-DAT (p < 0.01) and 192.7 ± 19.9 nmol (p = 0.07) for the DAT that only underwent self-cross-linking without heparin (Table 1). These results suggest that while self-cross-linking within the DAT did not significantly alter the average free amine groups from the original DAT, there was a significant reduction of free amine groups in the Hep-DAT, suggesting the cross-linked heparins were occupying the free amine groups.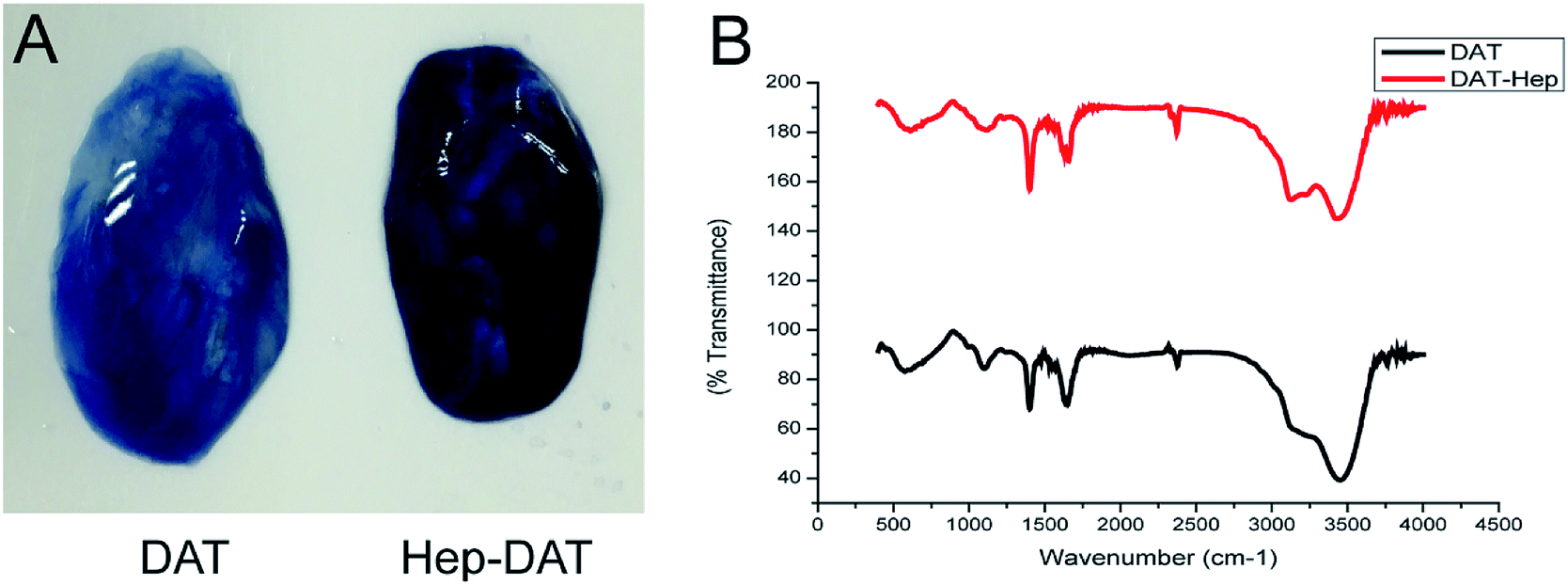 | ||
| Fig. 2 (A) Toluidine blue staining of DAT (left) and Hep-DAT (right). (B) Infrared maps of DAT and Hep-DAT. | ||
| Type of DAT | Cross-linked with EDC/NHS | Addition of heparins | Amine group content | Cross-linked heparins | Encapsulated VEGF |
|---|---|---|---|---|---|
| (nmol/100 mg wet weight) | (μg/100 mg wet weight) | (ng/100 mg wet weight) | |||
| a Quantification of amine group content, cross-linked heparins and encapsulated VEGF in DAT scaffolds between treatment groups. n = 3 in each group, *p < 0.05, **p < 0.01 (compared to control DAT). | |||||
| Control DAT | — | — | 261.5 ± 14.2 | — | 42.7 ± 5.0 |
| Cross-linked DAT | + | — | 192.7 ± 19.9 | — | — |
| Hep-DAT | + | + | 150.8 ± 25.9* | 43.8 ± 3.7 | 121.2 ± 12.4** |
3.3 In vitro binding and release VEGF
To further investigate the VEGF binding and release ability of Hep-DAT an in vitro release assay was performed. The encapsulation test demonstrated that the Hep-DAT could capture more quality VEGF than control DATs (42.7 ± 5.0 ng vs. 121.2 ± 12.4 ng) per 100 mg wet weight (Fig. 3A, Table 1). The cumulative release of VEGF from Hep-DATs was significantly higher than from control DATs at all tested time points (Fig. 3B). When normalized to total encapsulated VEGF, the percentages of released VEGF from the Hep-DAT were lower than those from the control DAT in the first 4 days (Fig. 3C). Compared to the Hep-DAT, the control DAT released VEGF at very fast rate (more than 40% encapsulated VEGF was released within 24 hours) and quickly reached a release plateau by 48 hours (Fig. 3B and C). These results demonstrate that Hep-DATs were able to encapsulate a significantly higher amount of VEGF and better control VEGF release in vitro than non-heparinized DATs.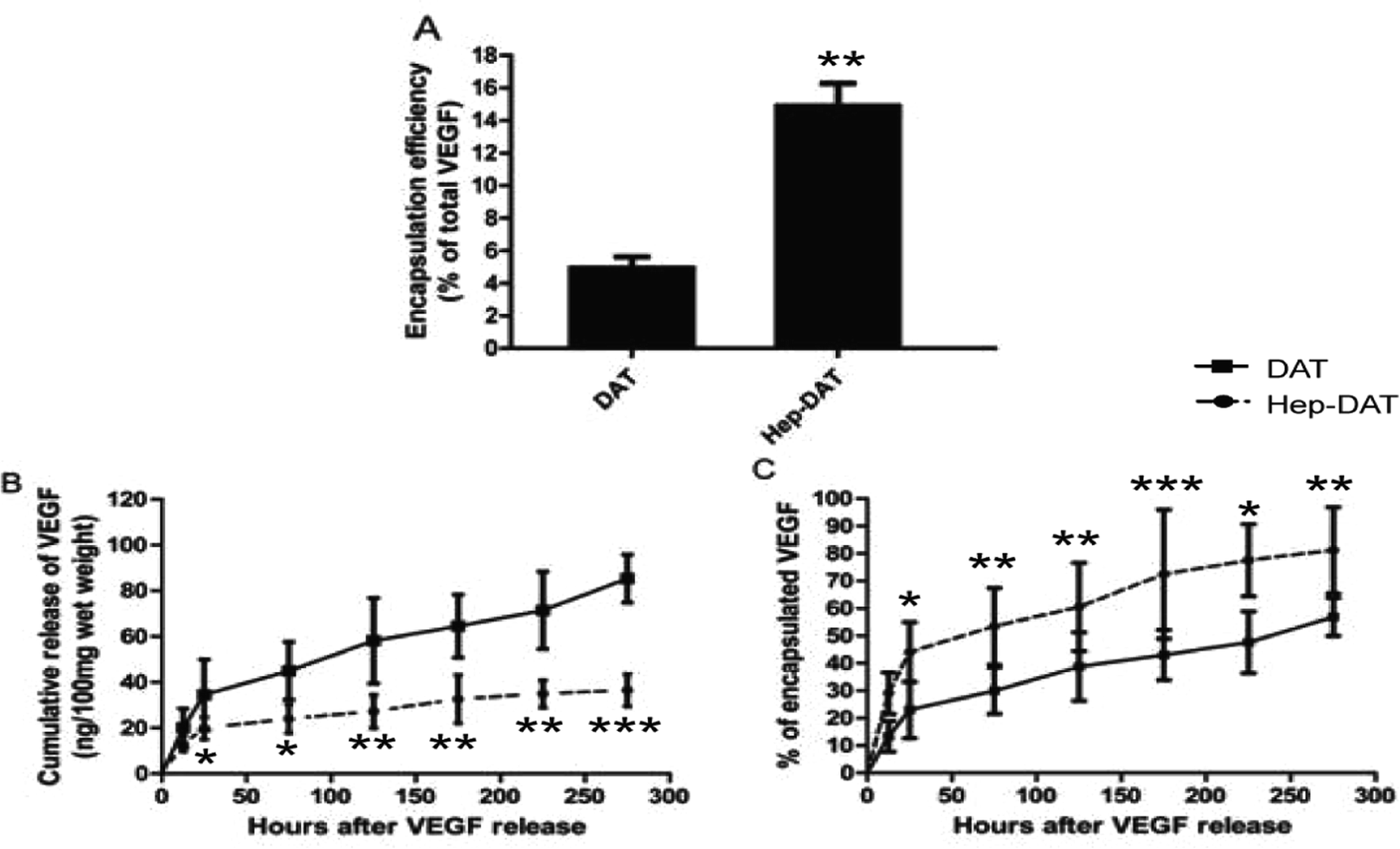 | ||
| Fig. 3 (A) VEGF encapsulation rate for DAT and Hep-DAT (B) VEGF cumulative release curve for DAT (dashed line) and Hep-DAT (solid line). (C) VEGF release percentage curve for DAT (dashed line) and Hep-DAT (solid line) at various time points. *p < 0.05, **p < 0.01. | ||
3.4 Culture and identification of hBMSC
Cultured hBMSCs are typically spindle-shaped and exhibit fibroblast-like morphology (Fig. 4A). hBMSCs express molecular markers on their surface, including CD90 and CD105, but lack the hematopoietic markers such as CD14 and CD34. In order to determine the differentiation potential of hBMSCs, we induced 5th generation hBMSCs into osteocytes and adipocytes. The adipogenic differentiation potential of hBMSCs could be confirmed by the accumulation of red lipid droplets in the cytoplasm within 2 weeks post-differentiation (Fig. 4B) and after 2 weeks of incubation in the osteogenic medium, red mineralized nodules could be detected in the cells (Fig. 4C), suggesting that these cells are a pure pluripotent population of BMSCs.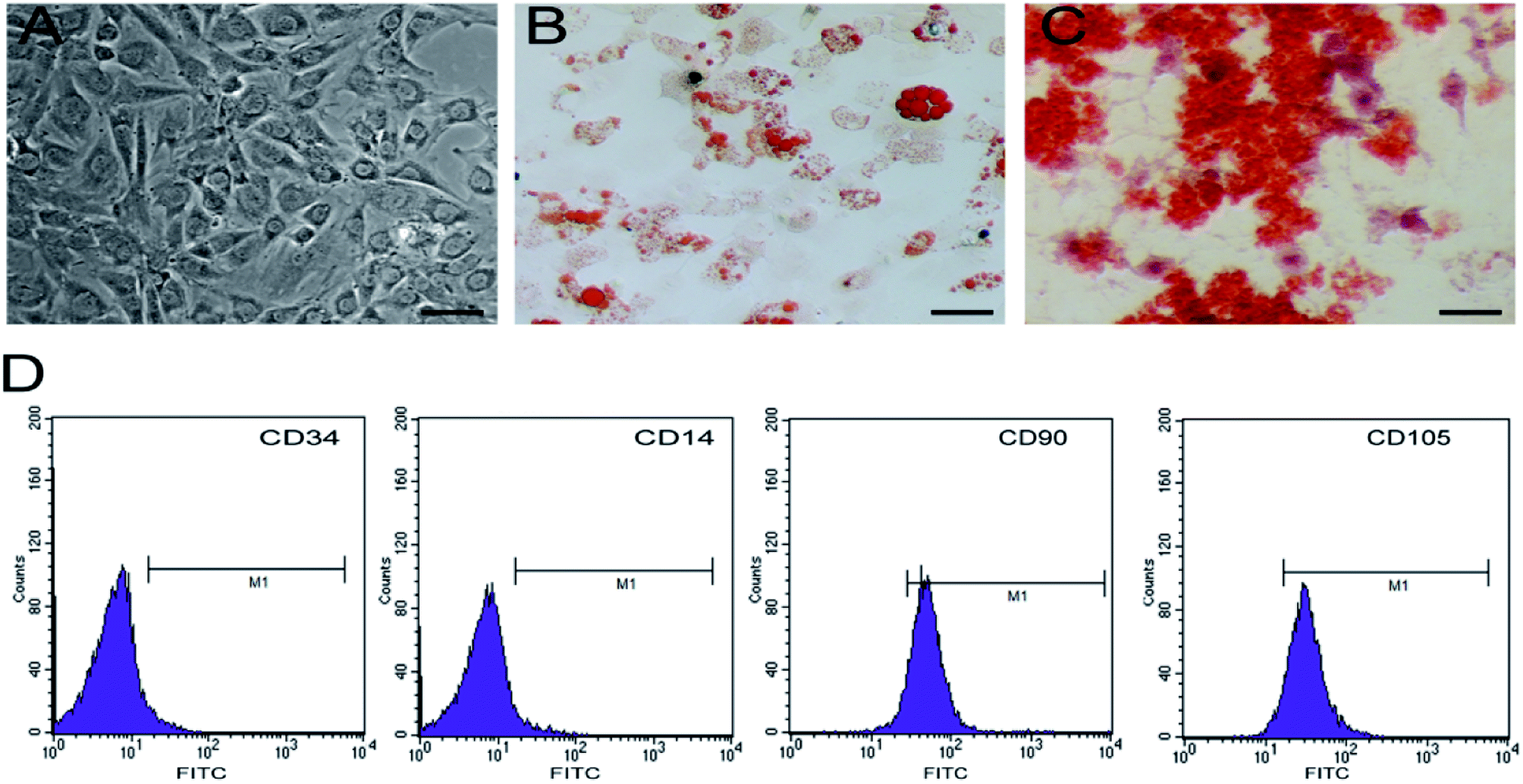 | ||
| Fig. 4 Characterization of cultured rat bone marrow mesenchymal stem cells (hBMSCs). The cultured hBMSCs showed spindle-shaped fibroblast morphology (A) and were capable of differentiating into adipocytes (B) and bone cells (B). HBMSCs were cultured in adipogenic medium and the accumulation of lipid into the intracellular vesicles was observed by staining with oil red O dye (B). HBMSCs were cultured in osteogenic medium and stained with Alizarin Red S to show calcium deposition and mineralization in the monolayer (C). The expression of hBMSCs markers was determined by flow cytometry (D). Scale bar: 100 μm. | ||
3.5 Observation of growth of hBMSCs on VEGF–Hep-DAT scaffolds
In order to track the growth of hBMSCs grown on the scaffold, we used CMFDA to label the transplanted cells. After 7 days of culture, the fluorescent intensity of the cells on the Hep-DAT scaffold (Fig. 5A) was significantly lower than the fluorescent intensity of the cells on the VEGF–Hep-DAT scaffold (Fig. 5B). Some of the transplanted cells on VEGF–Hep-DAT scaffold were adhered to the EMC network (shown by the yellow arrow in Fig. 5B). After 14 days of culture the number of cells on the Hep-DAT (Fig. 5C) and the VEGF–Hep-DAT scaffolds (Fig. 5D) was significantly higher than what was observed at 7 days. While the transplanted cells on the Hep-DAT scaffold were partially adhered to the EMC network, most of the cells were still in suspension (as shown by the yellow arrows in Fig. 5C). However, most of the transplanted cells on the VEGF–Hep-DAT scaffold not only adhered to the ECM network (as indicated by the yellow arrows in Fig. 5D) but also showed adhered growth trends (as indicated by the yellow arrows), indicating that the cell growth status of the VEGF–Hep-DAT scaffold was significantly better than that of the HEP-DAT. As revealed by scanning electron microscope, the scaffold consisted of a crisp cross-sectional network structure, the cells transplanted on the scaffold were oval-shaped, and the adherent cells observed on the VEGF–Hep-DAT scaffold were significantly more than those on the Hep-DAT scaffold (as indicated by the yellow arrows in Fig. 5E and F). Cell proliferation experiments confirmed that cells on the VEGF–Hep-DAT scaffolds grew fastest on the fourth day of in vitro culture, and may be associated with the significant VEGF release peaks in the first 48 hours of culture of the VEGF–Hep-DAT scaffold (Fig. 3G) The proliferation rate of the VEGF–Hep-DAT scaffold cells was significantly higher than that of cells on the Hep-DAT scaffold, indicating that the growth environment for the cells on the VEGF–Hep-DAT scaffold is superior to that of the Hep-DAT.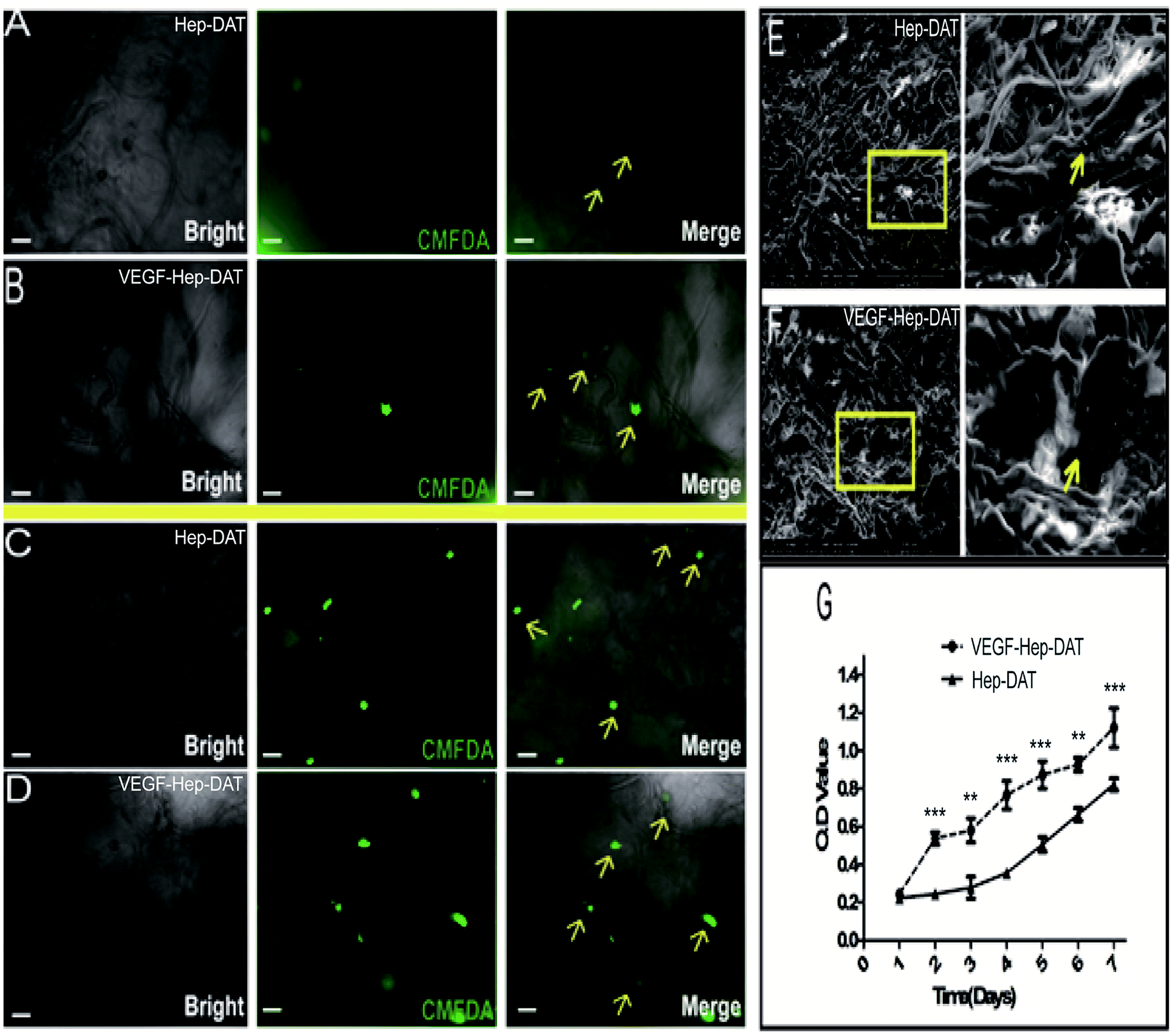 | ||
| Fig. 5 (A and C) BMSCs were seeded on Hep-DAT for 7 and 14 days, respectively. (B and D) BMSCs were seeded on VEGF–Hep-DAT for 7 and 14 days, respectively. (E and F) Scanning electron microscope of frozen sections obtained from scaffolds. (G) Cytoxicity test for cell growth curve. | ||
3.6 The differentiation of hBMSCs on VEGF–Hep-DAT scaffolds
An in vitro model was established to demonstrate the effect of VEGF–Hep-DAT sustained-release VEGF on endothelial cell differentiation. We performed IF for the vascular endothelial cell marker VWF on the scaffold grafted cells.28 The results showed that the green fluorescent labeled stem cells on the Hep-DAT scaffold did not co-express VWF (Fig. 6A) protein labeled with red fluorescent cy3. However, the green fluorescent labeled stem cells on VEGF–Hep-DAT scaffold co-expressed VWF (Fig. 6 B) protein labeled with red fluorescent cy3 (co-expressing cells were yellow) after 21 days of culture. qPCR results demonstrated that VWF mRNA expression levels were increased more than 5-fold from cells obtained from the Hep-DAT scaffolds as compared to cells obtained from control DATs (Fig. 6C). These results showed that VEGF–Hep-DAT scaffolds are capable of inducing hBMSCs to differentiate into vascular endothelial cells through sustained release of VEGF and stimulate the expression of vascular endothelial cell marker VWF in hBMSCs, thus accelerating the angiogenesis of transplanted tissue.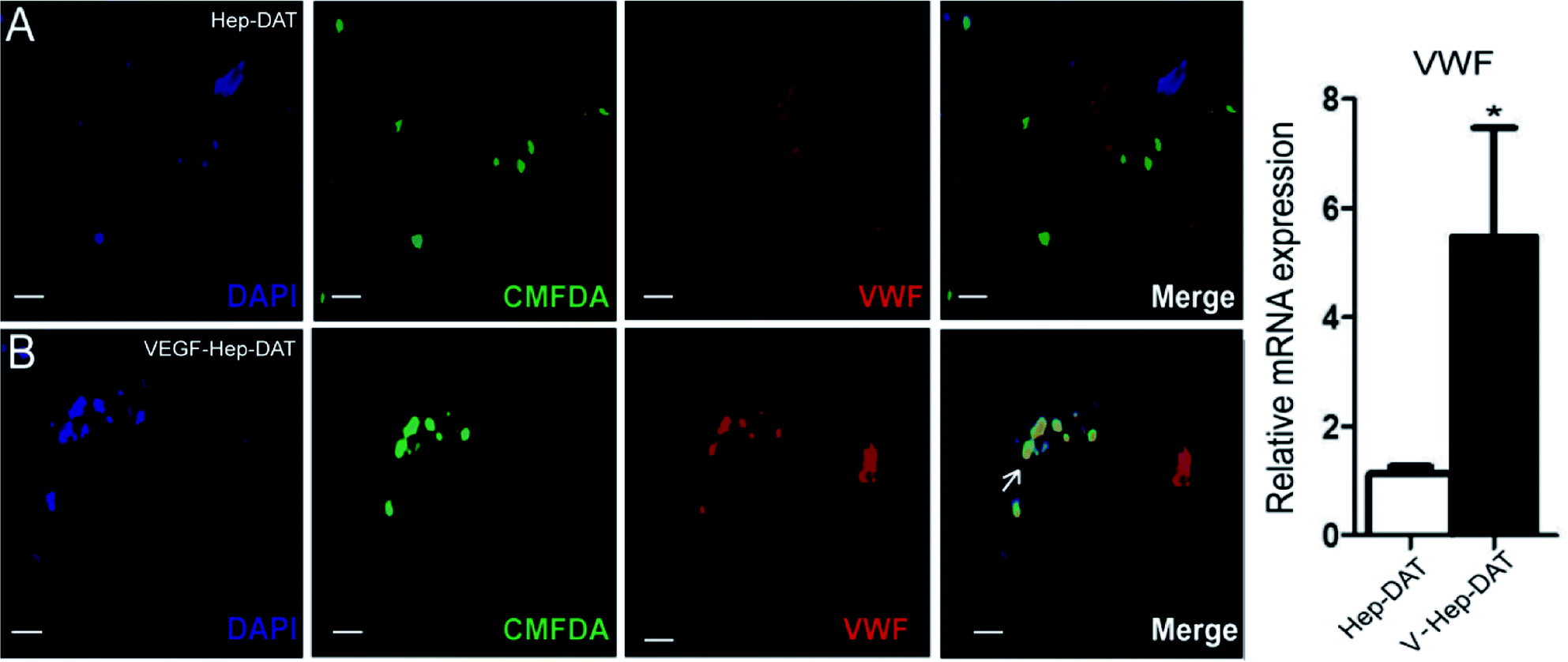 | ||
| Fig. 6 VEGF–Hep-DAT scaffold induces hBMSCs to differentiate into vascular endothelial cells in vitro. Cultured cells were labeled with cell track reagent CMFDA (green) and immunostained with anti-VWF (red). Nuclei were counterstained with DAPI (blue) (A and B). Values are expressed as mean S.D. (n = 3). VWF mRNA levels of the hBMSCs as analyzed by qPCR. | ||
3.7 In vivo DATs transplantation and histological analysis
To investigate the in vivo angiogenic potentials of hBMSCs and VEGF–Hep-DAT, we transplanted the scaffolds with seeded hBMSC subcutaneously on the back of animals. After 2 weeks, transplanted VEGF–Hep-DAT could be clearly identified without obvious volume shrinkage (Fig. 7B), whereas the volume of the control Hep-DAT was dramatically reduced (Fig. 7A) at the same time, the similar results was obverse at 4 weeks after transplant the scaffold into animals. The volume of VEGF–Hep-DAT scaffolds were greatly preserved compared to the control (Fig. 7H and I). In addition, hematoxyl in staining on the VEGF–Hep-DAT samples revealed that an abundant quantity of cells (purple dots, Fig. 7D) infiltrated into the scaffold compared to Hep-DAT scaffolds(purple dots, Fig. 7C). In addition, these dispersed cells started to form early vascular-like structures (Fig. 7D, indicated by red arrows)at first 2 weeks transplant. At 4 weeks, the collected VEGF–Hep-DAT scaffold were recellularized and displayed highly vascularized tissue-like morphology (Fig. 7K, indicated by red arrows) compared to Hep-DAT scaffolds(Fig. 7J, indicated by red arrows). To further examine angiogenesis, we carried out IF for the vascular endothelial cell marker VWF on the scaffolds. VWF expressing hBMSCs were widely detected within the VEGF–Hep-DAT scaffold (Fig. 7F), while very few VWF positive cells were detected in Hep-DAT (Fig. 7E). At 4 weeks post-transplantation, more cells were found in the VEGF–Hep-DAT scaffolds (Fig. 7L) being positive for VWF (Fig. 7N and O) as compared to those at 2 weeks post-transplantation. Some VWF-expressing endothelial cells were aligned together to form the vascular (Fig. 7M). | ||
| Fig. 7 In vivo adipogenic studies and histological analysis. Morphology of control DAT (A and H) and VEGF–Hep-DAT (B and I) 2 weeks or 4 weeks post-transplantation, respectively. (C, D, J and K) H&E staining of frozen sections from recellularized control DAT (C and J) and VEGF–Hep-DAT (D and K) samples (40× magnification). (E and M) Comparison of VWF stained area (% of total image area) between VEGF–Hep-DAT and control DAT samples. (L) Comparison of angiogenesis count between VEGF–Hep-DAT and control DAT samples. Cultured cells were labeled with the cell track reagent CMFDA (green) and immunostained with anti-VWF (red). Nuclei were counterstained with DAPI (blue) (F, G, N and O). Values are expressed as mean ± S.D. (n = 3). | ||
4. Discussion
At present, the study of vascularization of engineered tissue mainly focuses on the mechanism of angiogenesis. Achievement of vascularization of the tissue organ is thought to be accomplished through construction of the cell-material-growth factor complex and mimicking the microenvironment in vitro.29 DAT is a recently developed scaffold that has wide-ranging applications for adipose tissue engineering. Previous techniques based on direct transplantation of cells on DAT scaffolds failed to induce vascular formation at a high level, suggesting that simply implanting adipose-derived stem cells (ADSC)on DAT scaffolds may not be sufficient to induce large amounts of angiogenesis. This is most likely due to complex environmental signals or hypoxia conditions in the body that limit the differentiation of ADSC into vascular endothelial cells. In contrast to the transplantation cells, we developed a VEGF-controlled release system based on Hep-DAT to induce angiogenesis. The VEGF–Hep-DAT scaffold achieved sustained and stable angiogenesis by controlling the slow release of growth factors in vitro subsequently inducing differentiation of hBMSCs to an endothelial cell fate where they expressed the vascular endothelial marker VWF, thereby accelerating the vascularization of transplanted tissue.Stem cells are an early undifferentiated cell with pluripotent differentiation potential and self-replicating ability. Stem cells can be divided into embryonic stem cells (ESC) and adult stem cells using genetic techniques.30 Embryonic stem cells are a pluripotent cell line derived from human or animal embryonic cell masses or primitive reproductive crests. They can differentiate into all cells types from the inner, middle and outer layers, and can remain undifferentiated when cultured in vitro.31 Adult stem cells are undifferentiated cells present in various tissues and organs, which have the potential to differentiate into a variety of cells and tissues, but lose their ability to differentiate in vitro over time. hBMSC belong to a kind of adult stem cell that can not only differentiate into hematopoietic stromal cells, but can also differentiate into a variety of other hematopoietic cells including vascular endothelial cells, thereby promoting vascularization in vitro. hBMSCs not only have high proliferative, self-renewal and multi-directional differentiation potential, they are also easy to isolate, cultivate and proliferate. hBMSCs also easily facilitate the introduction and expression of foreign genes, and maintain the potential of multi-directional differentiation in the process of long-term culture. Multi-directional differentiation potential is considered to be one of the most important biological characteristics of MSCs. In vitro differentiation and production of a large number of vascular endothelial (precursor) cells is a key link for the treatment of clinical cell transplantation, repairing damaged vascular intima, promoting vascular regeneration of ischemic injury organs or constructing tissue engineered blood vessels and complicated organ tissue engineering vascularization.32
According to our results, DAT was effectively generated and had good preservation of ECM proteins like collagen IV and laminin (Fig. 1), which are proteins that constitute the main portions of the basement membrane in biological adipose tissues.33 The presence of ECM proteins (like collagens) suggest that there would be abundant free amine groups for heparin cross-linking.16,24 As a natural VEGF-binding molecule, heparin can serve as binding and release platforms for VEGF.24 Even DAT may contain a certain amount of endogenous heparin-like molecules (such as heparan-sulfate).34 However, the endogenous growth factor binding sites of DAT alone may not be enough to retain sufficient amounts of VEGF for inducing effective angiogenesis. Hence, we cross-linked exogenous heparin to the DAT to further enhance its VEGF retention and controlled release ability. After cross-linking with NHS and EDC, attached heparin could be directly visualized by toluidine blue staining (Fig. 2A).17,35
As shown in the VEGF release assay, Hep-DAT was able to significantly absorb VEGF (Fig. 2B) and release VEGF more sustainably in vitro than non-heparinized DAT (Fig. 3A and B). These improved growth factor binding and controlled release capacities are critical for inducing effective angiogenesis, as studies have shown that the effectiveness of tissue vascularization depends to a large extent on the availability of cytokines and their in vivo control release mode.
We further evaluated the in vitro vascularization of the system. The endothelial cell marker VWF was observed 14 days after hBMSCs were cultured on the ECM network of the VEGF–Hep-DAT, indicating that VEGF–Hep-DATs in vitro had a significantly higher potential for promoting tissue vascularization than control DATs.
In conclusion, our in vivo studies have shown that VEGF–Hep-DAT are not only biocompatible, but have the potential for promoting tissue vascularization during adipose tissue engineering. This system is highly applicable for clinical use because it can be conveniently constructed through a simple chemical modification on adipose tissue collected from fat aspiration.36 In addition, it can be used directly without the need for transplanting exogenous stem cells, which further reduces the cost of production and potential tumor formation associated with long-term expansion of stem cells.37
5. Conclusion
In this study we demonstrated that VEGF cross-linked to Hep-DAT is effective in promoting tissue vascularization in vitro. The system can be easily produced by degellation of adipose tissue and subsequent heparin cross-linking. In vitro, due to the affinity and protective effect of heparin on growth factors, the biological activity of VEGF can be protected by grafting heparin to the surface of the DAT scaffold followed by loading of VEGF. This method ensures that the sustained release of VEGF was achieved in an in vitro microenvironment. Hep-DAT was able to encapsulate very high amounts of VEGF in vitro due to its propensity for strong water absorption and porosity. Thus, Hep-DAT bound VEGF more strongly and controlled release of VEGF greater than non-heparinized DAT. Slow release of VEGF from the Hep-DAT could also effectively induce stem cells to form new blood vessels without causing a significant host response, thus inducing within the scaffold sustained and stable angiogenesis. VEGF-bound Hep-DAT are also effective as scaffolds for engineered tissues in vitro, as they are biocompatible and injectable angiogenesis system. These studies demonstrate that the use of a sustained-release VEGF scaffold system can promote tissue engineered vascularization to greater effect, thus enhancing their clinical applicability. In the future, we plan to utilize this technique to construct entire artificial tissues and organs in vitro.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (grant no. 81401612) and the Key Project of the “12th Five-Year Plan” for Medical Science and Technology of PLA (grant no. BWS11C061).References
- A. B. Mohd Hilmi and A. S. Halim, World J. Stem Cells., 2015, 7, 428–436 CrossRef PubMed.
- L. J. Bonassar and C. A. Vacanti, J. Cell. Biochem., 2015, 72, 297–303 CrossRef.
- M. Lovett, K. Lee, A. Edwards and D. L. Kaplan, Tissue Eng., Part B, 2009, 15, 353–370 CrossRef CAS PubMed.
- Q. Lu, M. Li, Y. Zou and T. Cao, J. Controlled Release, 2014, 174, 43–50 CrossRef CAS PubMed.
- S. J. Lee, G. J. Lim, J. W. Lee, A. Atala and J. J. Yoo, Biomaterials, 2006, 27, 3466–3472 CrossRef CAS PubMed.
- F. B. Stillaert, C. Di Bartolo, J. A. Hunt, N. P. Rhodes, E. Tognana, S. Monstrey and P. N. Blondeel, Biomaterials, 2008, 29, 3953–3959 CrossRef CAS PubMed.
- B. P. Chan and K. W. Leong, Eur. Spine J., 2008, 17(suppl. 4), 467–479 CrossRef PubMed.
- Y. Zhang, Y. He, S. Bharadwaj, N. Hammam, K. Carnagey, R. Myers, A. Atala and M. Van Dyke, Biomaterials, 2009, 30, 4021–4028 CrossRef CAS PubMed.
- L. E. Flynn, Biomaterials, 2010, 31, 4715–4724 CrossRef CAS PubMed.
- C. Yu, J. Bianco, C. Brown, L. Fuetterer, J. F. Watkins, A. Samani and L. E. Flynn, Biomaterials, 2013, 34, 3290–3302 CrossRef CAS PubMed.
- S. F. Badylak, D. Freytes and T. W. Gilbert, Acta Biomater., 2009, 5, 1–13 CrossRef CAS PubMed.
- V. Nikolova-Krstevski, M. Bhasin, H. H. Otu, T. Libermann and P. Oettgen, BMC Genomics, 2008, 9, 240 CrossRef PubMed.
- Q. Wu, Y. Li, Y. Wang, L. Li, X. Jiang, J. Tang, H. Yang, J. Zhang, J. Bao and H. Bu, J. Biomed. Mater. Res., Part A, 2016, 104, 3021–3030 CrossRef CAS PubMed.
- W. Liu, J. Xu, M. Wang, Q. Wang, Y. Bi and M. Han, Int. J. Oncol., 2011, 39, 1213–1220 CAS.
- A. H. Zisch, M. P. Lutolf, M. Ehrbar, G. P. Raeber, S. C. Rizzi, N. Davies, H. Schmokel, D. Bezuidenhout, V. Djonov, P. Zilla and J. A. Hubbell, FASEB J., 2003, 17, 2260–2262 CrossRef CAS PubMed.
- S. T. Nillesen, P. J. Geutjes, R. Wismans, J. Schalkwijk, W. F. Daamen and T. H. van Kuppevelt, Biomaterials, 2007, 28, 1123–1131 CrossRef CAS PubMed.
- S. Singh, B. M. Wu and J. C. Dunn, Biomaterials, 2011, 32, 2059–2069 CrossRef CAS PubMed.
- G. C. Steffens, C. Yao, P. Prevel, M. Markowicz, P. Schenck, E. M. Noah and N. Pallua, Tissue Eng., 2004, 10, 1502–1509 CrossRef CAS PubMed.
- O. Jeon, S. J. Song, S. W. Kang, A. J. Putnam and B. S. Kim, Biomaterials, 2007, 28, 2763–2771 CrossRef CAS PubMed.
- G. V. Silva, S. Litovsky, J. A. Assad, A. L. Sousa, B. J. Martin, D. Vela, S. C. Coulter, J. Lin, J. Ober, W. K. Vaughn, R. V. Branco, E. M. Oliveira, R. He, Y. J. Geng, J. T. Willerson and E. C. Perin, Circulation, 2005, 111, 150–156 CrossRef CAS PubMed.
- M. Abedin, Y. Tintut and L. L. Demer, Circ. Res., 2004, 95, 671–676 CrossRef CAS PubMed.
- K. Hamano and T. S. Li, Journal of Japan Surgical Association, 2004, 105, 464–468 CAS.
- S. Fuchs, R. Baffour, Y. F. Zhou, M. Shou, A. Pierre, F. O. Tio, N. J. Weissman, M. B. Leon, S. E. Epstein and R. Kornowski, J. Am. Coll. Cardiol., 2001, 37, 1726–1732 CrossRef CAS PubMed.
- M. J. Wissink, R. Beernink, J. S. Pieper, A. A. Poot, G. H. Engbers, T. Beugeling, W. G. van Aken and J. Feijen, Biomaterials, 2001, 22, 2291–2299 CrossRef CAS PubMed.
- J. L. Kelly, M. W. Findlay, K. R. Knight, A. Penington, E. W. Thompson, A. Messina and W. A. Morrison, Tissue Eng., 2006, 12, 2041–2047 CrossRef CAS PubMed.
- M. Mark, M. Teletin, C. Antal, O. Wendling, J. Auwerx, S. Heikkinen, K. Khetchoumian, C. A. Argmann and M. Dgheem, Histopathology in Mouse Metabolic Investigations, John Wiley & Sons, Inc., 2007 Search PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- A. M. Muller, M. I. Hermanns, C. Skrzynski, M. Nesslinger, K. M. Muller and C. J. Kirkpatrick, Exp. Mol. Pathol., 2002, 72, 221–229 CrossRef PubMed.
- M. W. Laschke and M. D. Menger, Eur. Surg. Res., 2012, 48, 85–92 CrossRef CAS PubMed.
- G. Q. Daley, M. A. Goodell and E. Y. Snyder, Hematology. American Society of Hematology. Education Program, 2003, pp. 398–418 Search PubMed.
- J. A. Thomson, J. Itskovitz-Eldor, S. S. Shapiro, M. A. Waknitz, J. J. Swiergiel, V. S. Marshall and J. M. Jones, Science, 1998, 282, 1145–1147 CrossRef CAS PubMed.
- R. Ogawa, Curr. Stem Cell Res. Ther., 2006, 1, 13–20 CrossRef CAS PubMed.
- Y. Kubo, S. Kaidzu, I. Nakajima, K. Takenouchi and F. Nakamura, In vitro cellular & developmental biology. Animal, 2000, Vol. 36, pp. 38–44 Search PubMed.
- T. F. Gesteira, V. J. Coulson-Thomas, A. Taunay-Rodrigues, V. Oliveira, B. E. Thacker, M. A. Juliano, R. Pasqualini, W. Arap, I. L. Tersariol, H. B. Nader, J. D. Esko and M. A. Pinhal, J. Biol. Chem., 2011, 286, 5338–5346 CrossRef CAS PubMed.
- S. Murugesan, J. Xie and R. J. Linhardt, Curr. Top. Med. Chem., 2008, 8, 80–100 CrossRef CAS PubMed.
- T. T. Han, S. Toutounji, B. G. Amsden and L. E. Flynn, Biomaterials, 2015, 72, 125–137 CrossRef CAS PubMed.
- A. M. Bailey, Sci. Transl. Med., 2012, 4, 147 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |