
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient and divergent synthesis of polyfunctionalized 2-pyridones from β-keto amides†
Baochang Gao *ab,
Yufeng Sunb,
Jun Wanga,
Zhigang Yuanb,
Liwu Zua,
Xu Zhangb and
Wenbin Liu*a
*ab,
Yufeng Sunb,
Jun Wanga,
Zhigang Yuanb,
Liwu Zua,
Xu Zhangb and
Wenbin Liu*a
aInstitute of Composite Materials, Key Laboratory of Superlight Material and Surface Technology of Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin 150001, P. R. China. E-mail: chemgbc@hrbeu.edu.cn; liuwenbin@hrbeu.edu.cn
bDaqing Branch of Heilongjiang Academy of Sciences, Daqing 163319, P. R. China
First published on 1st October 2018
Abstract
Efficient and divergent one-pot synthesis of polyfunctionalized 2-pyridones from β-keto amides based on reaction condition selection was developed. The methodology offers several significant advantages including mild conditions, ease of handling, high yields, and a relatively broad range of substrates. Based on various experiments and observations, a plausible mechanism for the selective synthesis of 2-pyridones was proposed.
Introduction
Molecularly diverse and complex heterocycles make a vital contribution to the organic synthesis and discovery of new pharmaceutical reagents.1 2-Pyridone is one of the most significant heteroaromatic rings in natural products, bioactive molecules, and pharmaceutical agents.2–5 Such well-known molecules possess a wide spectrum of biological properties such as antimalarial,6 anti-hepatitis B,7 vasorelaxant,8 anti-fungal,9 anti-epilepsy,10 anti-fibrosis,11 anti-HIV,12 MEK-1 inhibitor,13 antitumor,14 anti-ulcer,15 antioxidant, and antituberculosis activities.16 Moreover, molecules bearing 2-pyridone derivatives are also used as important structural units in the synthesis of nitrogen-containing heterocycles, such as pyridines,17 piperidines,18 β-lactams,19 indolizidines, and quinolizidines.20 Thus, the synthesis of 2-pyridone derivatives has attracted considerable attention in recent years. Owing to their biologically and structurally interesting properties, a number of approaches for their construction have been reported. The classical methods for synthesis of 2-pyridones include pyridinium salt chemistry and N-alkylation,21,22 Guareschi–Thorpe reaction,23 Dieckmann-type condensation,24 Diels–Alder reaction,25 and halogenated 2-pyridones under Vilsmeier conditions,26 and other methods from the synthesis of 2-pyridones under metal catalyst.27 Each of these approaches represents an important progress for the synthesis of 2-pyridones, however, the development of an efficient and facile strategies are desirable for synthesis of the valuable heterocycles. The development of an efficient and facile methodology for the construction of diverse 2-pyridone derivatives still remains a great challenge.β-Keto amide and their derivatives have been proven to be the important starting materials and reagents in the construction of heterocyclic systems since they possess six reactive sites in the same molecule.28–35 They have also been used as building blocks for the construction of biologically interesting heterocycles.36 So far, a number of natural product-like heterocyclic compounds have been successfully synthesized based on β-keto amide and their derivatives. Their synthetic methodology and associated medicinal activity should be engaged in the chemistry community.37 As part of our efforts to discover novel and practical synthesis methods for the construction of heterocyclic compounds, we recently developed an efficient synthesis of substituted thieno[2,3-b]pyridines from β-keto amides.38 We are interested in the fields of cleavage or construction of C–C and C–N bonds because of its varied applications.39 The development of a catalytic process for simultaneous cleavage and building of C–N and C–C bonds is a significant challenge in synthetic chemistry.40 Liang et al. reported a new series of compounds via three-component one-pot reactions of 1-acetyl-1-carbamoyl cyclopropanes, malononitrile and cyclic secondary amines.41 Given that the approach of Liang involved the tandem reaction of β-keto amide reactants to the corresponding fully substituted 2-pyridones, we supposed that the direct reaction of β-keto amides with malononitrile through control reaction conditions would afford 2-pyridones with diverse structures through the control of reaction conditions. When piperidine was replaced with other bases, the polysubstituted 2-pyridones with different substituted patterns could be obtained by using β-keto amides as raw material.
Inspired by these findings, we envisioned the construction of the 2-pyridone core from β-keto amides through intramolecular nucleophilic cyclization. As the result of our continued interest in this area, we have provided an efficient and selective synthesis methodology for the construction of two libraries of 2-pyridones 3 and 4 via a tandem annulation of β-keto amides 1 and malononitrile 2 in CH2Cl2 and DMF, respectively (Scheme 1). Herein, we wish to report our results and the possible mechanism involved was discussed.
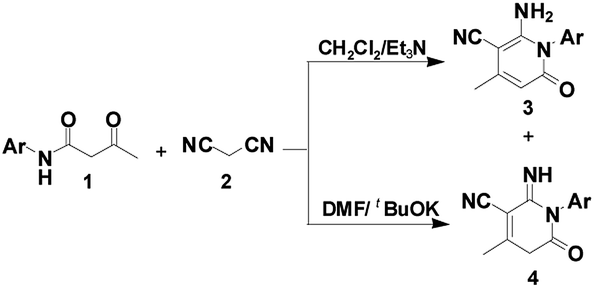 | ||
| Scheme 1 Proposed mechanism for the construction of 2-pyridones 3 and 4. | ||
Results and discussion
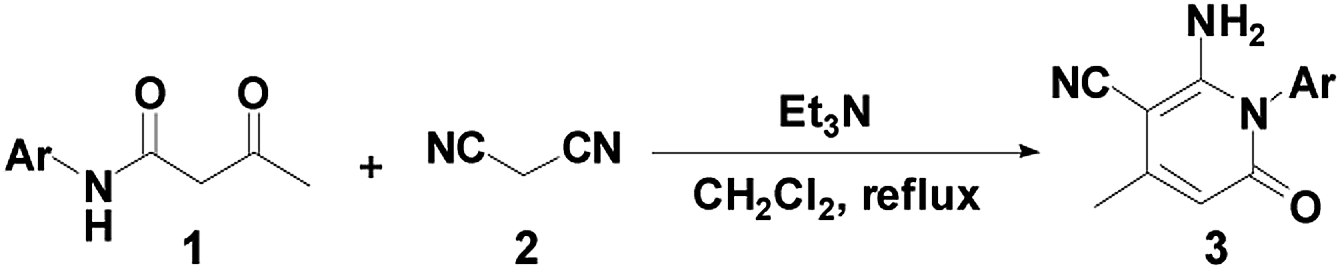
To reduce this idea to practice, we examined the reactivity of the β-keto amides 1 and 2. We then selected 3-oxo-N-phenyl-butyramide 1a as the model substrate to screen its reaction conditions. The reaction was executed with EtONa in anhydrous ethanol at room temperature. Unfortunately the reactions was unsuccessful, no reaction occurred even till 2 h as indicated by TLC (Table 1, entry 1). When 1a was heated with 2 (1.1 equiv.) in EtOH at reflux for 6 h, we were delighted to have the desired 2-pyridone 3a, although the yield was very low (38%, Table 1, entry 2). Surprisingly the transformation was observed using EtONa to generate the reaction. The product was characterized as 2-amino-4-methyl-6-oxo-1-phenyl-1,6-dihydro-pyridine-3-carbonitrile 3a on the basis of its NMR spectra and analytical data (Table 1, entry 2).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Mediatedb | Temp. (°C) | Yieldc (%) | |
| 3a | 4a | ||||
| a The reaction was performed with 1a (1.0 mmol), 2 (1.1 mmol), and the solvent (10 mL).b Mediated (1.0 mmol) was added to the reaction.c Isolated yields were based on β-keto amide 1a. | |||||
| 1 | EtOH | EtONa | rt | n.d. | n.d. |
| 2 | EtOH | EtONa | Reflux | 38 | n.d. |
| 3 | EtOH | DBU | Reflux | 50 | n.d. |
| 4 | EtOH | Et3N | Reflux | 55 | n.d. |
| 5 | CH2Cl2 | Cs2CO3 | Reflux | 63 | n.d. |
| 6 | CH2Cl2 | DBU | Reflux | 72 | n.d. |
| 7 | CH2Cl2 | Et3N | Reflux | 88 | n.d. |
| 8 | DMF | Piperidine | rt | 32 | 10 |
| 9 | DMF | Cs2CO3 | 70 | 45 | n.d. |
| 10 | DMF | K2CO3 | 70 | 41 | n.d. |
| 11 | DMF | tBuOK | rt | n.d. | 85 |
| 12 | DMF | tBuOK | 70 | n.d. | 87 |

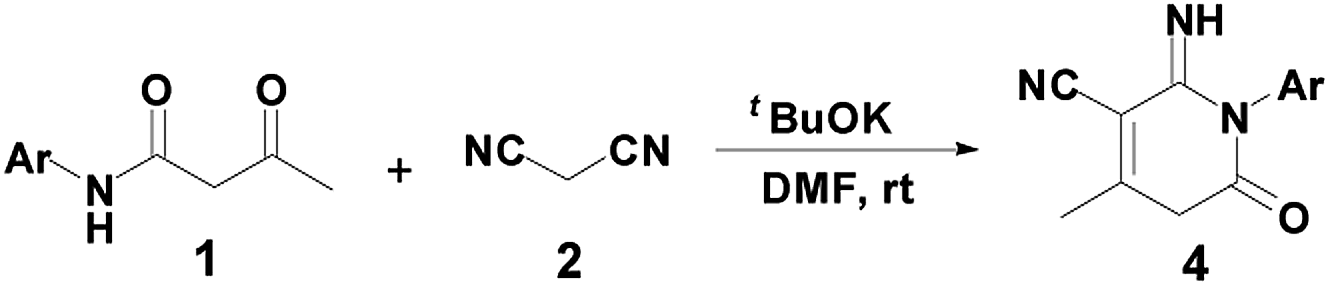
To optimize the reaction conditions, the reaction of β-keto amide derivative 1a with 2 was investigated using various bases and solvent (Table 1). Initially, different organic bases, such as DBU and Et3N, were employed as in EtOH. When EtONa was replaced by DBU or Et3N, the conversion of (2-amino-4-methyl-6-oxo-1-phenyl-1,6-dihydro-pyridine-3-carbonitrile) 3a was increased, affording compound 3a in yields of 50% and 55%, respectively (Table 1, entries 3–4). The reaction conditions were then optimized by screening several solvents. Hence, treatment of 1a with 2 in the presence of Cs2CO3 or DBU in CH2Cl2 at reflux, product 3a was formed in 63 and 72% yield, respectively (Table 1, entries 5–6). The best yield was achieved with Et3N in refluxing CH2Cl2, the yield of desired product 3a reached 88% yield (Table 1, entry 7). When the reaction was performed in the presence of piperidine in DMF at room temperature, a new compound 4a was successfully isolated in 10% yield (Table 1, entry 8). In order to efficiently obtain a single final product 4a, additional attempts were made. The introduction of carbonate such as Cs2CO3 or K2CO3 did not improve the selectivity and the conversion (Table 1, entries 9–10). However, product 4a was obtained in 85% yield by using tBuOK in DMF at room temperature (Table 1, entry 11). Thus, dichloromethane was the best medium for selectively obtaining product 3a. Conversely, the best conditions for the preparation of 4a were conducted in DMF at room temperature.
Under the optimal reaction conditions (Table 1, entry 7), the 2-pyridone 3 was tested by using a range of readily available β-keto amides and malononitrile. In the following work, the scope of the substrate was investigated (Table 2). Under the conditions, a series of reactions of β-keto amides with different substitution groups on the phenyl ring were examined, and the results are listed in Table 2. As shown in Table 2, it was observed that the substrate with various electron-donating and -withdrawing groups 1b–1h could easily be converted into the corresponding 2-pyridones 3b–3h in high yields (85–95%) in 6–7.5 hours (Table 2, entries 2–8). Furthermore, the positions of the substituents on the phenyl ring did not show any effects on the reaction. It is noteworthy that all the crude products 3 could be purified simply by recrystallization from ethanol.
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | Ar | 3 | Temp. (°C) | Yieldb (%) |
| a All reactions were carried out with 1 (1.0 mmol), 2 (1.1 mmol), Et3N (1.0 equiv.), in CH2Cl2 (5.0 mL) at reflux for 6 hours.b Isolated yield. | |||||
| 1 | 1a | Ph | 3a | Reflux | 88 |
| 2 | 1b | 4-ClC6H4 | 3b | Reflux | 95 |
| 3 | 1c | 4-MeOC6H4 | 3c | Reflux | 85 |
| 4 | 1d | 2-ClC6H4 | 3d | Reflux | 88 |
| 5 | 1e | 2,4-Me2C6H3 | 3e | Reflux | 91 |
| 6 | 1f | 4-MeC6H4 | 3f | Reflux | 85 |
| 7 | 1g | 2-MeOC6H4 | 3g | Reflux | 87 |
| 8 | 1h | 2-MeC6H4 | 4h | Reflux | 85 |
Next, with the aim to make product 4 to be the main product (Scheme 1), the selective synthesis of 2-pyridone 4a under appropriate conditions may be realized. Basic conditions play an important role in the transformation process. The use of Et3N selectively afforded compound 3 in high conversion. After several trials, we found that when Et3N was replaced by a stronger base such as tBuOK, product 4a could be obtained in a yield of 85% (Table 1, entry 11). Effect of temperature showed no improvement in the yield of product 4a at room temperature and 70 °C (Table 1, entry 12). Under the optimal reaction conditions, a series of reactions of β-keto amides with different substitution groups on the phenyl ring were examined, affording 2-pyridones 4b–4h in good-to-high yields (85–95%). Notably, all the crude products 4 could be purified simply by recrystallization from DMF (Table 3).
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | Ar | 4 | Temp. (°C) | Yieldb (%) |
| a All reactions were carried out with 1 (1.0 mmol), 2 (1.1 mmol), tBuOK (1.0 equiv.), in CH2Cl2 (5.0 mL) at room temperature.b Isolated yield. | |||||
| 1 | 1a | Ph | 4a | rt | 88 |
| 2 | 1b | 4-ClC6H4 | 4b | rt | 95 |
| 3 | 1c | 4-MeOC6H4 | 4c | rt | 85 |
| 4 | 1d | 2-ClC6H4 | 4d | rt | 88 |
| 5 | 1e | 2,4-Me2C6H3 | 4e | rt | 91 |
| 6 | 1f | 4-MeC6H4 | 4f | rt | 85 |
| 7 | 1g | 2-MeOC6H4 | 4g | rt | 87 |
| 8 | 1h | 2-MeC6H4 | 4h | rt | 85 |
On the basis of the obtained results and previously reported work,41,42 a plausible mechanism for the syntheses of 2-pyridones 3 and 4 are presented, as depicted in Scheme 2. First, the β-keto amide 1 reacts with malononitrile 2 accompanying the loss of one molecule of H2O to generate intermediate 5, and affords intermediate 6. Then, the isomerization of I in the presence of Et3N forms final product 3 under the control experiments conditions, which is cyclized by intramolecular nucleophilic process. Likewise, the isomerization of I′ in the presence of the tBuOK affords final product 4.
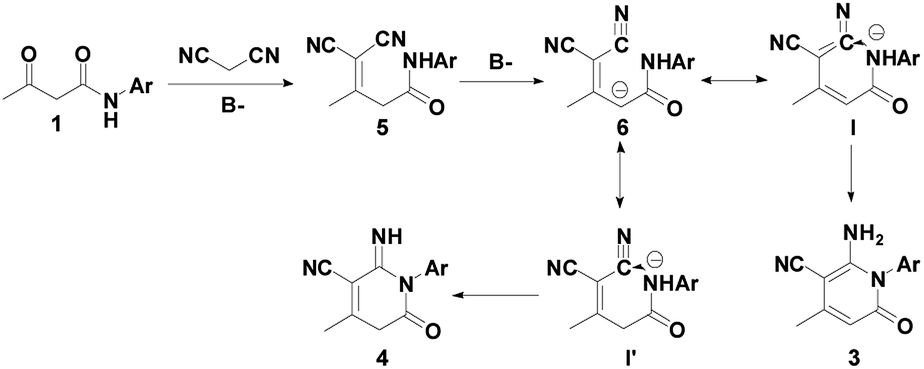 | ||
| Scheme 2 Plausible mechanism for the construction of 2-pyridones 3 and 4. | ||
Conclusions
A simple and efficient one-pot methodology for the divergent synthesis of polysubstituted 2-pyridones 3 and 4 in high yield has been developed. This method illustrates a practical protocol using simple and inexpensive starting materials. Furthermore, the methodology offers several significant advantages, such as economic availability, metal catalyst-free, ease of handling, a relative broad range of substrates, and environmental benignity under mild reaction condition. Further expanding the scope of this reaction and application the products are currently on-going in our laboratory.Experimental
General
All reagents were purchased from commercial sources and used without treatment, unless otherwise indicated. The products were purified by recrystallization. 1H and 13C NMR spectra were recorded on a Bruker AVANCE-300 spectrometer at 25 °C using TMS as an internal standard and deuterated DMSO (DMSO-d6) as the solvent. Petroleum ether (PE) used was the fraction boiling in the range 30–60 °C. Elemental analyses were obtained on a Vario EL analyzer.General procedure for the synthesis of compounds 3a–h
To a solution of β-keto amides 1 (1.0 mmol) and malononitrile (1.1 mmol) in CH2Cl2 (10 mL) was added Et3N (1.0 mmol) in one portion. The mixture was well stirred for 6 h at reflux. When consumption of 1 was complete (TLC), the mixture was washed with saturated sodium chloride solution (10 mL × 3) and dried over anhydrous MgSO4. The organic solvent CH2Cl2 was then removed under reduced pressure and the residue was purified by recrystallization from ethanol to give product 3a–h.General procedure for the synthesis of compounds 4a–h
To a solution of β-keto amides 1 (1.0 mmol) and malononitrile (1.1 mmol) in DMF (10 mL) was added tBuOK (1.0 mmol) in one portion. The mixture was well stirred for 4 hours at room temperature. When consumption of 1 was complete (TLC), the mixture was poured into sat. aq. NaCl (50 mL) under stirring. The precipitated solid was collected by filtration, washed with water (3 × 20 mL) and dried in vacuo to afford the product 4a–h.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly appreciated the financial supports from the National Natural Science Foundation of China (Project No. 0973022), International Science & Technology Cooperation Program of China (No. 2015DFR50260).Notes and references
- (a) J. D. Sunderhaus, C. Dockendorff and S. F. Martin, Org. Lett., 2007, 9, 4223 CrossRef CAS PubMed; (b) F. Liéby-Muller, T. Constantieux and J. Rodriguez, J. Am. Chem. Soc., 2005, 127, 17176 CrossRef PubMed; (c) C. Hulme and V. Gore, Curr. Med. Chem., 2003, 10, 51 CrossRef CAS PubMed; (d) T. Liu, Y. Sun, C. Yang, C. Huang and X. Chen, RSC Adv., 2018, 8, 12635 RSC.
- (a) Q. Li, L. A. Mitscher and L. L. Shen, Med. Res. Rev., 2000, 20, 231 CrossRef CAS PubMed; (b) M. Nagarajan, X. S. Xiao, S. Antony, G. Kohlhagen, Y. Pommier and M. Cushman, J. Med. Chem., 2003, 46, 5712 CrossRef CAS PubMed.
- (a) R. E. Dolle and K. C. Nicolaou, J. Am. Chem. Soc., 1985, 107, 1691 CrossRef CAS; (b) R. J. Cox and D. J. Hagan, J. Chem. Soc., Perkin Trans. 1, 1991, 1, 2537 RSC.
- (a) G. Jones and S. P. Stanforth, in Organic Reactions, Wiley, New York, 1997, vol. 49, pp. 1–330 Search PubMed; (b) E. F. V. Scriven, in Comprehensive Heterocyclic Chemistry, Pergamon Press, Oxford, 1984, vol. 2 Search PubMed; (c) G. Jones, Org. React., 1967, 15, 204 CAS.
- S. M. B. Maezono, T. N. Poudel, L. k. Xia and Y. R. Lee, RSC Adv., 2016, 6, 82321 RSC.
- M. J. Capper, O. Neil, P. M. S. A. Moss and N. G. Ward, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 755 CrossRef CAS PubMed.
- Z. Lv, C. Sheng, T. Wang, Y. Zhang, J. Liu, J. Feng, H. Sun and H. Zhong, J. Med. Chem., 2010, 53, 660 CrossRef CAS PubMed.
- T. Przygodzki, B. Grobelski, P. Kazmierczak and C. Watala, J. Physiol. Biochem., 2012, 68, 329 CrossRef CAS PubMed.
- (a) J. Breinholt, S. Ludvigesen, B. R. Rassing and C. N. Rosendahl, J. Nat. Prod., 1997, 60, 33 CrossRef CAS PubMed; (b) K. Otsubo, S. Morita, M. Uchida, K. Yamasaki, T. Kanbe and T. Shimizu, Chem. Pharm. Bull., 1991, 39, 2906 CrossRef CAS PubMed.
- L. Ceolin, Z. A. Bortolotto, N. Bannister, G. L. Collingridge, D. Lodge and A. Volianskis, RSC Adv., 2014, 4, 44141 RSC.
- S. Mirkovic, A. M. L. Seymour, A. Fenning, S. B. Strachan, S. M. Margolin, S. M. Taylor and L. Br. Brown, J. Pharmacol., 2002, 135, 961 CAS.
- J. L. Medina-Franco, K. Mayorga, C. Gordiano and R. Castillo, ChemMedChem, 2007, 2, 1141 CrossRef CAS PubMed.
- E. M. Wallace, J. Lyssikatos, J. Blake, F. J. Seo, H. W. Yang, C. Perrier, M. Jarski, H. V. Marsh, G. Poch, M. G. Livingston, J. G. Otten, G. Hingorani, R. Woessner, P. Lee, J. Winkler and K. Koch, J. Med. Chem., 2006, 49, 441 CrossRef CAS PubMed.
- (a) Z. Lv, Y. Zhang, M. Zhang, H. Chen, Z. Sun, D. Geng, C. Niu and K. Li, Eur. J. Med. Chem., 2013, 67, 447 CrossRef CAS PubMed; (b) M. T. Cocco, K. C. Congiu and V. Onnis, Eur. J. Med. Chem., 2000, 35, 545 CrossRef CAS PubMed.
- K. Otsubo, S. Morita, M. Uchida, K. Yamasaki, T. Kanbe and T. Shimizu, Chem. Pharm. Bull., 1991, 39, 2906 CrossRef CAS PubMed.
- (a) P. S. Ng, U. H. Manjunatha, S. P. Rao, L. R. Camacho and N. L. Ma, Eur. J. Med. Chem., 2015, 106, 144 CrossRef CAS PubMed; (b) H. Tomioka, Curr. Pharm. Des., 2006, 12, 4047 CrossRef CAS PubMed.
- H. Zhang, B. C. Chen, B. Wang, S. T. Chao, R. Zhao, N. Lim and B. Balasubramanian, Synthesis, 2008, 1523 CrossRef.
- E. Klegraf, M. Follmann, D. Schollmeyer and H. Kunz, Eur. J. Org. Chem., 2004, 3346 CrossRef CAS.
- K. Tanaka, T. Fujiwara and Z. Urbanczyk-Lipkowska, Org. Lett., 2002, 4, 3255 CrossRef CAS PubMed.
- (a) S. M. Sheehan and A. Padwa, J. Org. Chem., 1997, 62, 438 CrossRef CAS PubMed; (b) S. S. P. Chou, C. L. Lu and Y. H. Hu, J. Chin. Chem. Soc., 2012, 59, 365 CrossRef CAS.
- (a) H. Decker, Chem. Ber., 1982, 25, 443 CrossRef; (b) D. L. Comins and J. K. Saha, J. Org. Chem., 1996, 61, 9623 CrossRef CAS; (c) W. Du, Tetrahedron, 2003, 59, 8649 CrossRef CAS.
- (a) D. L. Comins and J. L. Gao, Tetrahedron Lett., 1994, 35, 2819 CrossRef CAS; (b) T. Hu and C. Li, Org. Lett., 2005, 7, 2035 CrossRef CAS PubMed; (c) H. J. Cristau, P. P. Cellier, J. F. Spindler and M. Taillefer, Chem. - Eur. J., 2004, 10, 5607 CrossRef CAS PubMed.
- (a) H. Baron, H. Renfry and J. Thorpe, J. Chem. Soc., 1904, 85, 1726 RSC; (b) T. Aggarwal, G. Sing, H. Ila and H. Junjappa, Synthesis, 1982, 214 CrossRef; (c) L. R. Gibson, L. Hitzel, R. J. Mortishire-Smith, U. Gerhard, R. A. Jelley, A. J. Reeve, M. Rowley, A. Nadin and A. P. Owens, J. Org. Chem., 2002, 67, 9354 CrossRef PubMed.
- (a) A. Robin, K. Julienne, J. C. Meslin and D. Deniaud, Tetrahedron Lett., 2004, 45, 9557 CrossRef CAS; (b) F. Sainta, B. Serckx-Poncin, A. M. Hesbain-Frisque and L. Ghosez, J. Am. Chem. Soc., 1982, 104, 1428 CrossRef.
- (a) J. A. Varela and C. Saa, Chem. Rev., 2003, 103, 3787 CrossRef CAS PubMed; (b) Y. Yamamoto, G. Takagishi and K. Itoh, Org. Lett., 2001, 3, 2117 CrossRef CAS PubMed; (c) A. Padwa, S. M. Sheehan and C. S. Straub, J. Org. Chem., 1999, 64, 8648 CrossRef CAS.
- D. Xiang, Y. Yang, R. Zhang, Y. Liang, W. Pan, J. Huang and D. Dong, J. Org. Chem., 2007, 72(22), 8593 CrossRef CAS PubMed; W. Pan, D. Dong, J. Wang, R. Zhang, E. Wu, D. Xiang and Q. Liu, Org. Lett., 2007, 9, 2421 CrossRef PubMed.
- (a) B. Stanovnik and J. Svete, Chem. Rev., 2004, 104, 2433 CrossRef CAS PubMed; (b) P. Wan and Y. Gao, Chem. Rec., 2016, 16, 1164 CrossRef PubMed.
- (a) T. Sengupta, K. S. Gayen, P. Pandit and D. K. Maiti, Chem.–Eur. J., 2012, 18, 1905 CrossRef CAS PubMed; (b) J. B. Pierce, Z. S. Ariyan and G. S. Ovenden, J. Med. Chem., 1982, 25, 131 CrossRef CAS PubMed; (c) H. L. Wang, Z. Li, G. W. Wang and S. D. Yang, Chem. Commun., 2011, 47, 11336 RSC; (d) B. Lu and D. Ma, Org. Lett., 2006, 8, 6115 CrossRef CAS PubMed; (e) L. Tan, P. Zhou, C. Chen and W. Liu, Beilstein J. Org. Chem., 2013, 9, 2681 CrossRef PubMed.
- (a) I. Couto, I. Tellitu and E. Domínguez, J. Org. Chem., 2010, 75, 7954 CrossRef CAS PubMed; (b) A. M. Farag, N. A. Kheder and Y. N. Mabkhot, Heterocycles, 2009, 78, 1787 CrossRef CAS; (c) L. Xia and Y. R. Lee, Org. Biomol. Chem., 2013, 11, 5254 RSC.
- R. J. Clemens, Chem. Rev., 1986, 86, 241 CrossRef CAS.
- N. Nishiwaki, Y. Nakaike and M. J. Ariga, J. Oleo Sci., 2008, 57, 53 CrossRef CAS PubMed.
- R. Jayalakshmi, P. Neelakatan, S. N. Rao, D. S. Iyengar and U. T. Bhalerao, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1979, 18B, 366 CAS.
- W. Ehm and J. Liebigs, Ann. Chem., 1977, 1642 CAS.
- Y. Wang, X. Xin, Y. Liang, Y. Lin, H. Duan and D. Dong, Adv. Synth. Catal., 2009, 351, 2217 CrossRef CAS.
- M. Han, K. D. Nam, H. G. Hahn and D. Shin, Tetrahedron Lett., 2008, 49, 5217 CrossRef CAS.
- (a) S. M. Hussain, A. M. El-Reedy and S. A. El-Sherabasy, J. Heterocycl. Chem., 1988, 25, 9 CrossRef CAS; (b) T. Kato, Acc. Chem. Res., 1974, 7, 265 CrossRef CAS; (c) R. J. Clemens, Chem. Rev., 1986, 86, 241 CrossRef CAS; (d) M. Han, K. D. Nam, H. G. Hahn and D. Shin, Tetrahedron Lett., 2008, 49, 5217 CrossRef CAS; (e) B. Zaleska and S. Lis, Synth. Commun., 2001, 31, 189 CrossRef CAS.
- F. Liu, T. Sun, Y. Yang, C. Huang and X. Chen, RSC Adv., 2018, 8, 12635 RSC.
- B. Gao, D. Dong, J. Zhang, C. Ding, C. m. Dong, Y. Liang and R. Zhang, Synthesis, 2012, 44, 201 CrossRef CAS.
- (a) T. Koreeda, T. Kochi and F. Kakiuchi, J. Am. Chem. Soc., 2009, 131, 7238 CrossRef CAS PubMed; (b) X. Zhao, D. Liu, H. D. Guo, Y. Liu and W. Zhang, J. Am. Chem. Soc., 2011, 133, 19354 CrossRef CAS PubMed; (c) W. L. Man, J. Xie, Y. Pan and W. W. Y. Lam, J. Am. Chem. Soc., 2013, 135, 5533 CrossRef CAS PubMed; (d) S. Khamarui, R. Maiti and D. K. Maiti, Chem. Commun., 2015, 51, 384 RSC.
- K. Hirano and M. Miura, Chem. Sci., 2018, 9, 22 RSC.
- F. Liang, X. Cheng, J. Liu and Q. Liu, Chem. Commun., 2009, 3636 RSC.
- For similar spiroannulated structures, see: P. Langer and G. Bose, Angew. Chem., Int. Ed., 2003, 42, 4033 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all products. See DOI: 10.1039/c8ra05709e |
| This journal is © The Royal Society of Chemistry 2018 |