
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic–inorganic hybridization for the synthesis of robust in situ hydrophobic polypropylsilsesquioxane aerogels with fast oil absorption properties†
Ze Wu,
Lei Zhang,
Ji Li,
Xiaolu Zhao and
Chunhui Yang *
*
MIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin, 150001, China. E-mail: kennywu@hit.edu.cn; yangchh@hit.edu.cn
First published on 2nd February 2018
Abstract
In situ hydrophobic polypropylsilsesquioxane aerogels (PSAs) were successfully synthesized via an organic–inorganic hybridization method by a sol–gel process, in which propyltriethoxysilane (PTES) and tetraethylorthosilicate (TEOS) were used as co-precursors. 29Si NMR and FTIR analyses indicated the high degree of condensation of the precursors and proved the attachment of propyl (–C3H7) groups in PSAs, respectively. By means of incorporating propyl groups, both mechanical robustness and in situ hydrophobicity were obtained. Meanwhile, the mechanical strength, contact angle and density obviously increased with the increase in propyl groups. Under optimized conditions, as-prepared PSA could endure up to a 70% maximum linear compression with few cracks. Benefiting from the robust structure and in situ hydrophobicity, PSAs showed high absorption capacities (8–10 times that of its own weight) and fast absorption properties (<20 s) for a wide range of organic solvents and could be reused at least 5 times.
1. Introduction
Silica aerogels are a kind of lightweight porous material that have been extensively used as catalyst supports,1 thermal and acoustic insulators,2,3 absorbents,4–6 membranes,7 drug delivery media,8 and energy storage materials9,10 due to their unique properties, such as low density, high porosity, low thermal conductivity and high specific surface area.11 However, most of those applications are limited because silica-based aerogels collapse easily due to the poor mechanical properties caused by their brittle skeleton and fragile internal “pearl-like” structure.12,13 To overcome those shortcomings, many efforts have been put forth to strengthen the mechanical robustness of silica aerogels, including ageing,14 fibre reinforcing15,16 and organic polymers crosslinking.17,18Organic–inorganic hybridization, occurring only in the polysiloxane-based materials due to the high stability of Si–C bonds over metal–carbon bonds, shows superior advantages towards improving the mechanical strength and flexibility of silica-based aerogels.19 Compared to fiber or polymer reinforcement, the mechanical strength and hydrophobicity of silica aerogels are improved in situ in this approach, and therefore avoid the use of any other additives (TMCS, HDMS or monomers).20 The hybridization is realized by using trifunctional organosilanes (R′Si(OR)3, where R′ = alkyl, aryl or vinyl groups, R = methyl or ethyl groups) as the precursor or co-precursor with TMOS or TEOS.21–23 Owing to the presence of non-hydrolysable R′ groups, the inter-chain bonding of hybrid aerogels is reduced resulting in a robust matrix. Among those hybrid aerogels, methyltrimethoxysilane (MTMS)-based silica aerogels (MSAs) have attracted more attention due to its easier gelling property compared to other organic hybrid aerogels. Rao first reported the synthesis of MSA by adding MTMS to the synthesis formulation of TMOS-based aerogel. The resultant aerogel showed no net increase in weight even after placing it directly on the surface of water.24 Moreover, they also synthesized flexible MSAs with the Young's modules ranging from 0.1411 MPa to 0.0343 MPa by varying the molar ratio of MeOH/MTMS from 14 to 35.23 Likewise, transparent MSAs with the highest bulk module up to 7.41 MPa was reported. More importantly, the sample could endure up to 80% linear compression and spring back to more than 95% of its original shape.25 Though, numerous supercritical or ambient pressure dried hybrid aerogels were reported, most of which were methyl-based because of the unavoidable phase separation of other organotrialkoxysilanes-based ones.26–28 For those precursors, cyclization reactions played an important role in the silsesquioxane polymerization process and produced crystalline oligomers, resinous materials, or precipitates.29 A surfactant assisted acid–base two-step process is a promising way to control phase separation and form a homogeneous gel. By this method, ethyl- and vinyl-modified silica aerogels with good mechanical properties were successfully synthesized. As-prepared ethyl-modified aerogels showed an elastic behavior on uniaxial compression up to 50% without collapse.30,31 Oweini also reported the synthesis of a variety of organic group hybrid (methyl, ethyl, propyl, phenyl, etc.) aerogels by using supercritical CO2 drying. However, the mechanical properties and hydrophobicity of those aerogels were not strengthened on account of the low organic group incorporation (20%).32,33
Our group has concentrated on the synthesis of propyl hybrid silica aerogels and has previously reported the synthesis of nanowire-reinforced PSAs at 60% molar incorporation of PTES.34 The Young's modulus of the nanowire-reinforced PSA was 0.35 MPa, which showed a 67% increase over un-reinforced PSA. In this work, we increased the molar fraction of PTES by changing the hydrolysis/polycondensation conditions (higher acidity and higher basicity). Through this mean, the macroscopic phase separation is retarded and the solution is solidified earlier by the sol–gel transition. Moreover, as-synthesized aerogel in this paper showed a comparable modulus (∼0.56 MPa) to that of nanowire-reinforced aerogels (0.35 MPa). Therefore, a complicated nanowire reinforcing process was avoided. Refer to the superhydrophobic and oleophilic properties, those mechanically robust aerogels were further used in oily water separation and showed fast absorption properties for a wide range of organic solvents, such as alkanes, ethers, esters, alcohols, amides, aromatic hydrocarbons, and kerosene.
2. Materials and methods
2.1 Materials
Propyltriethoxysilane (98% purity) and TEOS (98% purity) were supplied by Hongbai Chemical Co, China. Hydrogen chloride (HCl), ammonia hydroxide (NH3·H2O), ethanol, hexane and heptane were purchased from Xilong Chemical Co, China. Cetyltrimethylammonium bromide (CTAB), trimethylchlorosilane (TMCS), kerosene, dimethylbenzene, acetone, petroleum ether, ethyl acetoacetate, dibutyl phthalate (DBP, 97%) and Sudan III were purchased from Aladdin Chemical Co, China. All reagents were AR grade.2.2 Synthesis process of PSAs
PSAs were synthesized through a two-step acid–base catalysed sol–gel process, as shown in Fig. 1. The synthetic procedure of a typical sample PSA-7 is as follows: PTES (0.07 mol) and TEOS (0.03 mol) were pre-mixed under vigorous stirring in a glass bottle. After 5 min of stirring, ethanol (0.5 mol), water (0.3 mol), HCl (1 M, 1.0 mL) and CTAB (5 × 10−3 mol) were added to the mixture and then hydrolysed at 90 °C for 5 h with continuous stirring. After hydrolysation, the sol (15 mL) was transferred to a sealed glass vial and gelled by NH3·H2O (12.4 M, 0.5 mL) in an oven at 50 °C. Gelation usually occurred within 5–6 h. Afterwards, the obtained gel was aged by ethanol (15 mL) for 1 day and subsequently soaked in hexane (15 mL) for 6 h (three times) at 50 °C. Finally, the alcogel was ambient pressure dried (APD) in a temperature gradient of 60, 80, 100 and 120 °C and left at each temperature for 2 h. For comparison, sample without the incorporation of PTES (SA) and TMCS modified PSA-7 (PSA-7-T) were synthesized using a similar process to that of the PSA-7 system (see in ESI†). For samples synthesized at higher amount of PTES than PSA-7, a macroscopic phase separation phenomenon was observed (Fig. S1†).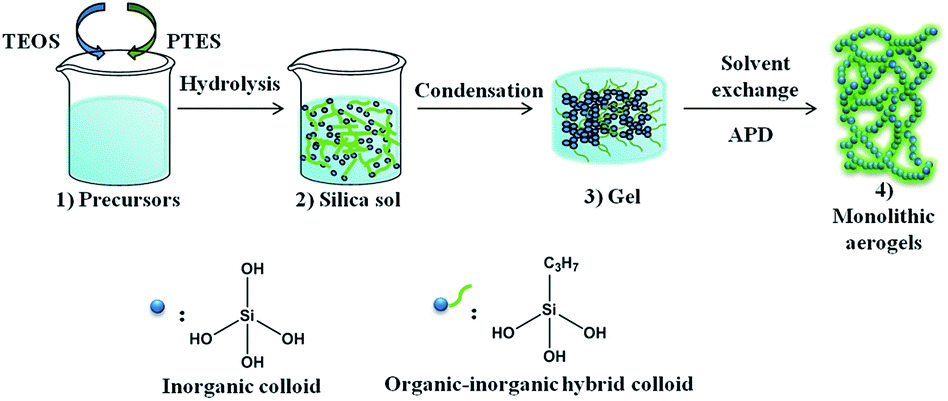 | ||
| Fig. 1 Synthesis process of propyl functionalized silica aerogels. | ||
2.3 Characterization
The bulk density (ρ) of PSA was measured by electronic densimeter (AU-300PM, China), while that of SA was calculated by measuring the dimensions and mass of the monolithic sample.The morphology of the Au-coated PSA was characterized by scanning electron microscopy (SEM, Supra, Germany). The surface profile of PSA-7 was detected by a non-contact surface profiler (SmartWLI, Germany). The static contact angle (θ) was measured at four varied plots on the surface of each sample, and the average value was taken as the contact angle (JC2000C, China).
The functional groups of PSA were investigated by Fourier transform infrared spectroscopy (FTIR, Bruker-TENSOR27, Germany) and 29Si solid-state NMR (AVANCE III HD 400M, Bruker).
The pore size distribution and Brunauer–Emmett–Teller (BET) specific surface area were obtained from the N2-sorption isotherm at 77 K (TriStar 3000, America), all samples were pre-dried at 120 °C.
A universal material tester (Instron-5965, America) was used to test the mechanical properties of PSAs. For the compression load–strain test, an as-prepared cylindrical aerogel (40 mm × 40 mm × 10 mm) was placed between testing plates and compressed with a speed of 0.5 mm min−1 until the strain reached 50%. For the fracture strain test, the sample (40 mm × 40 mm × 10 mm) was compressed with a speed of 0.5 mm min−1 until the sample broke.
The oil absorption capacity was detected by wholly dipping the pre-weighed PSA (W0) in a solvent, and the weight was obtained every 0.5 min over a 3 min period (Wt). The absorption capacity of each solvent was measured for five times and the error bars were automatically fitted in origin by the mean value and SD value.
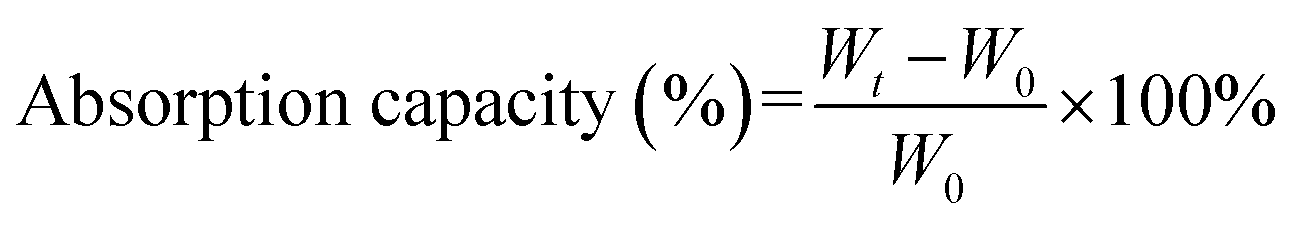
3. Results and discussion
The physicochemical properties of PSAs are shown in Table 1. The density of PSAs rose from 0.1552 to 0.1811 g cm−3 with the increase in the molar fraction of PTES. That can be attributed to the increasing higher amount of propyl groups in the framework of PSAs which is similar to the reported MTMS/TMOS system.35 The densities of PSAs were higher than that of pure silica aerogels (0.03–0.10 g cm−3)36,37 but comparable to that of the mechanically robust aerogels (0.10–0.30 g cm−3).38–40 Notably, PSA-7-T showed the highest density (0.1825 g cm−3), which is about a 125% increase over SA (0.0811 g cm−3). However, on the positive side, the high incorporation of PTES also brought a qualitative leap to the mechanical strength and hydrophobicity of PSAs.| Samplesa | Density (g cm−3) | SBET (m2 g−1) | Pore sizeb (nm) | E (MPa) | CA (°) |
|---|---|---|---|---|---|
| a The number after PSA indicates the molar fraction of PTES; T after the number means the surface is modified by TMCS.b Mean pore size of PSAs.c Compression modulus and contact angle of supercritical CO2 dried SA were undetectable. | |||||
| SA | 0.0811 | 838.35 | 12.41 | —c | —c |
| PSA-4 | 0.1552 | 794.58 | 10.44 | 0.0150 | 146.21 |
| PSA-6 | 0.1765 | 560.00 | 15.42 | 0.2745 | 152.49 |
| PSA-7 | 0.1811 | 215.09 | 30.21 | 0.5568 | 155.22 |
| PSA-7-T | 0.1825 | 181.39 | 25.13 | 0.4611 | 152.05 |
FTIR analysis was employed to study the characteristic groups of as-prepared aerogels (Fig. 2). It was found that compared to SA, peaks located at 3500 and 965 cm−1 dramatically decreased in PSA-7, which meant there were rarely –OH groups or absorbed water located in PSA-7.41 Peaks at 1080 cm−1 and 780 cm−1 confirmed the typical Si–O–Si network of silica-based aerogels.42 More importantly, according to the peaks of the C–H stretching vibrations43 at approximately 2957 cm−1 and 1460 cm−1 and the characteristic absorption peak of the Si–C bond at 1223 cm−1, the attachment of propyl groups in PSA-7 was confirmed. Furthermore, the spectrum of PSA-7 showed no significant difference compared with that of PSA-7-T, which was a consequence of the steric effect and decreased reaction sites in PSA-7. In general, during the surface modification process, the –OH group was substituted by –O–Si(CH3)3 groups through the followed reaction:44
| –SiOH + (CH3)3SiCl → Si–O–Si(CH3)3 + HCl | (1) |
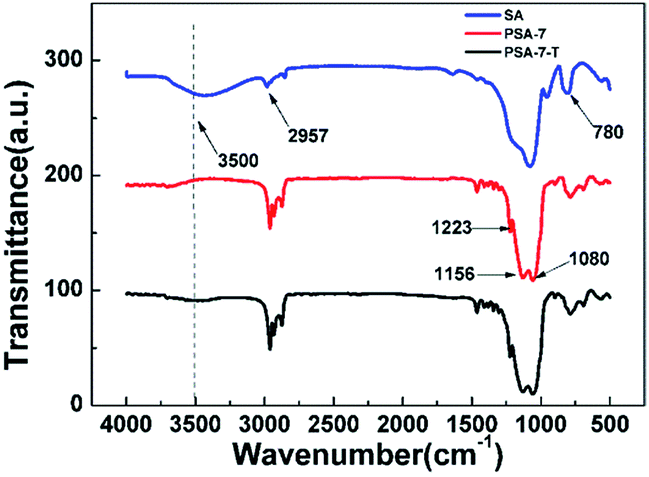 | ||
| Fig. 2 FTIR spectrums of SA, PSA-7 and PSA-7-T. | ||
However, in PSA-7 this reaction was largely restricted, because the steric effect caused by –C3H7 can hinder the entry of TMCS, leading to few TMCS participating in the reaction. Moreover, PSA-7 possessed fewer –OH groups than SA because of the high degree of condensation of the precursors (Fig. 3) and the existence of propyl groups, which meant there were few reaction sites for TMCS. Thus, a similar spectrum was observed for PSA-7 and PSA-7-T.
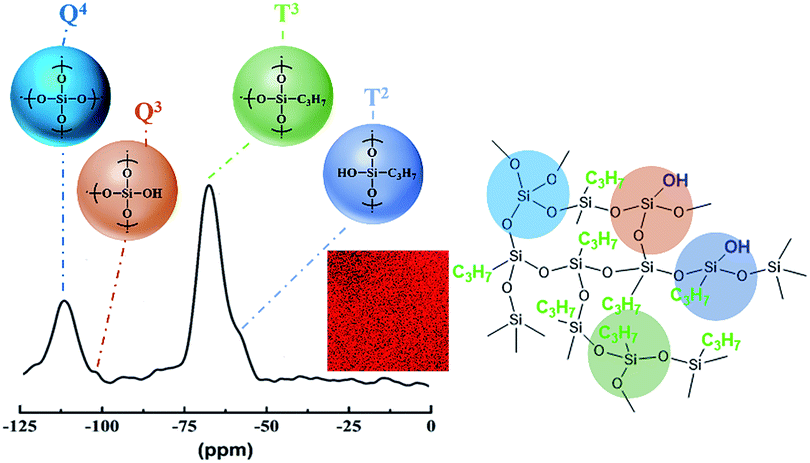 | ||
| Fig. 3 Solid-state 29Si NMR spectra and the assumed molecular structure of PSA-7. The inset middle red graph is the EDS mapping of the C atom. | ||
In addition to the FTIR test, we further used 29Si solid-state NMR to investigate the building blocks and the degree of condensation of the precursors in PSA-7. As shown in Fig. 3, the NMR spectra exhibited two broad peaks located at −64 ppm (T3 peak) and −110 ppm (Q4 peak). The T3 peak is corresponding to the C3H7–Si–(OSi)3 structure with one Si atom crosslinks with three siloxane bonds, and the Q4 peak relates to the Si–(OSi)4 structure with one Si atom crosslinks with four siloxane bonds.29 The strong T3 and Q4 peaks, combined with the weak Q3 and T2 peaks, revealed a high degree of condensation of PTES and TEOS.45 Based on the 29Si NMR and FTIR analyses, the molecular structure of PSA-7 was proposed. As assumed, –C3H7 groups distributed uniformly in the network of PSA-7 with few uncondensed –OH groups bonded to Si atoms. The EDS mapping of the C atom also proved this hypothesis.
To investigate the effects of the propyl content on the difference in microstructure of PSAs, SEM characterization was employed. All samples showed a uniform three-dimensional reticulated structure with few defects (Fig. 4 and S2†). Notably, PSA-7 revealed a finer microstructure than other samples because of the high number of propyl groups in the network. Unlike the hydroxyl groups, the propyl groups did not condense with each other during the APD process and therefore avoided the aggregation of silica particles.46 Hence, a finer microstructure was observed. In addition, SA also showed a fine microstructure regardless of the existence of abundant hydroxyl groups. This result was ascribed to the change in the drying method in which supercritical CO2 drying eliminated the capillary force and consequently the uniform microstructure of the hydrogel was retained.47 Meanwhile, in the APD process, the capillary force (Pr) was unavoidable when hexane within the pores was replaced by air: causing the collapse of three-dimensional network.48 For a certain pore and solvent, Pr was definite and was exerted on the pore simultaneously with the solvent evaporation. This force usually resulted in the shrinkage and even collapse of the pore. Whereas, for PSAs, substituted propyl groups could sustain this force by repulsive reactions, as previously reported in MSAs.25 Therefore, PSAs showed uniform three-dimensional structures and could be derived by ambient pressure drying without surface treatment.
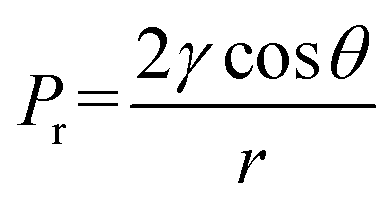 | (2) |
 | ||
| Fig. 4 SEM images of propyl functionalized silica aerogels: (1) SA; (2) PSA-4; (3) PSA-6; (4) PSA-7. | ||
All samples showed a type IV curve with a hysteresis loop at P/P0 > 0.8 (Fig. 5), which illustrates the existence of mesopores (pore diameters between 2–50 nm).49 It could be noted that the specific surface area decreased significantly with the increase of PTES, and the corresponding BET surface area of SA, PSA-4, PSA-6 and PSA-7 was 838, 794, 560 and 215 m2 g−1, respectively. That was a consequence of the increase in the number of macropores (pore diameter > 50 nm) in higher PTES incorporated aerogels and was further indicated by the rapid increase of N2 absorption at high relative pressure.50 The BJH pore distribution also proved the increase of macropores along with the number of propyl groups used. While propyl groups enlarged the pores and decreased the specific surface area of the PSAs, they also improved the mechanical strength of PSAs and avoided aggregating of inter silica particles.
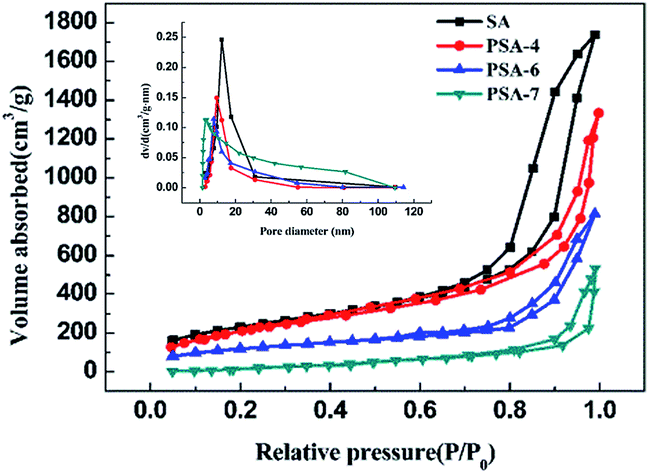 | ||
| Fig. 5 BJH pore size distribution (inset) and N2 absorption–desorption isotherms of PSAs with different amount of incorporated PTES. | ||
The inherent low mechanical strength of SA was strengthened by introducing propyl groups into the network. The Young's modulus of PSA-7, PSA-7-T, PSA-6 and PSA-4 was 0.5568, 0.5311, 0.2745 and 0.0150 MPa, respectively (Fig. 6). The mechanical strength of PSAs (i.e., Young's modulus and maximum compression load) are enhanced with the increase in the number of propyl groups. Furthermore, PSA-7 was able to endure up to 70% linear compression and rebounded to nearly 95% of its original shape with only a few cracks (Fig. S3†). However, PSA-4 still showed a similar poor mechanical property with SA, and a lower Young's modulus was detected (0.0150 MPa). This can be attributed to the weak repulsive interactions in PSA-4 because it owned the smallest moiety of propyl groups. Upon compression, external force was exerted on the entire aerogel body. To offset this stress, the aerogel skeleton shrink, causing the internal pores to compress. During this process, repulsive interactions inside the propyl-terminated pores could protect the network from breaking. Thus, a more robust skeleton was formed when a high PTES molar fraction was incorporated.35 Meanwhile, the relatively low density also lowered the mechanical strength of PSA-4 as most lightweight materials followed the general law for conventional materials. Their compressive modulus tended to decrease dramatically with decreasing the density, i.e., (E/ES) ∝ (ρ/ρS)n=2or3 or (α/αS) ∝ (ρ/ρS)n=2or3, where E is the Young's modulus and α is the yield strength.51 Therefore, improvement in the mechanical strength of PSAs were a consequence of the increase in density and repulsive interactions.
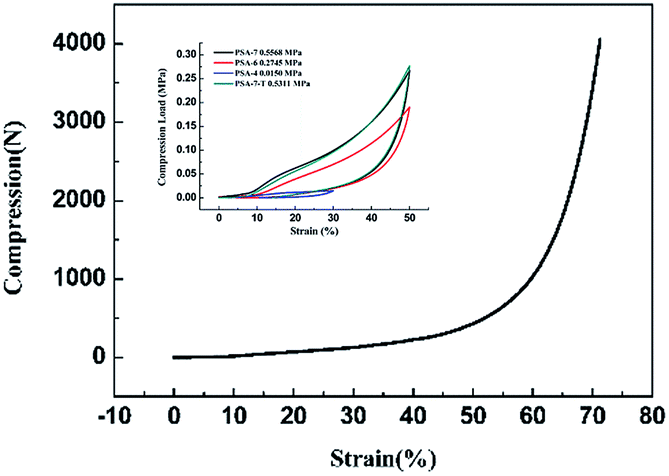 | ||
| Fig. 6 Fracture strain curve of PSA-7 and compression load–strain circle curves of aerogels prepared from different PTES amounts. | ||
It was known that to obtain superhydrophobic silica aerogels, surface modification is an unavoidable process regardless of the drying method.52,53 Nevertheless, our research showed that PTES-crosslinked PSAs possessed an in situ superhydrophobic property (Fig. 7). This property was mainly attributed to two aspects. The first was that the inherent mesoporous and macroporous structure of the PSAs constructed a micro–nanosurface on the PSAs and endowed them with a “lotus-effect”.54 A non-contact surface profiler was applied to detect the surface morphology of PSA-7 (Fig. S4†). The resultant surface roughness Ra was 128.82, which illustrates a crude surface structure. The second was that propyl groups formed a low-energy surface on PSA-7 and hindered the absorption of water. Owing to those two aspects, PSAs showed in situ hydrophobicity. The advancing and receding contact angles of PSA-7 were 150.35° and 160.49°, respectively (Fig. 7-(2) and (3)), which means PSA-7 possessed a contact angle hysteresis of 10.14°.
 | ||
| Fig. 7 (1) The static contact angle, (2) advancing contact angle and (3) receding contact angle of PSA-7. | ||
Considering the highly porous, mechanically robust and in situ superhydrophobic properties of PSA-7, its application in fast oil absorption was presented. PSA-7 showed absorption capacities approximately 8–10 times greater than its own weight for a wide range of organic solvents (Fig. 8). The maximum absorption capacity observed was for DBP (1114%) and the minimum was for hexane (867%). According to Gurav,55 for a certain material, the absorption capacity is proportional to the surface tension of organic solvents. Likewise, our research showed a similar result (Fig. 9), in which the absorption capacity almost linearly increased with the surface tension between 15 N m−1 and 40 N m−1. Thus, it was not surprising that DBP absorbed the most organic solvents considering its highest surface tension (37.9 N m−1).
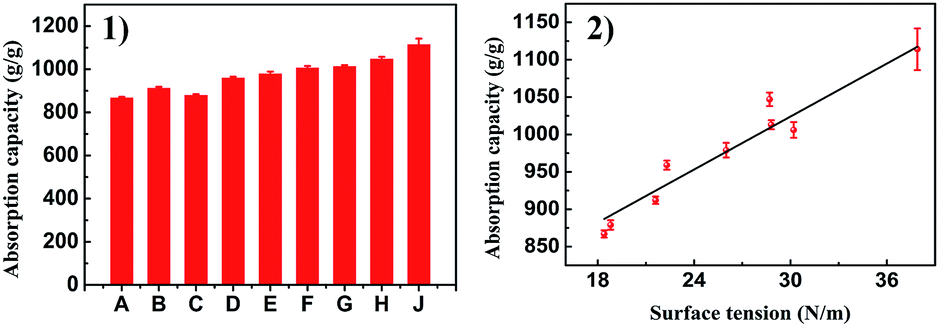 | ||
| Fig. 8 (1) Absorption capacities of PSA-7 and (2) its relationship with the surface tension of tested organic solvents. A: hexane; B: heptane; C: acetone; D: EtOH; E: kerosene; F: ethyl acetoacetate; G: petroleum ether; H: dimethylbenzene; J: DBP. | ||
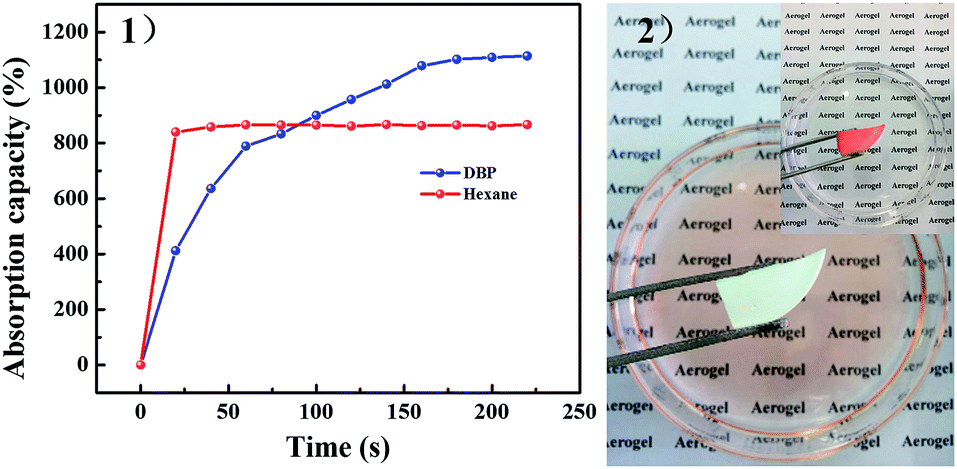 | ||
| Fig. 9 (1) Absorption efficiency of PSA-7 for two model solvents and (2) absorption process for Sudan dyed hexane. | ||
The absorption rate is a key index for the emergency treatment of organic solvents leakage. Here, we investigated the absorption rate of PSA-7 by using hexane and DBP as model solvents. Hexane was wholly absorbed in less than 20 s owing to the highly porous interconnected pore structure of PSA-7 and the low viscosity of hexane. DBP was totally absorbed over 3 min (Fig. 9) because of the higher viscosity (9.72 mPa s−1, 35 °C) and higher molecule weight (278.34 g mol−1) than hexane. High viscosity means poor mobility, which suppresses the fast entrance of DBP into the internal pores of PSA-7. In contrast, hexane was more likely to be absorbed due to its high fluidity.56 Furthermore, as discussed above, propyl groups inside the pores may inhibit the reaction between –OH and TMCS due to the steric effect. Comparably, the absorption efficiency of DBP was largely restricted because of its large steric configuration. Thus, a longer absorption equilibrium time was observed for DBP than hexane. Moreover, we found that PSA-7 could be reused at least 5 times by simple ambient pressure drying without the decline of its fast absorption property for hexane (Fig. S5†), and only a 7% decrease in the absorption capacity was observed. 29Si NMR and FTIR were further applied to characterize the structure transformation of PSA-7 after hexane was absorbed. The hexane saturated PSA-7 (PSA-7-H) presented the NMR and FTIR spectrums as shown in Fig. S6.† Nothing significant difference can be identified in the spectrums with that of PSA-7. The NMR spectrum revealed two sharp peaks (Q4 and T3) with two small ones (Q3 and T2) similar to that of PSA-7, which meant that the internal building blocks of PSA-7 remained unchanging during the absorption process. Likewise, the FTIR test gave a comparable result that the characteristic groups located in PSA-7 were not broken by hexane, and therefore retained the microstructure of PSA-7.
4. Conclusions
Mechanically robust PSA was successfully synthesized by an organic–inorganic hybridization method, and its application in oil absorption was presented. The whole process was time saving and easy to operate. 29Si NMR confirmed the high degree of condensation of precursors and FTIR analysis proved the attachment of propyl groups in the PSAs. Propyl incorporation was vital for the realization of in situ superhydrophobicity and protected the PSAs from collapsing during the APD process. The PSAs exhibited properties of low densities, high modulus, in situ superhydrophobicity and high surface areas. At an optimized condition, as-synthesized PSA can endure up to 70% maximum linear compression with few cracks. Benefiting from the robust structure and in situ hydrophobicity, PSA-7 showed high absorption capacities (8–10 times that of its own weight) and fast absorption properties (<20 s) for a wide range of organic solvents, which made it a promising material for emergency treatment of organic solvent leakage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding support from the National Science Fund for Distinguished Young Scholars of China (Grant No. 51325201), National Natural Science Foundation of China (Grant No. 51302052), the Fundamental Research Funds for the Central Universities (Grant No. HIT. NSRIF. 2017026), and the Program of Heilongjiang Province (Project No. GC13A105).Notes and references
- G. Zu, J. Shen, W. Wang, L. Zou and Y. Lian, ACS Appl. Mater. Interfaces, 2015, 7, 5400–5409 CAS.
- C. Buratti and E. Moretti, Appl. Energy, 2012, 98, 396–403 CrossRef CAS.
- G. Tao, B. P. Jelle, T. Ihara and A. Gustavsen, Appl. Energy, 2014, 128, 27–34 CrossRef.
- Y. Shan, H. Luo and Y. Gao, RSC Adv., 2013, 4, 4535–4542 Search PubMed.
- S. Zong, W. Wei, Z. Jiang, Z. Yan, J. Zhu and J. Xie, RSC Adv., 2015, 5, 55579–55587 RSC.
- M. Shi, C. Tang, X. Yang, J. Zhou, F. Jia, Y. Han and Z. Li, RSC Adv., 2017, 7, 4039–4045 RSC.
- P. Veres, A. M. López-Periago, I. Lázár, J. Saurina and C. Domingo, Int. J. Pharm., 2015, 496, 360–370 CrossRef CAS PubMed.
- S. K. Rajanna, D. Kumar, M. Vinjamur and M. Mukhopadhyay, Ind. Eng. Chem. Res., 2015, 54, 949–956 CrossRef CAS.
- X. Huang, Z. Liu, W. Xia, R. Zou and R. P. S. Han, J. Mater. Chem. A, 2015, 3, 1935–1940 CAS.
- D. Liu, G. Cheng, H. Zhao, C. Zeng, D. Qu, L. Xiao, H. Tang, Z. Deng, Y. Li and B. L. Su, Nano Energy, 2016, 22, 255–268 CrossRef CAS.
- S. S. Kistler, Nature, 1931, 127, 741 CrossRef CAS.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2003, 102, 4243–4265 CrossRef.
- N. Hüsing and U. Schubert, Angew. Chem., Int. Ed., 1998, 37, 22–45 CrossRef.
- S. Haereid, J. Anderson, M. A. Einarsrud, D. W. Hua and D. M. Smith, J. Non-Cryst. Solids, 1995, 185, 221–226 CrossRef.
- S. Zhao, Z. Zhang, G. Sebe, R. Wu and R. V. R. Virtudazo, Adv. Funct. Mater., 2015, 25, 2326–2334 CrossRef CAS.
- T. Zheng, A. Li, Z. Li, W. Hu, L. Shao, L. Lu, Y. Cao and Y. Chen, RSC Adv., 2017, 7, 34461–34465 RSC.
- S. Mulik, C. Sotiriou-Leventis, G. Churu, H. Lu and N. Leventis, Chem. Mater., 2008, 20, 5035–5046 CrossRef CAS.
- J. Wang and X. Zhang, ACS Nano, 2015, 9, 11389–11397 CrossRef CAS PubMed.
- T. Shimizu, K. Kanamori and K. Nakanishi, Chem.–Eur. J., 2017, 23, 5176 CrossRef CAS PubMed.
- H. Maleki, L. Durães and A. Portugal, J. Non-Cryst. Solids, 2014, 385, 55–74 CrossRef CAS.
- S. D. Bhagat, C. S. Oh, Y. H. Kim, Y. S. Ahn and J. G. Yeo, Microporous Mesoporous Mater., 2007, 100, 350–355 CrossRef CAS.
- T. Zhang, H. Yuan, S. Wang, X. Guo, R. Hu, Y. Li and G. Yang, RSC Adv., 2017, 7, 32861–32865 RSC.
- A. V. Rao, S. D. Bhagat, H. Hirashima and G. M. Pajonk, J. Colloid Interface Sci., 2006, 300, 279–285 CrossRef CAS PubMed.
- A. V. Rao and D. Haranath, Microporous Mesoporous Mater., 1999, 30, 267–273 CrossRef CAS.
- K. Kanamori, M. Aizawa, K. Nakanishi and T. Hanada, Adv. Mater., 2007, 19, 1589–1593 CrossRef CAS.
- S. He and X. Chen, J. Non-Cryst. Solids, 2017, 463, 6–11 CrossRef CAS.
- A. V. Rao, M. M. Kulkarni, D. P. Amalnerkar and T. Seth, J. Non-Cryst. Solids, 2003, 330, 187–195 CrossRef CAS.
- A. V. Rao, G. M. Pajonk and D. Haranath, Mater. Sci. Technol., 2013, 17, 343–348 Search PubMed.
- D. A. Loy, B. M. Baugher, C. R. Baugher, D. A. Schneider and K. Rahimian, Chem. Mater., 2000, 12, 3624–3632 CrossRef CAS.
- T. Shimizu, K. Kanamori, A. Maeno, H. Kaji, C. M. Doherty, P. Falcaro and K. Nakanishi, Chem. Mater., 2016, 28, 6860–6868 CrossRef CAS.
- T. Matias, C. Varino, H. C. D. Sousa, M. E. M. Braga, A. Portugal, J. F. J. Coelho and L. Durães, J. Mater. Sci., 2016, 51, 6781–6792 CrossRef CAS.
- R. Al-Oweini and H. El-Rassy, Appl. Surf. Sci., 2010, 257, 276–281 CrossRef CAS.
- R. Al-Oweini and H. El-Rassy, J. Mol. Struct., 2009, 919, 140–145 CrossRef CAS.
- S. Li, L. Zhang, J. Li, Z. Wu and C. Yang, J. Sol-Gel Sci. Technol., 2017, 1–10 Search PubMed.
- A. V. Rao, G. M. Pajonk and D. Haranath, Met. Sci. J., 2013, 17, 343–348 Search PubMed.
- F. Zou, P. Yue, X. Zheng, D. Tang, W. Fu and Z. Li, J. Mater. Chem. A, 2016, 4, 10801–10805 CAS.
- S. K. Hong, M. Y. Yoon and H. J. Hwang, J. Am. Ceram. Soc., 2011, 94, 3198–3201 CrossRef CAS.
- D. Sanli and C. Erkey, ACS Appl. Mater. Interfaces, 2013, 5, 11708–11717 CAS.
- S. Zhao, W. J. Malfait, A. Demilecamps, Y. Zhang, S. Brunner, L. Huber, P. Tingaut, A. Rigacci, T. Budtova and M. M. Koebel, Angew. Chem., Int. Ed., 2015, 54, 14282–14286 CrossRef CAS PubMed.
- S. Yun, H. Luo and Y. Gao, J. Mater. Chem. A, 2015, 3, 3390–3398 CAS.
- J. He, X. Li, D. Su, H. Ji and X. Zhang, J. Mater. Chem. A, 2016, 4, 5632–5638 CAS.
- Y. Huang, L. Gong, Y. Pan, C. Li, T. Zhou and X. Cheng, RSC Adv., 2018, 8, 2350–2356 RSC.
- W. F. Huang, G. C. P. Tsui, C. Y. Tang and M. Yang, Composites, Part B, 2016, 95, 272–281 CrossRef CAS.
- Z. Li, X. Cheng, S. He, X. Shi and H. Yang, J. Sol-Gel Sci. Technol., 2015, 76, 138–149 CrossRef CAS.
- L. Wa, L. Fengyun, Z. Fanlu, C. Mengjing, C. Qiang, H. Jue, Z. Weijun and M. Mingwei, Appl. Surf. Sci., 2015, 353, 1031–1036 CrossRef CAS.
- L. Duffours, T. Woignier and J. Phalippou, J. Non-Cryst. Solids, 1995, 186, 321–327 CrossRef CAS.
- N. Leventis, C. Sotiriouleventis, A. Guohui Zhang and A. M. M. Rawashdeh, Nano Lett., 2002, 2, 957–960 CrossRef CAS.
- A. Parvathy Rao and A. V. Rao, J. Non-Cryst. Solids, 2008, 354, 10–18 CrossRef.
- H. Zheng, H. Shan, Y. Bai, X. Wang and L. Liu, RSC Adv., 2015, 5, 91813–91820 RSC.
- H. Yu and S. L. Brock, ACS Nano, 2008, 2, 1563–1570 CrossRef CAS PubMed.
- S. Chabi, V. G. Rocha, E. Garcia-Tunon, C. Ferraro and E. Saiz, ACS Nano, 2016, 10, 1871–1876 CrossRef CAS PubMed.
- S. W. Hwang, H. H. Jung, S. H. Hyun and Y. S. Ahn, J. Sol-Gel Sci. Technol., 2007, 41, 139–146 CrossRef CAS.
- C. E. Kim, J. S. Yoon and H. J. Hwang, J. Sol-Gel Sci. Technol., 2009, 49, 47 CrossRef CAS.
- H. Y. Erbil, A. L. Demirel, Y. Avci and O. Mert, Science, 2003, 299, 1377–1380 CrossRef CAS PubMed.
- J. L. Gurav, A. V. Rao, D. Y. Nadargi and H. H. Park, J. Mater. Sci., 2010, 45, 503–510 CrossRef CAS.
- Y. Pan, W. Wang, C. Peng, K. Shi, Y. Luo and X. Ji, RSC Adv., 2013, 4, 660–669 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13165h |
| This journal is © The Royal Society of Chemistry 2018 |