
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharged porous organic frameworks bearing heteroatoms with enhanced isosteric enthalpies of gas adsorption†
Guolong Xing,
Tingting Yan,
Saikat Das,
Ling Ye and
Kaiqi Ye *
*
State Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: yekq@jlu.edu.cn
First published on 5th June 2018
Abstract
Charged porous organic frameworks containing heteroatoms were synthesized via a Yamamoto-type Ullmann coupling reaction using potassium tetrakis(4-bromopyrazolyl)borate and tetrakis(4-chlorophenyl)phosphonium bromide as monomers. For the first time, a monomer containing boron atoms was successfully homocoupled to obtain a 3D charged porous organic framework. The heteroatoms and charges in the porous organic frameworks help to increase the interaction between the frameworks and the gases. Therefore the charged porous organic frameworks bearing heteroatoms synthesized in the present study exhibit high isosteric enthalpies of gas adsorption which surpass those of many other porous organic materials.
Introduction
Porous organic frameworks have lately attracted interest from scientists for applications such as gas separation, gas storage, catalysis and others because they have outstanding surface areas, high microporosities and remarkable physicochemical stabilities.1–6 Porous organic frameworks can be categorized into crystalline and amorphous frameworks. Crystalline porous organic frameworks such as covalent organic frameworks7–9 (COFs) retain structural order and well-defined porosities and incorporate building units linked by covalent bonds. Porous aromatic frameworks (PAFs) are porous networks with remarkably high surface areas and superb stabilities.10–13 Porous organic frameworks used for gas adsorption/storage and selective gas separation are routinely characterized by their isosteric enthalpies of gas adsorption since they provide an understanding of the affinity of the frameworks for gases. The rapid increase of carbon dioxide in the environment due to industrialization has necessitated its adsorption, while hydrogen and methane are stored due to their use as clean fuels. Porous organic frameworks are being extensively used for the adsorption and storage of the aforementioned gases so it is important to know their isosteric enthalpies of gas adsorption.As we know, a nitrogen atom in the framework is beneficial for CO2 adsorption and charges can make it easier for gas polarization to take place, therefore enhancing the interactions between the frameworks and gases.14–16 From considering this, integrating charges and heteroatoms into porous organic frameworks to synthesize charged porous organic frameworks bearing heteroatoms may help to increase the isosteric enthalpies of gas adsorption. Herein we introduce a series of charged porous organic frameworks, namely, B-POF, P-POF and BP-POF, bearing heteroatoms and synthesized via a Yamamoto-type Ullmann cross-coupling reaction from potassium tetrakis(4-bromopyrazolyl)borate and tetrakis(4-chlorophenyl)phosphonium bromide as monomers in different molar ratios. The heteroatoms and charges in these porous organic frameworks increase their isosteric enthalpies of gas adsorption. Thus, the charged porous organic frameworks show outstanding isosteric enthalpies for gas adsorption and outperform many other typical porous organic materials, such as PAF-1, COF-102, HCP-1 etc.
Experimental
Materials and methods
All of the starting materials were purchased from Aldrich and J&K. N,N-Dimethylformamide (DMF) and 1,5-cyclooctadiene (COD) were dried over CaH2 and distilled under reduced pressure prior to their use. The FT-IR experiments with the charged porous organic frameworks began with the mixing of the charged porous organic frameworks and KBr which was followed by the forming of pellets from the mixture upon the application of pressure. Then a Shimadzu IRAffinity-1 FT-IR spectrophotometer was used to obtain the FT-IR spectra. The FT-IR measurements under vacuum were performed in a Shimadzu IRTracer-100 Fourier transform infrared spectrophotometer with a vacuum system by pressing the mixture of the charged porous organic frameworks and KBr into the porous alumina crucibles. To carry out TGA of the samples, the samples were loaded in an alumina pan and inserted inside a Shimadzu DTG-60 thermal analyzer which was heated to 900 °C at a heating rate of 10 °C min−1 in a dry air atmosphere with an air flow rate of 30 mL min−1. A PANalytical B.V. Empyrean powder diffractometer using Cu-Kα radiation, 40 kV, 40 mA was employed to perform PXRD with the samples over the range 2θ = 4.0–40.0°. The SEM images were taken with a JEOL JSM 6700 scanning electron microscope on silicon wafer. The TEM images were obtained with a JEOL JEM-2100F transmission electron microscope. N2 sorption isotherm experiments were carried out at 77 K using a MicroMeritics Tristar II 3020 surface area and pore size analyzer. The samples were degassed at 150 °C overnight under ultra-high vacuum prior to the N2 (99.999% purity) sorption measurements. H2 (99.999% purity), CO2 (99.999% purity) and CH4 (99.99% purity) sorption isotherm measurements were carried out using the same analyzer that was used for N2 sorption isotherm measurements. To measure the free space, He of 99.999% purity was used and it was presumed that He was not adsorbed at any temperature in which the experiments were carried out. H2 isotherm measurements at 77 K were carried out in a liquid nitrogen bath, H2 isotherm measurements at 87 K were carried out in a liquid argon bath and H2 isotherms measurements at 195 K were carried out in an acetone/dry ice bath. CO2 and CH4 isotherm measurements at 273 K and 285 K were carried out in an ice-water bath and a 1,4-dioxane/dry ice bath, respectively.Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 3:1 and 0:1 mol/mol) were dissolved in N,N-dimethylformamide (DMF, 33 mL, 0.422 mol) to prepare the monomer mixture. After this, 1,5-cyclooctadiene (0.224 mL, 1.824 mmol) was added to a solution of bis(1,5-cyclooctadiene)nickel(0) (500.4 mg, 1.824 mmol) and 2,2′-bipyridyl (284.9 mg, 1.824 mmol) in anhydrous DMF (5 mL). The mixture was subsequently heated at 80 °C for 1 h. Next the monomer mixture solution was added to the resulting solution at 80 °C. The whole mixture was stirred at 80 °C for 3 d. Then the mixture was cooled down to room temperature following which an aqueous solution of HCl (pH = 3) was added to it. The solution was then stirred for 36 h (keep pH = 3), filtered and the residue washed with distilled water, ethanol and chloroform and dried under vacuum to obtain the charged porous organic frameworks, namely, B-POF, P-POF and BP-POF.
:0, 3:1 and 0:1 mol/mol) were dissolved in N,N-dimethylformamide (DMF, 33 mL, 0.422 mol) to prepare the monomer mixture. After this, 1,5-cyclooctadiene (0.224 mL, 1.824 mmol) was added to a solution of bis(1,5-cyclooctadiene)nickel(0) (500.4 mg, 1.824 mmol) and 2,2′-bipyridyl (284.9 mg, 1.824 mmol) in anhydrous DMF (5 mL). The mixture was subsequently heated at 80 °C for 1 h. Next the monomer mixture solution was added to the resulting solution at 80 °C. The whole mixture was stirred at 80 °C for 3 d. Then the mixture was cooled down to room temperature following which an aqueous solution of HCl (pH = 3) was added to it. The solution was then stirred for 36 h (keep pH = 3), filtered and the residue washed with distilled water, ethanol and chloroform and dried under vacuum to obtain the charged porous organic frameworks, namely, B-POF, P-POF and BP-POF.Results and discussion
Synthesis
The synthesis of the charged porous organic frameworks was carried out via a Yamamoto-type Ullmann coupling reaction using potassium tetrakis(4-bromopyrazolyl)borate and tetrakis(4-chlorophenyl)phosphonium bromide as monomers. Scheme 1 illustrates the synthesis technique adopted to obtain B-POF, P-POF and BP-POF.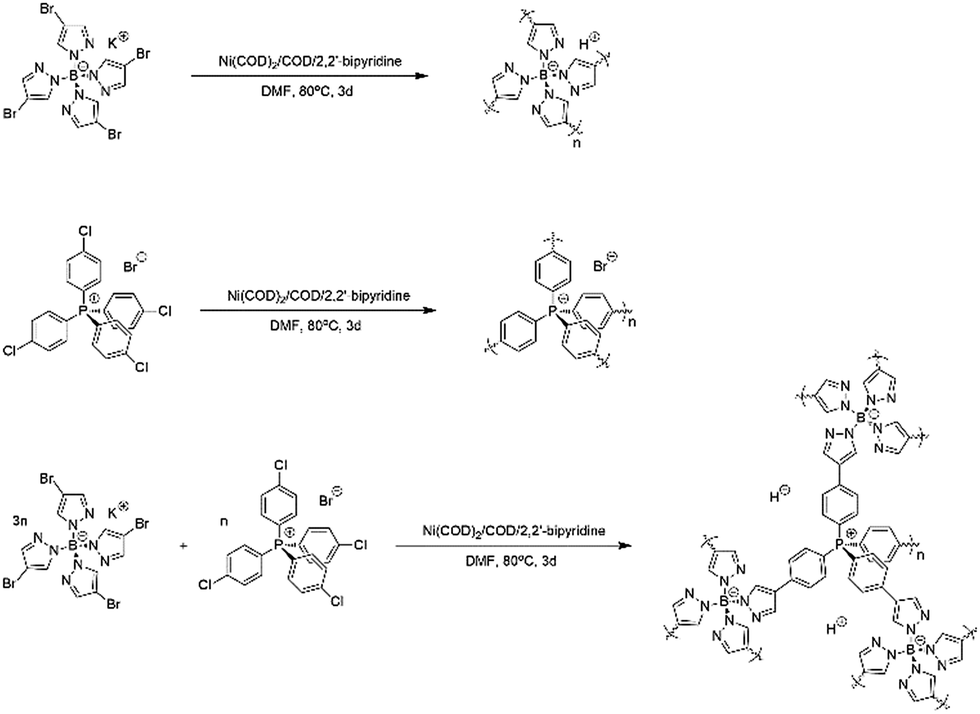 | ||
| Scheme 1 Schematic representation of the synthesis of the porous organic frameworks, namely, (top) B-POF, (middle) P-POF, and (bottom) BP-POF. | ||
Characterization
The structures of the charged porous frameworks were confirmed by their Fourier transform infrared (FT-IR) spectra, as shown in Fig. 1. The bands at about 611 and 760 cm−1 corresponding to the stretching vibrations of the C–Br and C–Cl bonds, respectively, decreased in the FTIR spectra of the porous organic frameworks, as compared to the FTIR spectra of the monomers. This substantiates the fact that the coupling reaction had occurred. Compared with the FT-IR spectra measured under vacuum (Fig S1–S3†), with the vacuum time increasing, the stretching bands appearing around ∼3406 cm−1 are less intense, which can be attributed to the presence of water molecules. The bands which appear around ∼3030 cm−1 in the FT-IR spectra of BP-POF, P-POF and the monomer tetrakis(4-chlorophenyl)phosphonium bromide can be assigned to the vibration of the C–H bond in the benzene ring, while the bands at about ∼3134 cm−1 and ∼3423 cm−1 which appear in the spectra of B-POF, BP-POF and the monomer potassium tetrakis(4-bromopyrazolyl)borate correspond to the C–H bond and the N–H bond in the pyrazole ring.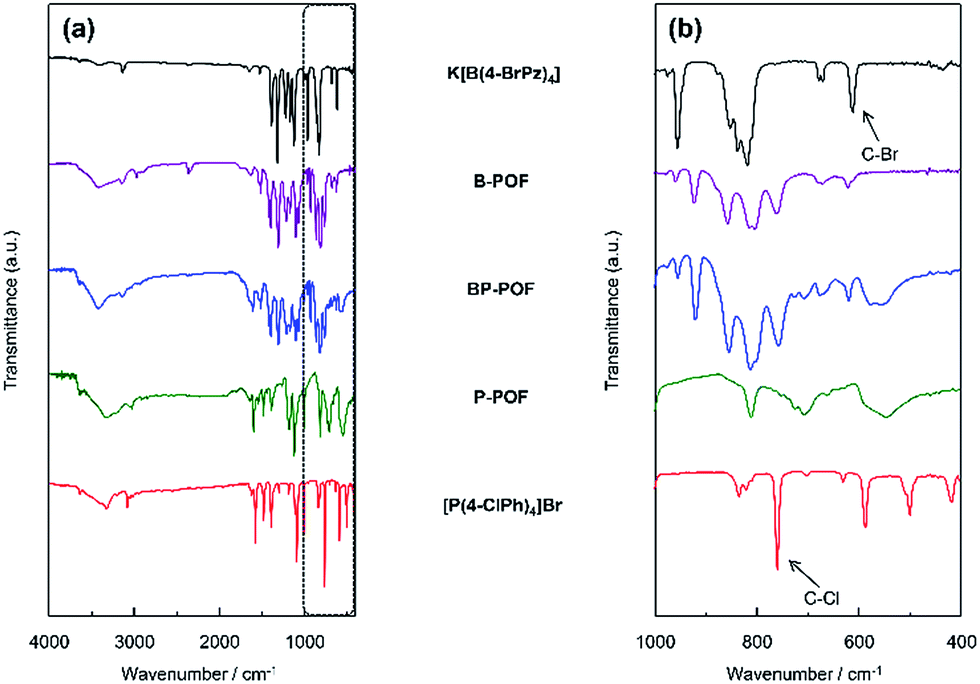 | ||
| Fig. 1 (a) FTIR spectra of K[B(4-BrPz)4], B-POF, BP-POF, P-POF and [P(4-ClPh)4]Br; (b) FT-IR spectra in detail from 400 to 1000 cm−1. | ||
To further investigate the structure, the charged porous organic frameworks were characterized using solid-state 11B and 31P NMR. As shown in Fig S4–S5,† the 31P NMR spectrum of BP-POF shows two signals at 23.2 ppm and 8.5 ppm while that of P-POF shows two signals at 22.8 ppm and 9.1 ppm, which can be attributed to the quaternary phosphonium and tertiary phosphine atoms, respectively. The tertiary phosphine atoms can be generated by the cleaving of the P–C bond during polymerization.18 However, unfortunately 11B NMR spectra can’t be obtained owing to the low content of boron in the frameworks.
The scanning electron microscopy (SEM) images of the porous organic frameworks are shown in Fig S6.† They show that the porous frameworks do not have a well-defined shape. The transmission electron microscopy (TEM) images of the porous organic frameworks are shown in Fig. 2. They reveal the amorphous nature of the porous frameworks.
 | ||
| Fig. 2 TEM images of (a) B-POF, (b) BP-POF and (c) P-POF. | ||
In order to explore the short-range and long-range orders of the samples, powder X-ray diffraction (PXRD) patterns of the samples were obtained. As shown in Fig. S7,† the PXRD patterns of the porous frameworks corroborate the amorphous nature (and thereby the lack of long-range order) of the materials. This is in accordance with the results from TEM. However, B-POF, for which a broad diffraction peak at 10.7° was revealed, likely has some short-range order.
The thermal and chemical stabilities of the porous organic frameworks were also investigated. Fig S8† shows the thermogravimetric analysis (TGA) plots of the samples. Of the materials, P-POF showed the highest stability with a 5% weight loss at 460 °C followed by B-POF and BP-POF with weight losses of 5% at 312 °C and 269 °C in an air atmosphere, respectively. Furthermore, the samples also exhibited high chemical stabilities, and were insoluble in water and common organic solvents such as hexanes, chloroform, ethanol and acetone.
Gas adsorption
The specific surface areas and porosities of the samples were explored using nitrogen sorption measurements at 77 K. As shown in Fig. 3a, in the low pressure region (p/p0 < 0.1), the isotherms show steep adsorption which confirms the presence of micropores. The three isotherms show a hysteresis loop, which means that mesopores are present in the frameworks. However, it can also be observed that the hysteresis loop extends to a low relative pressure. This may be attributed to swelling in the porous organic framework.18 The Brunauer–Emmett–Teller (BET) surface areas of the porous frameworks were calculated from the sorption isotherms to be 515 m2 g−1, 629 m2 g−1 and 223 m2 g−1 for B-POF, P-POF, and BP-POF, respectively (Table 1). Fig. 3b shows the pore size distribution of the samples calculated by the density functional theory (DFT) method. It shows dominant pore diameters centered at about 1.18 nm, 1.18 nm and 0.93 nm corresponding to B-POF, BP-POF, and P-POF, respectively. The total pore volumes for B-POF, BP-POF, and P-POF are 0.13 cm3 g−1, 0.08 cm3 g−1 and 0.18 cm3 g−1. BP-POF shows a low surface area value and a low pore volume which may be attributed to a low level of polymerization caused by the different reactivity of Br and Cl.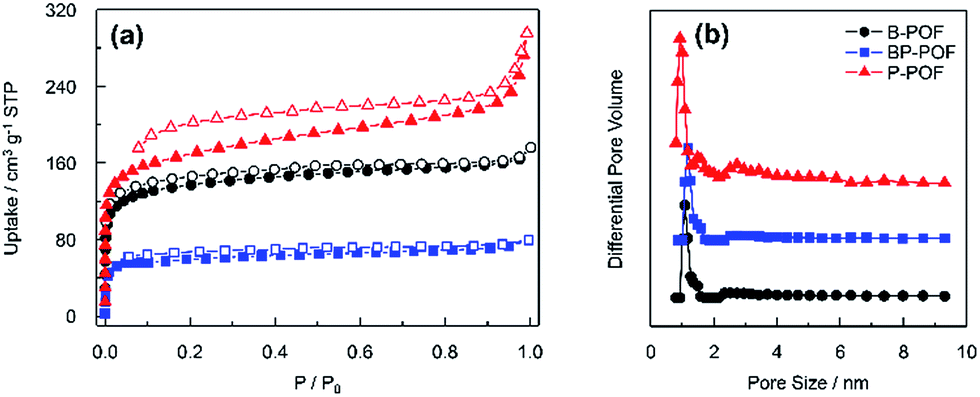 | ||
| Fig. 3 (a) N2 sorption isotherms and (b) corresponding pore size distribution of B-POF (circles), BP-POF (squares), and P-POF (triangles) at 77 K (solid symbols, adsorption; open symbols, desorption). | ||
| POFs | SBETa (m2 g−1) | Pore size (nm) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | Vtotal (cm3 g−1) | QstH2 (kJ mol−1) | QstCO2 (kJ mol−1) | QstCH4 (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|
| a The BET surface areas were determined in the partial pressure (p/p0) range 0.01–0.1. | ||||||||
| B-POF | 515 | 1.18 | 0.13 | 0.03 | 0.16 | 8.5 | 32.3 | 22.4 |
| BP-POF | 629 | 1.18 | 0.08 | 0.02 | 0.10 | 7.9 | 27.7 | 21.5 |
| P-POF | 223 | 0.93, 1.48 and 2.73 | 0.18 | 0.08 | 0.26 | 7.8 | 35.2 | 21.4 |
H2, CO2 and CH4 are apolar molecules with kinetic diameters 2.90, 3.30, and 3.80 Å, respectively. In addition, the order of polarizability is CO2 > CH4 > H2, which means that CO2 is easier to polarize than the other two. In order to investigate the affinities of the samples, adsorption isotherms were collected, and isosteric enthalpies of gas adsorption (Qst) were calculated using the Clausius–Clapeyron equation from the adsorption isotherms at different temperatures.
The H2 isotherms at 77 K and 1 bar exhibit maximum uptakes of 103.3 cm3 g−1, 102.8 cm3 g−1, and 100.0 cm3 g−1 for B-POF, P-POF, and BP-POF, respectively (Fig. 4a). The Qst values for H2 adsorption for B-POF, P-POF, and BP-POF were found to be 8.5, 7.8, and 7.9 kJ mol−1 (Fig. 4b). These values are higher than those for numerous porous polymers. PAF-1 with a high BET surface area of up to 5600 m2 g−1 exhibited a Qst value of 4.6 kJ mol−1 for H2.12 COF-102, with a BET surface area as high as 3620 m2 g−1, shows a Qst value of 3.9 kJ mol−1 for H2.19 JUC-Z7-10, which also has a high BET surface area, exhibited a Qst value in the range 5.6–6.9 kJ mol−1.20 GNF-1 with a similar BET surface area (679 m2 g−1) gave a Qst value of 7.7 kJ mol−1.21 Although H2 is not easy to polarize, the charged boron atoms and phosphorus atoms in the frameworks still enhance the polarization effects of the frameworks for gases, and therefore help to increase the interactions between the frameworks and H2, which thus exhibit high isosteric enthalpies.
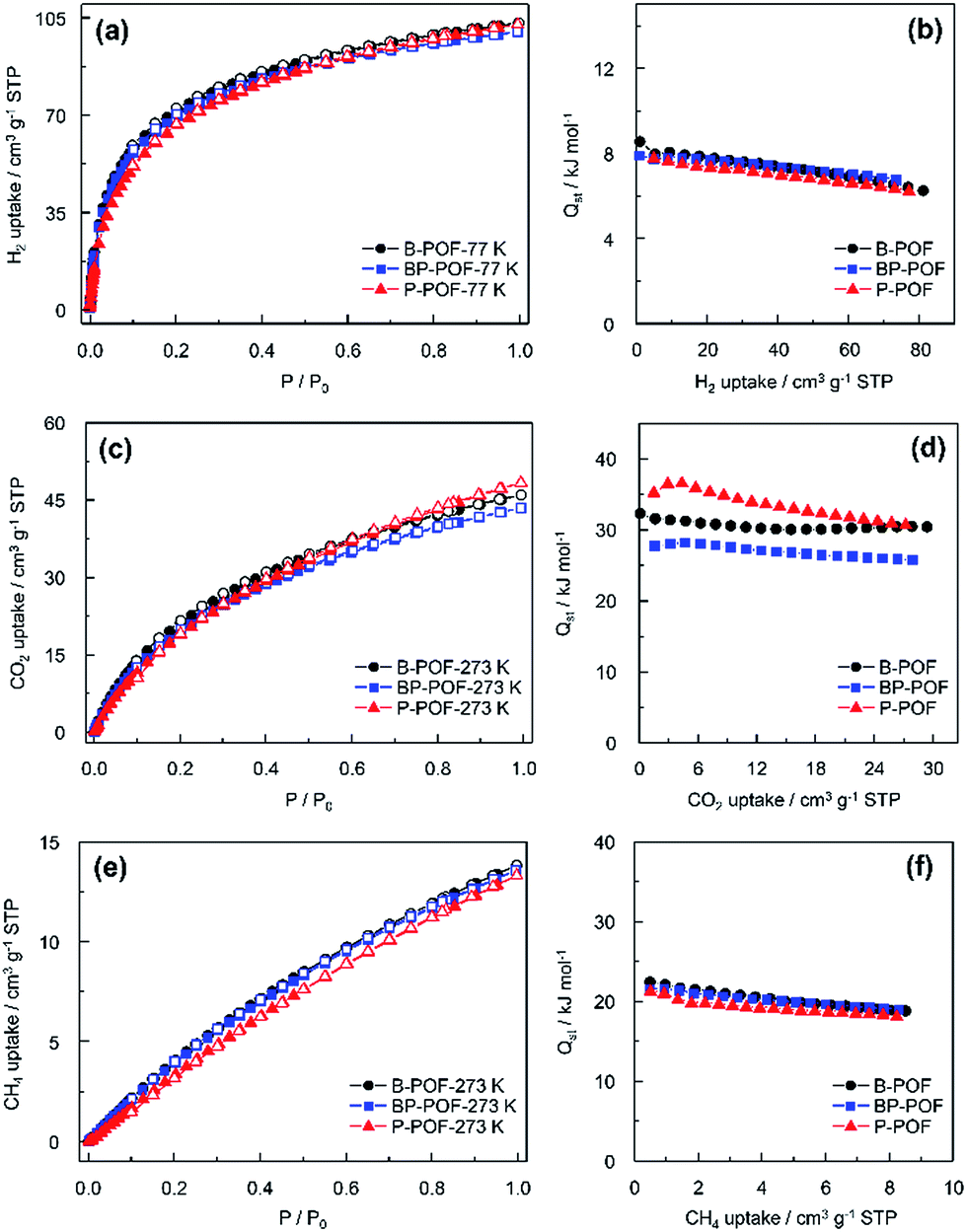 | ||
| Fig. 4 (a) H2 sorption isotherms and (b) QstH2 of B-POF (circles), BP-POF (squares), and P-POF (triangles) at 77 K; (c) CO2 sorption isotherms and (d) QstCO2 of B-POF (circles), BP-POF (squares), and P-POF (triangles) at 273 K; (e) CH4 sorption isotherms and (f) QstCH4 of B-POF (circles), BP-POF (squares), and P-POF (triangles) at 273 K (solid symbols, adsorption; open symbols, desorption). | ||
The CO2 isotherms of B-POF, P-POF, and BP-POF are shown in Fig. 4c. At 273 K, the CO2 uptake capacities of the porous organic frameworks are 46.0 cm3 g−1, 48.4 cm3 g−1 and 43.5 cm3 g−1 at 1 bar. The Qst values for CO2 were calculated from the adsorption data collected at 273 K, 285 K and 295 K. The three POFs exhibit high Qst values, especially B-POF and P-POF. These values are comparable to, if not greater than, the values reported for most porous polymers including HCP-1(∼23.5 kJ mol−1),22 CTF-1(∼27.5 kJ mol−1),23 BILP-1(26.5 kJ mol−1),24 PMOP-1(18.5 kJ mol−1),25 PCTF 1–7(25–30 kJ mol−1),26 N4CMP 1–4(25.5–35.1 kJ mol−1),27 polycarbazole CPOP-1(24.5–30.2 kJ mol−1)28 and P-THIDT(29 kJ mol−1).29 The high value for Qst for CO2 adsorption could perhaps be attributed to the presence of charges and nitrogen atoms in the POF. The coexistence of positive and negative charges in BP-POF are not beneficial for polarizing CO2 molecules, therefore BP-POF exhibits a relatively lower isosteric enthalpy than B-POF and P-POF.
As a promising energy source in the future, methane is highly popular due to its high ratio of hydrogen to carbon. Therefore, the CH4 sorption behavior of B-POF, P-POF, and BP-POF were also investigated. As shown in Fig. 4e, the CH4 isotherms are reversible and show maximum uptakes of 13.8 cm3 g−1, 13.3 cm3 g−1, and 13.6 cm3 g−1, respectively. The Qst of CH4 for B-POF, P-POF, and BP-POF were also calculated and the values are 22.4, 21.4 and 21.5 kJ mol−1. These Qst values are similar and surpass those of PAF-1(14.0 kJ mol−1),12 PMOP-1(∼11 kJ mol−1),25 JUC-Z7-Z10(15.7–16.0 kJ mol−1)20 and BILP-3 (16.6 kJ mol−1),30 and are similar to pre-GNF(22.7 kJ mol−1),21 P-DTBDT (25 kJ mol−1)29 and CTF-DCBT(22.8 kJ mol−1).31 These high Qst values can also be attributed to the charged boron atoms or phosphorus atoms existing in the frameworks which favor CH4 polarization. This therefore increases the interactions between the frameworks and CH4 molecules.
Isosteric enthalpies of gas adsorption (Qst) are useful values that characterize porous organic frameworks in their affinities for gases such as CO2, H2, CH4 etc. The porous organic frameworks synthesized in the present study possibly owe their outstanding isosteric enthalpies of gas adsorption to the heteroatoms and charges in the porous organic frameworks. The nitrogen atoms in the frameworks favor CO2 adsorption, and charges in the frameworks make the gas molecules much easier to polarize, which helps to increase the interactions between the frameworks and the gases. Thus, the POFs may have another potential application in adsorbing other gas molecules which are easier to polarize.
Conclusions
To summarize, porous organic frameworks which comprised heteroatoms were synthesized by a Yamamoto-type Ullmann coupling reaction using potassium tetrakis(4-bromopyrazolyl)borate and tetrakis(4-chlorophenyl)phosphonium bromide as monomers. These porous organic frameworks were found to exhibit high isosteric enthalpies of gas adsorption which may be attributed to the presence of heteroatoms and charges in the porous frameworks. Therefore, the charged porous organic frameworks bearing heteroatoms may effectively store the gases in ambient environments owing to their high affinities for the gases and release the gases when needed due to the reversible physical adsorption process. This study may motivate an exploration of the potential for more charged porous organic frameworks bearing heteroatoms for applications in gas sorption/storage and separation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2016YFB0401001).Notes and references
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef PubMed.
- T. Ben and S. Qiu, CrystEngComm, 2013, 15, 17–26 RSC.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O’Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef PubMed.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef PubMed.
- T. Ben, C. Pei, D. Zhang, J. Xu, F. Deng, X. Jing and S. Qiu, Energy Environ. Sci., 2011, 4, 3991–3999 Search PubMed.
- T. Ben, Y. Li, L. Zhu, D. Zhang, D. Cao, Z. Xiang, X. Yao and S. Qiu, Energy Environ. Sci., 2012, 5, 8370–8376 Search PubMed.
- C. Pei, T. Ben and S. Qiu, Mater. Horiz., 2015, 2, 11–21 RSC.
- S. Fischer, A. Schimanowitz, R. Dawson, I. Senkovska, S. Kaskel and A. Thomas, J. Mater. Chem. A, 2014, 2, 11825–11829 Search PubMed.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 Search PubMed.
- (a) M. Zhang, Z. Perry, J. Park and H.-C. Zhou, Polymer, 2014, 55, 335–339 CrossRef; (b) W. Lu, J. P. Sculley, D. Yuan, R. Krishna and H.-C. Zhou, J. Phys. Chem. C, 2013, 117, 4057–4061 CrossRef.
- M. Onishi, Y. Arikawa, M. Yamaguchi, T. Nagano, T. Inoue, A. Terasoba, S. Matsuo, M. Nakagawa, H. Kawano, J. Nagaoka, K. Umakoshi and M. Furukawa, J. Organomet. Chem., 2012, 700, 135–141 CrossRef.
- Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981–2988 CrossRef.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef PubMed.
- C. Pei, T. Ben, Y. Li and S. Qiu, Chem. Commun., 2014, 50, 6134–6136 RSC.
- Y. Byun and A. Coskun, Chem. Mater., 2015, 27, 2576–2583 CrossRef.
- C. F. Martin, E. Stöckel, R. Clowes, D. J. Adams, A. I. Cooper, J. J. Pis, F. Rubiera and C. Pevida, J. Mater. Chem., 2011, 21, 5475–5483 RSC.
- Y. Zhao, K. X. Yao, B. Teng, T. Zhang and Y. A. Han, Energy Environ. Sci., 2013, 6, 3684–3692 Search PubMed.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2011, 23, 1650–1653 CrossRef.
- J. Liu, Y. Liu, X. Jiang, Y. Luo and Y. Lyu, Microporous Mesoporous Mater., 2017, 250, 203–209 CrossRef.
- A. Bhunia, I. Boldog, A. Möller and C. Janiak, J. Mater. Chem. A, 2013, 1, 14990 Search PubMed.
- G. Li, L. Qin, C. Yao and Y. Xu, Sci. Rep., 2017, 7, 15394 CrossRef PubMed.
- Q. Chen, M. Luo, P. Hammershoj, D. Zhou, Y. Han, B. W. Laursen, C. G. Yan and B. H. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef PubMed.
- H. Liu, Q. Li, Q. Li, W. Jin, X. Li, A. Hameed and S. Qiao, Poly. Chem., 2017, 8, 6733–6740 RSC.
- M. G. Rabbani, T. E. Reich, R. M. Kassab, K. T. Jackson and H. M. El-Kaderi, Chem. Commun., 2012, 48, 1141–1143 RSC.
- K. Wang, Y. Tang, Q. Jiang, Y. Lan, H. Huang, D. Liu and C. Zhong, J. Energy Chem., 2017, 26, 902–908 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12860f |
| This journal is © The Royal Society of Chemistry 2018 |