
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe behavior of the aluminum trimer when combining with different superatom clusters†
Hui Yangab,
Di Wu a,
Hui-Min Hea,
Dan Yua,
Ying Li*a and
Zhi-Ru Lia
a,
Hui-Min Hea,
Dan Yua,
Ying Li*a and
Zhi-Ru Lia
aLaboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130023, P. R. China. E-mail: liyingedu@jlu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shanxi Datong University, Datong 037009, P. R. China
First published on 12th February 2018
Abstract
The interaction between the aluminum trimer and representative (super)halogens X (X = F, LiF2, BeF3, BF4) and (super)alkalis M (M = Li, FLi2, OLi3, NLi4) has been theoretically investigated at the MP2/6-311+(3df) level. Various geometrical structures were obtained for the resulting Al3–X and Al3–M superatom compounds, respectively. Natural bond orbital analysis reveals that the Al3 moiety exists in a cationic state in Al3–X while in an anionic state in Al3–M compounds. And the charge transfer between Al3 and (super)atoms is found to be enhanced in either polar or nonpolar solvent. The studied superatom compounds feature large bond energies, binding energies, and HOMO–LUMO gaps, which not only reflect their stability but indicate strong interactions between Al3 and (super)atoms. Although the solvent effect is not significant for the stability of Al3–X, the Al3–superalkali compounds can be better stabilized in the presence of solvent molecules. In addition, these superatom compounds exhibit aromaticity both in the gas phase and in solution.
1. Introduction
Clusters are extensively studied in physics because they represent the transition states between single atoms and bulk solid.1–8 On the one hand, clusters possess properties that are neither atomic-like nor solid-like but depend on their composition, size, geometry, charge state, etc. On the other hand, stable clusters can serve as basic building blocks in chemistry.9 Hence, the research of clusters is also of significance in developing novel cluster-assembled materials with tunable properties.One of the most exciting developments in the research area of clusters is the realization that specific clusters exhibit similar chemical behavior to atoms in the periodic table. Such clusters are consequently termed superatoms.10–13 Two well-known subsets of superatoms are superhalogens14–17 and superalkalis,18–21 which have been extensively studied for more than 30 years. Superhalogens have higher electron affinities (EAs) than atomic EA limit (Cl: 3.617 eV)22 while superalkalis are unique clusters possessing ionization potentials (IPs) lower than those of alkali atoms (5.39–3.89 eV).23 Lately, the idea of combining superalkali with superhalogen clusters has been theoretically proposed and the generated superatom compounds include Al13K3O and Al13Na3O,24 BF4–M (M = Li, FLi2, OLi3, NLi4),25 BLi6–X (X = F, LiF2, BeF3, BF4),26 and Li3O–X (X = BF4, BeF3, NO3),27 etc. It has been found that both superhalogens and superalkalis play the role of building block in the resulting ionic compounds that are named as “supersalts” by Jena et al.27 These inspiring results motivate us to think about the following questions: can superatoms combine with other clusters, especially metal clusters? If so, what are the preferred structures as well as bonding nature of such superatom compounds? Will the structural and electronic integrity of the metal cluster break when it interacts with superatoms? How does the metal cluster behave when combining with superalkalis and superhalogens, respectively?
During the last two decades, aluminum clusters have become a rich area of research in cluster physics and chemistry. In addition to providing a basic understanding of size-dependent physical and chemical properties of simple metal clusters, researches also bring out some special characteristics of aluminum clusters. These include the potentially multivalent character of the bonding in aluminum clusters, the free electron character of aluminum which makes aluminum clusters an archetypal example of the shell model, all-metal aromaticity found in small Al-based clusters, for example, Al3−, Al42−, and Al62−, etc.28–34 Besides, some pure or doped aluminum clusters, such as Al13,35 Al14,36 Al7−,37 Al12Be,38 Al12Cu,39 have been proven to show superatom features. Furthermore, small aluminum clusters share some properties in common with the more electronically complex transition metal clusters. Thus, the studies of p-block aluminum clusters are good complements to those of the less computationally tractable d-block metal clusters.40
As one of the smallest and thus most foundational components of aluminum clusters, aluminum trimer has been extensively studied and its electronic and geometrical structures are well understood.30,41–44 Hence, it has been chosen in our work as a representative metal cluster to interact with differently shaped (super)halogens X (X = F, LiF2, BeF3, BF4) and (super)alkalis M (M = Li, FLi2, OLi3, NLi4). The main objectives of this contribution are (1) to reveal different behaviors of Al3 when combining with different (super)atoms, (2) to examine stability of the resulting Al3–X and Al3–M compounds both in gas phase and in solution. Besides, aromaticity of these superatom compounds is analyzed as well. We hope that the results we provide in this work can further enrich our knowledge on superatoms and the principles obtained may work well for a variety of superatom compounds involving metal cluster building blocks, especially the Aln group.
2. Computational details
The minima on the potential-energy surfaces of the Al3–X (X = F, LiF2, BeF3, BF4) and Al3–M (M = Li, FLi2, OLi3, NLi4) compounds were explored by using two approaches. The first one is to construct initial geometries artificially by considering all the possible bonding orientations between Al3 cluster and (super)atoms X/M. The second one employs a random search procedure,38,45–47 in which structures were generated by randomly distributing all atoms inside a sphere with radius R = 5.0 Å. The resulting geometries were optimized at the B3LYP/3-21G level automatically. Then, all the geometries obtained by the first method and the minimum structures from the second method were optimized using the second order Møller–Plesset (MP2) method48 with the 6-311+G(3df) basis set, followed by vibrational frequency calculations. Note that only those minimum structures where the Al3 and superatom subunits retain their respective integrity are discussed in the present work since the interaction between Al3 and superatom clusters is the focus of our attention. Natural bond orbital (NBO)49 and atom in molecules (AIM)50,51 analyses were performed at the same level. The nucleus-independent chemical shifts (NICS)52 values were obtained employing the GIAO-B3LYP/6-311+G(3df) method.53The intramolecular interaction energies (Eint) between Al3 and X/M subunits and binding energy per atom (Ea) for these Al3–X and Al3–M species were calculated at the higher CCSD(T)//MP2/6-311+G(3df) level based on the MP2 geometries.25,38 We used the counterpoise (CP) procedure54 to eliminate the basis set superposition error (BSSE) effect given by eqn (1):55
| Eint = EAB(XAB) − EA(XAB) − EB(XAB) | (1) |
All calculations were performed using the GAUSSIAN 09 program package.56 The plots of molecular configurations and orbitals were generated by the GaussView program.57
3. Results and discussion
3.1. Geometrical structures
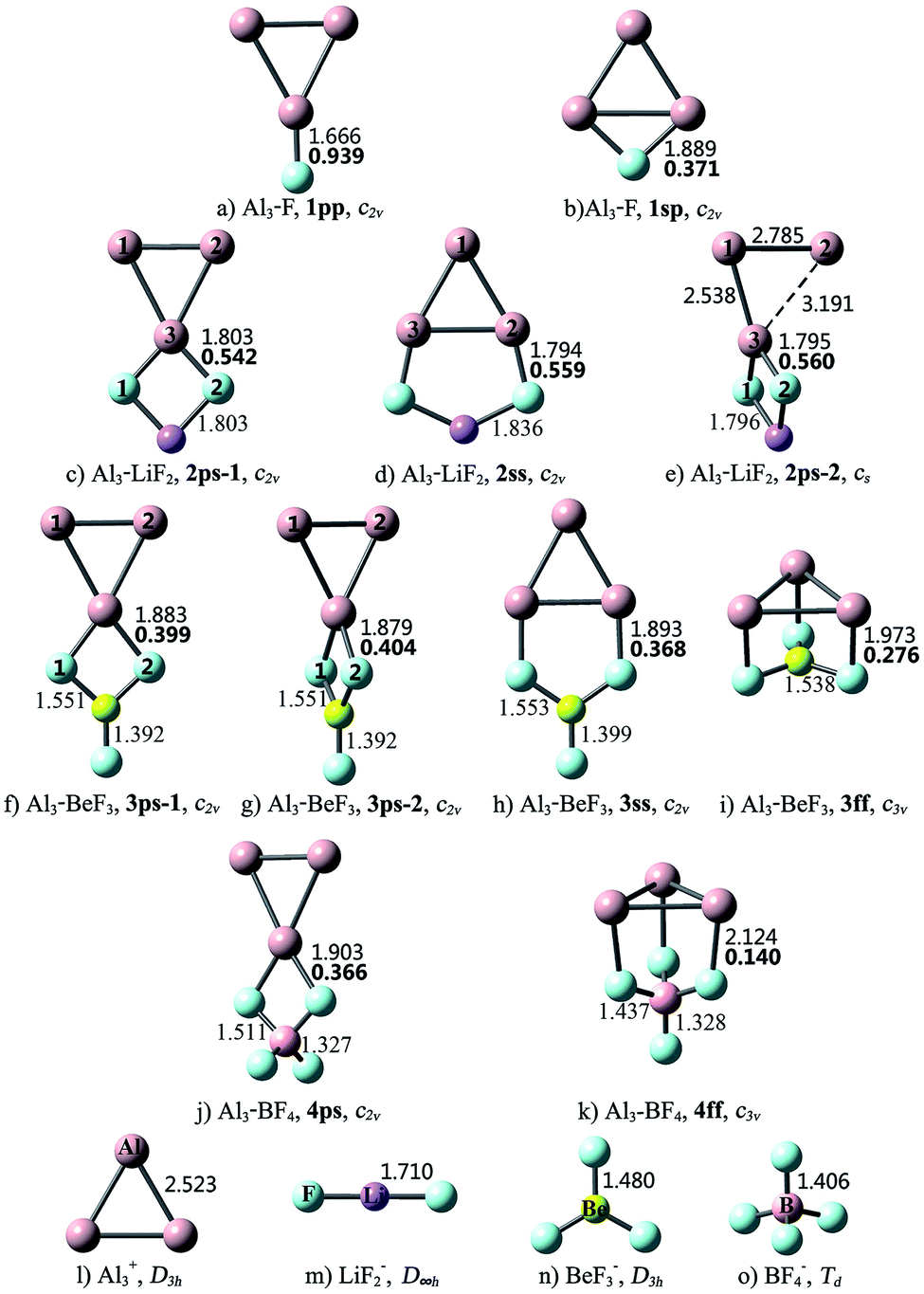 | ||
| Fig. 1 Optimized structures of the Al3–X compounds and Al3+, LiF2−, BeF3−, BF4− ions at the MP2/6-311+G(3df) level, bond lengths (Å) and Laplacian of the electron density at a bond critical point ∇2ρ(r) (in au., bold font) for the Al–F bonds that connect Al3 and X subunits. | ||
| Species | Orientation | Erel | ν1 | QAl3 | Gap | Ea | Eb | NICSmax | |
|---|---|---|---|---|---|---|---|---|---|
| Al3–F | 1pp | Point-to-point | 0.00 | 126 | 0.777 | 5.55 | 55.54 | 137.1 | −28.5 |
| 1sp | Side-to-point | 23.03 | 174 | 0.818 | 4.80 | 51.17 | 120.1 | −23.9 | |
| Al3–LiF2 | 2ps-1 | Point-to-side | 0.00 | 49 | 0.685 | 4.97 | 68.55 | 178.8 | −27.5 |
| 2ss | Side-to-side | 64.32 | 54 | 0.699 | 5.37 | 68.32 | 190.5 | −37.6 | |
| 2ps-2 | Point-to-side | 84.34 | 29 | 0.685 | 4.53 | 66.05 | 174.7 | — | |
| Al3–BeF3 | 3ps-1 | Point-to-side | 0.00 | 50 | 0.748 | 5.66 | 79.47 | 168.2 | −30.6 |
| 3ps-2 | Point-to-side | 2.61 | 39 | 0.756 | 5.40 | 78.99 | 164.6 | −24.4 | |
| 3ss | Side-to-side | 9.07 | 41 | 0.768 | 5.47 | 78.45 | 169.4 | −32.0 | |
| 3ff | Face-to-face | 12.51 | 134 | 1.375 | 6.34 | 78.31 | 180.8 | −13.9 | |
| Al3–BF4 | 4ps | Point-to-side | 0.00 | 38 | 0.757 | 5.68 | 86.87 | 166.4 | −30.8 |
| 4ff | Face-to-face | 19.99 | 89 | 0.824 | 5.64 | 85.38 | 166.7 | −12.5 | |
Different from linear diatomic molecules, the Al3–X compounds have a variety of structures (see Fig. 1). The eleven Al3–X structures can be classified into five types according to the relative orientation (bonding pattern) between Al3 and X, namely, point-to-point (pp), point-to-side (ps), side-to-point (sp), side-to-side (ss), and face-to-face (ff). Thereby the nomenclature employed for an Al3–X isomer designates the number of F atoms in Arabic numerals, followed by the bonding pattern. For example, 3ss represents an Al3–BeF3 structure with side-to-side bonding pattern.
As shown in Fig. 1, the structural integrity of superhalogens X is maintained in all the Al3–X compounds. For Al3–F, the F atom is either bound to an apex Al atom (1pp), or side-on bound to the Al3 triangle (1sp). From Table 1, the former is 23.03 kcal mol−1 more stable than the latter. There are two kinds of interaction orientations between Al3 and LiF2, namely, point-to-side (2ps-1 and 2ps-2) and side-to-side (2ss). From Fig. 1, the relative position between Al3 and LiF2 units in isomer 2ps-1 is different from that in 2ps-2. To be specific, line Al1Al2 is parallel to line F1F2 in 2ps-1, but is perpendicular to line F1F2 in 2ps-2. The Al3–Li distances are 2.629, 2.850 and 2.610 Å for the 2ps-1, 2ss and 2ps-2 structures, respectively. Note that these lengths are close to those of Al3–Li (2.854 and 2.653 Å for Ifp and Ipp, respectively), so there might also be Al–Li connections between Al3 and LiF2 units. The stability sequence is 2ps-1 > 2ss > 2ps-2 for the three Al3–LiF2 structures in accordance to the total energy order. Four isomers were found for the Al3–BeF3 compound. From Table 1, the point-to-side orientation (3ps-1, 3ps-2) is superior to the side-to-side orientation (3ss), and the least favorable structure is 3ff with the face-to-face orientation. Herein, the bonding pattern in 3ps-1 is similar to that in 2ps-1. It is worth to mention that, though 3ps-2 exhibits a similar bonding pattern to that for 2ps-2, the former has a higher symmetry (C2v) than the latter (Cs). The Al–Be distance of 2.460 Å for 3ff is close to that for the pyramidal Al3Be cluster (2.370 Å),58 hence structure 3ff can also be regarded as three F atoms side-on attached to an Al3Be unit. As to Al3–BF4, two structures were obtained with point-to-side (4ps) and face-to-face (4ff) bonding patterns, respectively. Isomer 4ff is 19.99 kcal mol−1 less stable than isomer 4ps.
According to the above results, when Al3 interacts with superhalogens, the preferred sequence of interaction site is apex Al atom > Al–Al side > Al3 ring plane, for the Al3 cluster. As to superhalogens, the F–F side is superior to the plane consisting of three F atoms. Therefore, the most beneficial bonding pattern for the Al3–X systems is point-to-side, while the least favorable one is face-to-face. The only exception is that 2ps-2 is 20.02 kcal mol−1 less stable than isomer 2ss, which may be attributed to the evidently distorted Al3 triangle in the former. In contrast, the Al3 ring is almost intact in the four structural isomers of Al3–BeF3, hence the 3ps-1 and 3ps-2 isomers with point-to-side orientation possess lower total energy than the others (3ss, 3ff).
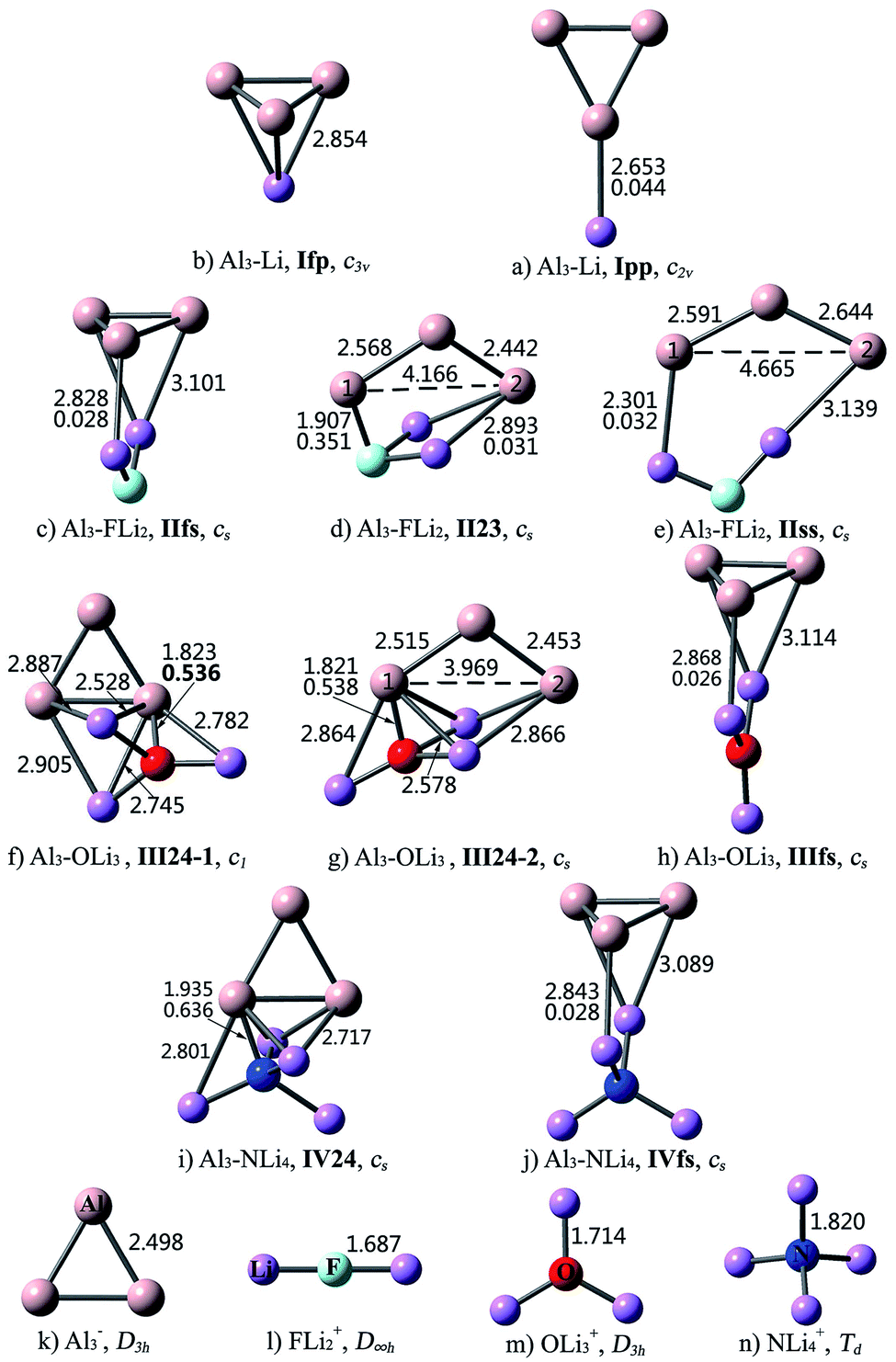 | ||
| Fig. 2 Optimized structures of the Al3–M compounds and Al3−, FLi2+, OLi3+, NLi4+ ions at the MP2/6-311+G(3df) level, bond lengths (Å) and Laplacian of the electron density at a bond critical point ∇2ρ(r) (in au., bold font) for the bonds that connect Al3 and M subunits. | ||
| Species | Orientation | Erel | ν1 | QAl3 | Gap | Ea | Eb | NICSmax | |
|---|---|---|---|---|---|---|---|---|---|
| Al3–Li | Ifp | Face-to-point | 0.00 | 180 | −0.506 | 5.55 | 33.17 | 48.6 | −39.0 |
| Ipp | Point-to-point | 11.99 | 80 | −0.675 | 4.93 | 30.00 | 37.2 | −29.6 | |
| Al3–FLi2 | IIfs | Face-to-side | 0.00 | 63 | −0.519 | 5.62 | 52.55 | 61.7 | −35.5 |
| II23 | Side-to-face | 16.28 | 119 | −0.295 | 5.36 | 49.75 | 75.7 | −17.9 | |
| IIss | Side-to-side | 35.64 | 43 | −0.422 | 4.95 | 47.84 | 58.0 | — | |
| Al3–OLi3 | III24-1 | Side-to-face | 0.00 | 60 | −0.266 | 4.08 | 60.71 | 92.4 | −34.8 |
| III24-2 | Side-to-face | 6.84 | 58 | −0.314 | 4.21 | 59.72 | 101.0 | −14.4 | |
| IIIfs | Face-to-side | 19.50 | 49 | −0.454 | 4.91 | 58.21 | 65.5 | −36.1 | |
| Al3–NLi4 | IV24 | Side-to-face | 0.00 | 24 | −0.361 | 4.11 | 54.08 | 94.7 | −26.1 |
| IVfs | Face-to-side | 31.05 | 25 | −0.350 | 4.90 | 50.58 | 55.0 | −34.7 | |
As can be seen from Fig. 2, the interaction between Al3 and superalkalis M is a bit complex. In some structures, the Al3 and M subunits are connected via Al–Li bonds, while in the other structures, the central nonmetal atom of superalkali M also takes part in the intramolecular interaction and directly binds to the Al3 unit. Accordingly, the nomenclature employed for the former kind of Al3–M structures designates the number of Li atoms in Roman numerals, followed by the bonding pattern. Differently, for the latter kind of structures, the Roman numerals are followed by the number of atoms participating in the intramolecular interaction, from Al3 and M, respectively. For example, IIfs represents an Al3–FLi2 structure with face-to-side bonding pattern, while II23 means that the interaction between Al3 and FLi2 involves two Al atoms, two Li atoms and the nonmetal F atom.
For Al3–Li, the Li atom may cap the Al3 triangle (Ifp) or bind with the apex Al atom (Ipp). Isomer Ifp with the face-to-point bonding pattern is more stable. There are three types of interactions between Al3 and FLi2. Herein, isomer IIfs with face-to-side bonding pattern is the lowest-energy structure, and isomer IIss with side-to-side bonding orientation is the least favorable one. As for isomer II23, the Al3 and FLi2 moieties are linked together by two Al–Li bonds and an Al–F bond. Three structures were identified for the Al3–OLi3 compound. Superalkali OLi3 is bound to Al3 by three Al–Li bonds in isomer IIIfs, where the Al3 and OLi3 planes are perpendicular to each other. In isomers III24-1 and III24-2, all the four atoms of OLi3 directly interact with the Al3 unit. From Table 2, the stability order is III24-1 > III24-2 > IIIfs. As to Al3–NLi4, two isomers were found and isomer IV24 is 31.05 kcal mol−1 more stable than IVfs. From Fig. 2, the bonding patterns in structures IV24 and IVfs are similar to those in structures III24-1 and IIIfs, respectively.
As shown in Fig. 2, intercluster fusion occurs when Al3 interacts with superalkali M, which leads to broken Al3 ring in the II23, IIss and III24-2 structures. Nevertheless, the structural integrity of the Al3 cluster and superalkali M are retained in the lowest-energy structure of each Al3–M compound.
The structural features of the Al3–M compounds indicate that Al3 does not interact with superalkali M through the apex Al atom as it does in superhalogen compounds. From Fig. 2, Al3 prefers to bind with M through the ring plane in the Al3–Li and Al3–FLi2 compounds, while in the other two species, it prefers to interact with M through the Al–Al edge. The isomer with more bonds between Al3 and M generally exhibits relatively higher stability. Take Al3–OLi3 as an example. The III24 isomer involving five Al–Li bonds and an Al–O bond is more stable than IIIfs with three Al–Li bonds. For two isomers with the same bonding mode, the one containing intact Al3 ring is more favorable. This is why III24-1 is 6.84 kcal mol−1 more stable than III24-2.
3.2. Stability and bonding nature
The HOMO–LUMO energy gap is considered to be an important index of electronic stability and chemical inertness of clusters. From Tables 1 and 2, the HOMO–LUMO gaps of the Al3–X and Al3–M compounds are comparable to each other, which are ranging from 4.53 to 6.34 eV and from 4.08 to 5.62 eV, respectively. These gap values are considerably large compared with that of superatom compound Al13K3O36 (1.24 eV), suggesting better stability of the studied compounds.The global chemical hardness (η),59 which can be approximately obtained as follows,
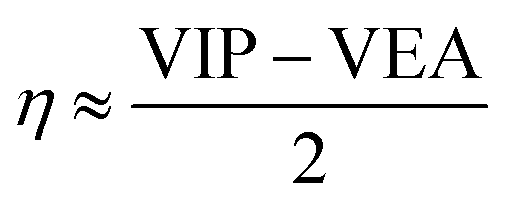
The relative stability of compounds can also be examined by binding energy per atom (Ea), and the larger the Ea value, the better the stability. It is found that the Ea values of the Al3–X compounds show an increasing tendency with increasing atom number. The lowest-energy structures can be taken as examples. From Table 1, the Ea values increase in the order 55.54 kcal mol−1 (Al3–F) < 68.55 kcal mol−1 (Al3–LiF2) < 79.47 kcal mol−1 (Al3–BeF3) < 86.87 kcal mol−1 (Al3–BF4). By contrast, among the Al3–M compounds, the Al3–OLi3 species exhibit the largest Ea values of 58.21–60.71 kcal mol−1. It is also noted that the Al3–superhalogen compounds possess larger Ea values than the Al3–superalkali compounds, which may reflect the superior stability of the former system.
The bond energies Eb of the Al3–X and Al3–M compounds are defined as the negative of Eint values. A larger Eb value implies a stronger interaction between Al3 and (super)atoms. As can be seen from Table 1, the Eb values of the Al3–X compounds are as large as 120.1–190.5 kcal mol−1, which are comparable to or much larger than traditional ionic bond energy of 133.5 kcal mol−1 for LiF and bond energies (117.5–128.45 kcal mol−1) of superatom compounds Al13K3O24 and Li3O–X (X = BF4, BeF3, NO3).27 Thus, the Al3 cluster can tightly bind with (super)halogen X. Note that the bond energy sequence is not completely consistent with the stability sequence of the isomers. For example, the total energy of 2ps-1 is much lower than that of 2ss, but the latter has a larger Eb value of 190.5 kcal mol−1. This is due to the fact that isomer 2ss contains one more Al–Li bond, and consequently, shows a stronger interaction between the Al3 and LiF2 moieties. Similarly, the 3ff isomer with Al–Be connections has the largest bond energy among the Al3–BeF3 species. For the other Al3–superhalogen compounds without Al–metal atom interactions, the Eb value varies in the 164.6–169.4 kcal mol−1 range. From Table 2, the bond energies of 37.2–101.0 kcal mol−1 for Al3–M are smaller compared with those of the Al3–X compounds, but are large enough to guarantee the strong interaction between Al3 and (super)alkali M. Besides, those Al3–M isomers involving nonmetal-atom–Al3 connections, namely II23, III24-1, III24-2, IV24, exhibit much larger Eb values than the others.
To better understand the structures and stability of compounds assembled by Al3 cluster and (super)atoms, we explored the bonding character of the Al3–X and Al3–M compounds on the basis of NBO and AIM analyses. Based on NBO analysis, the Al3 unit exists in cationic state in Al3–X while in anionic state in the Al3–M compounds.
As shown in Table 1, the sum of NBO charges (0.685–0.824|e|) on the Al3 subunit in each Al3–X compound is close to +1 (except for isomer 3ff), denoting that an electron transfers from Al3 to (super)halogen X. This is consistent with the recent work of Zhao et al., where Al3 has been indicated to be a superalkali cluster.60 Structure 3ff contains an Al3Be unit, and the electron sharing between Al3 and Be results in 1.375|e| NBO charge on the Al3 subunit. Different from the case of Al3–X, the Al3 subunits are negatively charged with −0.266 to −0.675|e| in the Al3–M compounds. It means that the (super)alkalis are capable of reducing the Al3 cluster. To be specific, (super)alkali M is apt to lose an electron while the Al3 cluster longs for an electron to achieve a closed-shell configuration. To clearly show the electron-shell structure and molecular orbital characteristics of the Al3–M compounds, isomer IIfs is taken as an example and its valence molecular orbitals (MOs) are illustrated in Fig. S1.† From the figure, the valence molecular orbitals of IIfs can be considered originated from Al3− and FLi2+ subunits, respectively. Obviously, both Al3 and FLi2 moieties obtain shell-closed electronic configurations (1s21p62s2 and 1s21p6, respectively, according to spherical jellium model61,62) by charge transfer. As a result, the IIfs structure achieve high stability from the Al3− and FLi2+ segments, respectively. This is the same case for other Al3–M compounds.
The Laplacian of the electron density at a bond critical point (BCP), ∇2ρ(r), is an important quantity based on the AIM theory for describing the chemical bonding nature.50,51 Hence, the ∇2ρ(r) values for dominant bonds that connect Al3 and X/M subunits were calculated, and are shown in Fig. 1 and 2, respectively. From Fig. 1, the ∇2ρ(r) values of Al–F bonds vary in the range of 0.140–0.939 au., indicating that the Al3 and (super)halogen subunits are connected by ionic bonds. These present a situation akin to that of superatom compounds BF4–M (M = Li, FLi2, OLi3, NLi4)25 and BLi6–X (X = F, LiF2, BeF3, BF4).26 The superhalogen and superalkali clusters are also ionically bonded in these compounds, and the ionic connections possess 0.106–0.361 au. ∇2ρ(r) values, which are comparable to those of the Al3–X compounds.
As can be seen from Fig. 2, the combination of Al3 and (super)alkali M involves one or more Al–Li metallic bonds. Besides, the ∇2ρ(r) values of 0.351–0.636 au. confirm the ionic bonding nature of the Al–F/O/N bonds in the II23, III24-1, III24-2, IV24 structures. Note that these compounds have much larger bond energies compared with the others, suggesting that the ionic bonds contribute a lot to the interaction between Al3 and superalkali M. Similarly, ionic bonds play an important role in higher stability (namely larger binding energy and bond energy values) of Al3–X compared with the Al3–M system, since the former series are typical ionic compounds. It can be seen that both 1pp and II23 structures contain an Al–F ionic bond. Whereas, the Al–F bond in 1pp is much stronger compared with that in II23, as reflected by shorter bond length and larger ∇2ρ(r) value of the former. Hence, the bond energy of 1pp is quite larger than that of II23. Besides, the preferred interaction site sequence of Al3 when interacting with superhalogens can also be explained by the strength of Al–X ionic bonds. To be specific, for each Al3–X compound, the Al–F bond is the strongest, reflected by the shortest bond length and largest ∇2ρ(r) value, when Al3 binds with superhalogens through an apex Al atom. The only exception is the Al3–LiF2 compound. Its three isomers have similar Al–F bond lengths and corresponding ∇2ρ(r) values. In contrast, the Al–F bond is the weakest, reflected by the longest bond length and smallest ∇2ρ(r) value, when Al3 interacts with superhalogens through its ring plane (see Fig. 1).
Since the aforementioned investigations were performed within the gas-phase approximation, one may wonder to what extent the calculations would be affected when solvent effects are taken into account. Besides, do Al3–X and Al3–M compounds behave differently upon including a solvent? To address these questions, we took Al3–BF4 (4ps) and Al3–NLi4 (IV-24) as examples and ran parallel calculations by employing a self-consistent reaction-field (SCRF) treatment with a polarizable continuum model (PCM).63,64 Thereby, their optimized structures were obtained in polar (ethanol) and nonpolar (cyclohexane) environments, respectively, and are displayed in Fig. S2.† The corresponding physicochemical properties of 4ps and IV-24 were also calculated by using the PCM model, and are listed in Table S2.†
Compared with the optimized structures in gas-phase, all the ionic bonds that connect Al3 and superatom subunits elongate in the presence of solvents. From Fig. S2,† the Al–Li metallic bonds of Al3–NLi4 elongate in polar solvent but shorten in nonpolar solvent. Nevertheless, it can be concluded that solvent effect on the geometrical structures of superatom compounds is not significant since the 4ps and IV-24 structures do not change much in solution.
To explore the solvent effect on infrared (IR) spectrum of the 4ps and IV-24 isomers, their characteristic vibrations with the largest IR intensity were selected and examined with the PCM model. The stretching movement of superhalogen BF4 toward Al3 cluster is the characteristic vibration of 4ps both in gas-phase and in solution (see Fig. S3a†). From Table S2,† the stretching frequency is red-shifted by 28.7 and 12.8 cm−1, and the corresponding IR intensity increases 213.2 and 93.8 km mol−1 in the presence of polar and nonpolar solvents, respectively. As to IV-24, its characteristic vibration is the stretching mode of superalkali NLi4 relative to Al3 no matter whether in gas phase or in solution (see Fig. S3b†). Meanwhile, the characteristic vibrational frequency of IV-24 also undergoes redshifts of 10.4 and 54.4 cm−1 in polar and nonpolar solvents, respectively. Moreover, it can be found that both polar and nonpolar solvents promote the charge transfer between Al3 and superatom clusters, especially superalkali NLi4. As a result, the stability of Al3–NLi4 is enhanced a lot in the presence of solvent molecules, which is reflected by the increased HOMO–LUMO gap, Ea, and Eb values. And this is particular the case when polar solvent (ethanol) is involved. For example, the bond energy of Al3–NLi4 reaches to 176.0 kcal mol−1 in ethanol environment. Note that this value is even larger than that of Al3–BF4. Hence, the Al3–superalkali compounds may be better stabilized in solvents than in gas phase. As far as Al3–BF4 is concerned, the HOMO–LUMO gap value becomes a bit larger according to the prediction of PCM solvation model. Apart from that, solvent effect hardly influences its stability.
3.3. Aromaticity
According to previous report, the Al3− anion has double aromaticity.30 From Fig. S4,† the σ-bonding HOMO orbital of Al3− renders σ-aromaticity, while the π-bonding HOMO−1 orbital renders π-aromaticity. The Al3+ ring, by contrast, is also expected to possess π-aromaticity arising from its π-bonding HOMO orbital. Since Al3− and Al3+ ions maintain their structural and electronic integrity in most Al3–M and Al3–X compounds, respectively, the resulting superatom compounds are supposed to be aromatic as well.The nucleus-independent chemical shift (NICS), proposed by Schleyer and coworkers, is an efficient method to probe aromaticity of a molecule. Negative and positive NICS values denote aromaticity and antiaromaticity, respectively.52 To examine the aromaticity of the studied superatom compounds, the NICS values were calculated at, above, and below the geometrical center of the Al3 subunits,53,65 and the spatial locations of the maximum NICS values are listed in Tables S3 and S4.† Because of the serious deformation of Al3 moiety in structures 2ps-2 and IIss, their aromaticity is not considered in this work. Although the Al3 moiety also undergoes severe deformation in isomers II23 and III24-2, the three Al atoms and two Li atoms are seen to form a metal cage which might have three-dimensional (3-D) aromaticity.
The maximum NICS values for the Al3–X and Al3–M compounds are shown in Tables 1 and 2, respectively. From the tables, the NICSmax values range from −12.5 to −37.6 ppm for Al3–X and from −14.4 to −39.0 ppm for Al3–M, confirming their aromatic nature. Nevertheless, it is worth noting that isomers 3ff and 4ff show considerably lower NICSmax values (−13.9 and −12.5 ppm, respectively) compared to isolated Al3+ ring (−31.4 ppm at the same computational level) and other Al3–X structures. To explore the reason behind this, isomers 4ff and 4ps are taken as examples. Their first four valence MOs are shown in Fig. 3. From the figure, the four MOs of 4ps originate from the Al3 subunit and look like duplicates of those of isolated Al3+ ring. As a result, 4ps exhibits π-aromaticity and its NICSmax value (−30.8 ppm) is close to that of isolated Al3+. This is the same case for isomers 1pp, 1sp, 2ps-1, 2ss, 3ps-1, 3ps-2, and 3ss. Interestingly, the MOs of the Al3 cluster seem to have been rearranged while it interacts with superhalogen BeF3 and BF4 in the face-to-face orientation. As shown in Fig. 3, the HOMO orbital of 4ff turns out to be a σ-bonding orbital formed from in-plane 3p orbital of Al atoms, which renders σ-aromaticity to this structure. The same holds true for the 3ff isomer. Thus, the Al3+ ring can exhibit different aromaticity depending on how it combined with superhalogen anions. Besides, the σ-aromaticity of the Al3+ subunit corresponds to a smaller NICS value compared with its π-aromaticity. In addition, isomers II23 and III24-2 do possess 3-D aromaticity although their NICSmax values of −17.9 and −14.4 ppm, respectively, are relatively low compared to other Al3–M compounds. Note that the aromaticity of these superatom compounds would reduce upon including solvent effect, which is reflected by decreased NICSmax values of 4ps and IV-24 in both polar and nonpolar environments (see Table S2†). It implies that the delocalized valence electron cloud of the Al3 subunit becomes less concentrated due to the interaction with solvent molecules.
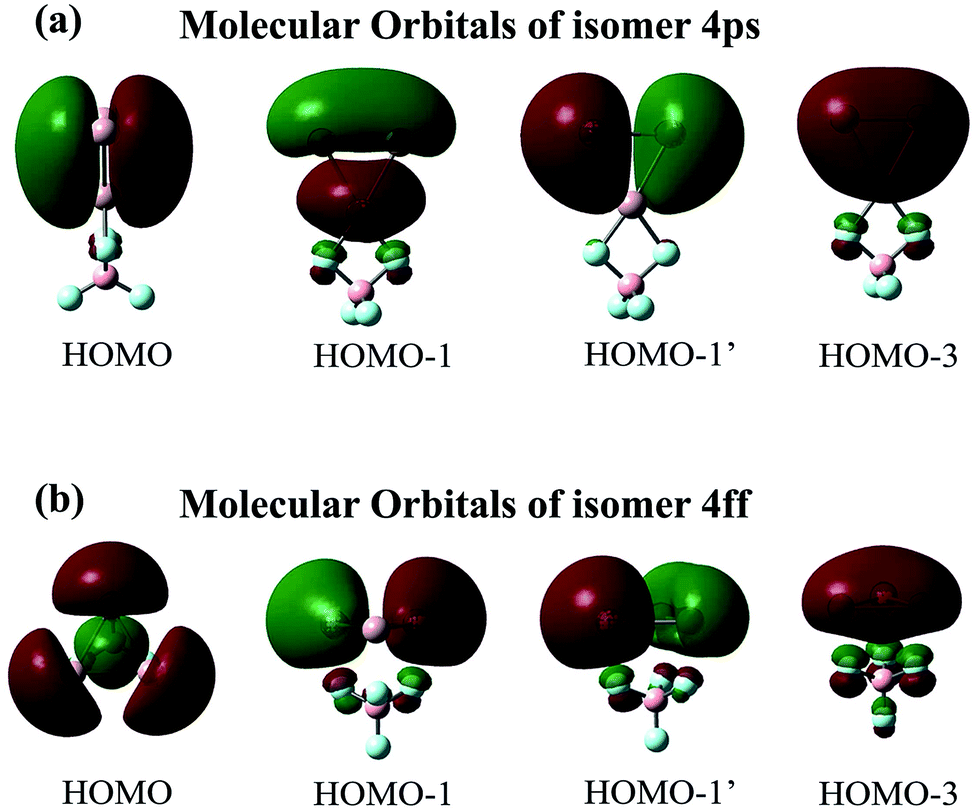 | ||
| Fig. 3 Valence molecular orbitals of isomers (a) 4ps and (b) 4ff. | ||
4. Conclusions
In summary, we have theoretically studied two types of superatom compounds by combining the Al3 trimer with different shaped (super)halogens X (X = F, LiF2, BeF3, BF4) or (super)alkalis M (M = Li, FLi2, OLi3, NLi4). NBO analysis reveals that the Al3 cluster donates electron to the former whereas gains electron from the latter species. Diverse structures have been obtained for the resulting Al3–X and Al3–M compounds. The most beneficial bonding pattern in the Al3–X systems is point-to-side, while the least favorable one is face-to-face. As for the Al3–M compounds, Al3 prefers to bind with Li and FLi2 through its ring plane, while prefers to interact with OLi3 and NLi4 through the Al–Al edge. All the studied superatom compounds possess large bond energies, indicating strong interactions between Al3 and (super)atoms. Although the geometrical structures of the studied compounds do not change much when solvent effects are taken into account, the stability of Al3–NLi4 is obviously enhanced in the presence of solvent molecules. As expected, the Al3 ring brings aromaticity to these superatom compounds no matter whether in gas phase or in solution. What is intriguing is that the Al3+ ring can exhibit different aromaticity (π or σ aromaticity) when combined with different superhalogen anions.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 21375017, 21603032) and State Key Development Program for Basic Research of China (Grant No. 2013CB834801).References
- J. Xiang, S. H. Wei, X. H. Yan, J. Q. You and Y. L. Mao, J. Chem. Phys., 2004, 120, 4251–4257 CrossRef CAS PubMed.
- Y. Li, D. Wu, Z. R. Li and C. C. Sun, J. Comput. Chem., 2007, 28, 1677–1684 CrossRef CAS PubMed.
- P. Jena, J. Phys. Chem. Lett., 2013, 4, 1432–1442 CrossRef CAS PubMed.
- Y. Negishi, W. Kurashige, Y. Niihori and K. Nobusada, Phys. Chem. Chem. Phys., 2013, 15, 18736–18751 RSC.
- J. Y. Liu, D. Wu, W. M. Sun, Y. Li and Z. R. Li, Dalton Trans., 2014, 43, 18066–18073 RSC.
- A. Fernando, K. D. M. Weerawardene, N. V. Karimova and C. M. Aikens, Chem. Rev., 2015, 115, 6112–6216 CrossRef CAS PubMed.
- X. Li, H. Wu, X.-B. Wang and L.-S. Wang, Phys. Rev. Lett., 1998, 81, 1909–1912 CrossRef CAS.
- T. Bergmann, H. Limberger and T. P. Martin, Phys. Rev. Lett., 1988, 60, 1767–1770 CrossRef CAS PubMed.
- S. Khanna and P. Jena, Phys. Rev. B, 1995, 51, 13705 CrossRef CAS.
- S. Khanna and P. Jena, Phys. Rev. Lett., 1992, 69, 1664 CrossRef CAS PubMed.
- V. M. Medel, J. U. Reveles, S. N. Khanna, V. Chauhan, P. Sen and A. W. Castleman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10062–10066 CrossRef CAS PubMed.
- V. Medel, J. U. Reveles and S. N. Khanna, J. Appl. Phys., 2012, 112, 064313–064319 CrossRef.
- V. Chauhana, V. M. Medelb, J. U. Revelesb, S. N. Khannab and P. Sena, Chem. Phys. Lett., 2012, 528, 39–43 CrossRef.
- I. Anusiewicz and P. Skurski, Chem. Phys. Lett., 2002, 358, 426–434 CrossRef CAS.
- S. Smuczynska and P. Skurski, Chem. Phys. Lett., 2008, 452, 44–48 CrossRef CAS.
- H. Yang, Y. Li, H.-M. He, J. Tong, D. Wu and Z.-R. Li, Chem. Phys. Lett., 2017, 684, 273–278 CrossRef CAS.
- G. L. Gutsev and A. I. Boldyrev, Chem. Phys., 1981, 56, 277–283 CrossRef CAS.
- A. N. Alexandrova and A. I. Boldyrev, J. Phys. Chem. A, 2003, 107, 554–560 CrossRef CAS.
- G. L. Gutsev and A. I. Boldyrev, Chem. Phys. Lett., 1982, 92, 262–266 CrossRef CAS.
- E. Rehm, A. I. Boldyrev and P. v. R. Schleyer, Inorg. Chem., 1992, 31, 4834–4842 CrossRef CAS.
- J. Tong, Y. Li, D. Wu, Z. R. Li and X. R. Huang, J. Chem. Phys., 2009, 131, 164307 CrossRef PubMed.
- H. Hotop and W. C. Lineberger, J. Phys. Chem. Ref. Data, 1985, 14, 731–750 CrossRef CAS.
- S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Homes, R. D. Levin and W. G. Mallard, J. Phys. Chem. Ref. Data, 1988, 17(Supp1), 1285–1363 Search PubMed.
- A. C. Reber, S. N. Khanna and A. W. Castleman, J. Am. Chem. Soc., 2007, 129, 10189–10194 CrossRef CAS PubMed.
- H. Yang, Y. Li, D. Wu and Z.-r. Li, Int. J. Quantum Chem., 2012, 112, 770–778 CrossRef CAS.
- Y. Li, D. Wu and Z. R. Li, Inorg. Chem., 2008, 47, 9773–9778 CrossRef CAS PubMed.
- S. Giri, S. Behera and P. Jena, J. Phys. Chem. A, 2014, 118, 638–645 CrossRef CAS PubMed.
- X. Li, A. E. Kuznetsov, H. F. Zhang, A. I. Boldyrev and L. S. Wang, Science, 2001, 291, 859–861 CrossRef CAS PubMed.
- A. E. Kuznetsov, A. I. Boldyrev, H. J. Zhai, X. Li and L. S. Wang, J. Am. Chem. Soc., 2002, 124, 11791–11801 CrossRef CAS PubMed.
- A. E. Kuznetsov and A. I. Boldyrev, Struct. Chem., 2002, 13, 141–148 CrossRef CAS.
- A. I. Boldyrev and L. S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed.
- N. He, H. B. Xie and Y. H. Ding, Microporous Mesoporous Mater., 2010, 130, 67–75 CrossRef CAS.
- J. M. Mercero, E. Matito, F. Ruipérez, I. Infante, X. Lopez and J. M. Ugalde, Chem.–Eur. J., 2015, 21, 9610–9614 CrossRef CAS PubMed.
- C.-G. Zhan, F. Zheng and D. A. Dixon, J. Am. Chem. Soc., 2002, 124, 14795–14803 CrossRef CAS PubMed.
- D. E. Bergeron, A. W. Castleman, T. Morisato and S. N. Khanna, Science, 2004, 304, 84–87 CrossRef CAS PubMed.
- D. E. Bergeron, P. J. Roach, A. W. Castleman, N. Jones and S. N. Khanna, Science, 2005, 307, 231–235 CrossRef CAS PubMed.
- J. U. Reveles, S. N. Khanna, P. J. Roach and A. W. Castleman Jr, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18405–18410 CrossRef CAS PubMed.
- W.-M. Sun, D. Wu, X.-H. Li, Y. Li, J.-H. Chen, C.-Y. Li, J.-Y. Liu and Z.-R. Li, J. Phys. Chem. C, 2016, 120, 2464–2471 CAS.
- J. Reveles, T. Baruah and R. R. Zope, J. Phys. Chem. C, 2015, 119, 5129–5137 CAS.
- S. R. Miller, N. E. Schultz, D. G. Truhlar and D. G. Leopold, J. Chem. Phys., 2009, 130, 024304 CrossRef PubMed.
- B. K. Rao and P. Jena, J. Chem. Phys., 2000, 113, 1508–1513 CrossRef CAS.
- J. Sun, W. C. Lu, H. Wang, Z.-S. Li and C.-C. Sun, J. Phys. Chem. A, 2006, 110, 2729–2738 CrossRef CAS PubMed.
- M. D. Deshpande and D. G. Kanhere, Phys. Rev. B, 2003, 68, 035428 CrossRef.
- L. G. M. Pettersson, C. W. Bauschlicher Jr and T. Halicioglu, J. Chem. Phys., 1987, 87, 2205–2213 CrossRef CAS.
- P. P. Bera, K. W. Sattelmeyer, M. Saunders, H. F. Schaefer and P. v. R. Schleyer, J. Phys. Chem. A, 2006, 110, 4287–4290 CrossRef CAS PubMed.
- J. Tong, Y. Li, D. Wu, Z.-R. Li and X.-R. Huang, J. Phys. Chem. A, 2010, 115, 2041–2046 CrossRef PubMed.
- M. Saunders, J. Comput. Chem., 2004, 25, 621–626 CrossRef CAS PubMed.
- C. Moller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. J. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- U. Kock and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef.
- P. L. A. Popelier, J. Phys. Chem. A, 1998, 102, 1873–1878 CrossRef CAS.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- F. Ma, R. Y. Li, Z. R. Li, M. M. Chen, H. L. Xu, Z. J. Li, D. Wu and Z. S. Li, J. Mol. Struct.: THEOCHEM, 2009, 913, 80–84 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- I. Alkorta and J. Elguero, J. Phys. Chem. A, 1999, 103, 272–279 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Coss, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. Dennington, K. Todd, J. Millam, K. Eppinnett, W. L. Hovell and R. Gilliland, GaussView, version 3.09 edn, Semichem, Inc, Shawnee Mission, KS, 2003 Search PubMed.
- W.-M. Sun, Y. Li, D. Wu and Z.-R. Li, Phys. Chem. Chem. Phys., 2012, 14, 16467–16475 RSC.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- T. Zhao, Q. Wang and P. Jena, Nanoscale, 2017, 9, 4891–4897 RSC.
- W. Ekardt, Phys. Rev. B, 1984, 29, 1558 CrossRef CAS.
- W.-D. Knight, K. Clemenger, W. A. de Heer, W. A. Saunders, M. Chou and M. L. Cohen, Phys. Rev. Lett., 1984, 52, 2141 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef CAS.
- F. F. Wang, Z. R. Li, D. Wu, X. Y. Sun, W. Chen, Y. Li and C. C. Sun, ChemPhysChem, 2006, 7, 1136–1141 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The valence molecular orbitals of IIfs, optimized structures and the corresponding physicochemical properties of the Al3–BF4 (4ps) and Al3–NLi4 (IV-24) compounds in the presence of solvents, The characteristic vibration mode of the (a) Al3–BF4 (4ps) and (b) Al3–NLi4 (IV-24) compounds in solvents and gas phase, electron-shell structures of Al3+ and Al3− ions, and the hardness (η, in eV) of the most stable Al3–X and Al3–M compounds, locations of the maximum negative NICS values, Cartesian coordinates and electronic states for the Al3–X and Al3–M compounds. See DOI: 10.1039/c7ra12852e |
| This journal is © The Royal Society of Chemistry 2018 |