DOI:
10.1039/C7RA12768E
(Paper)
RSC Adv., 2018,
8, 7494-7508
Sulfonated graphene oxide/Nafion composite membranes for high temperature and low humidity proton exchange membrane fuel cells†
Received
25th November 2017
, Accepted 10th February 2018
First published on 16th February 2018
Abstract
Iron oxide (Fe3O4) nanoparticles anchored over sulfonated graphene oxide (SGO) and Nafion/Fe3O4–SGO composites were fabricated and applied as potential proton exchange membranes in proton exchange membrane fuel cells (PEMFCs) operated at high temperature and low humidity. Fe3O4 nanoparticles bridge SGO and Nafion through electrostatic interaction/hydrogen bonding and increased the intrinsic thermal and mechanical stabilities of Nafion/Fe3O4–SGO composite membranes. Nafion/Fe3O4–SGO composite membranes increased the compactness of ionic domains and enhanced the water absorption and proton conductivity while restricting hydrogen permeability across the membranes. The proton conductivity of Nafion/Fe3O4–SGO (3 wt%) composite membrane at 120 °C under 20% relative humidity (RH) was 11.62 mS cm−1, which is 4.74 fold higher than that of a pristine recast Nafion membrane. PEMFC containing the Nafion/Fe3O4–SGO composite membrane delivered a peak power density of 258.82 mW cm−2 at a load current density of 640.73 mA cm−2 while operating at 120 °C under 25% RH and ambient pressure. In contrast, under identical operating conditions, a peak power density of only 144.89 mW cm−2 was achieved with the pristine recast Nafion membrane at a load current density of 431.36 mA cm−2. Thus, Nafion/Fe3O4–SGO composite membranes can be used to address various critical problems associated with commercial Nafion membranes in PEMFC applications.
1. Introduction
Proton exchange membrane fuel cells (PEMFCs) operating at high temperature and low humidity are considered as advanced electrochemical energy conversion systems owing to their advantages of fast electrode reaction kinetics, limited requirement for a precious metal (Pt) catalyst, improved electrocatalytic CO tolerance and simplified water and thermal management.1–4 One method to realize the practical application of such PEMFCs is to employ a proton exchange membrane (PEM) that is able to operate under high temperature and low humidity conditions. Until now, perflurorosulfonic acid (PFSA) membranes such as Nafion are considered state-of-the-art PEMs, which have long term durability, high electronic insulation, high proton conductivity and unique hydrophilic percolation;5–8 unfortunately, the water uptake and consequently the proton conductivity of a Nafion membrane decrease by orders of magnitude during PEMFC operation at high temperature (>80 °C) and low humidity (<50%), owing to its inability to retain water in ionic clusters.1,9 Besides, Nafion membrane exhibits high hydrogen permeability denoted to as high hydrogen crossover from the anode to cathode through PEM as a result of concentration gradient.10,11 Accordingly, researchers are developing modified PFSA membranes or designing alternative PEMs. Incorporation of inorganic fillers, such as SiO2, ZrO2, CeO2, ZnO2, TiO2 and zeolite nanoparticles and ZrO2, CeO2 and TiO2 nanotubes, into the Nafion matrix has been demonstrated to result in composite PEMs with promising water uptake and consequently proton conductivity at high temperature and low humidity.4,12–16 Although the addition of inorganic fillers into Nafion matrix led to reorganized ion conducting channels (consisting of pendent sulfonic acid groups and embedded hydrophilic inorganic fillers), inorganic fillers without any sulfonic acid group often reduces the proton conductivity at hydrated condition, owing to a decrease in net volume of sulfonic acid contents in composite PEMs.17
Carbon nanomaterials with sulfonic acid groups have drawn attention in fabrication of composites with Nafion as they can increase the proton conductivity by increasing number of sulfonate groups per cluster volume of each domain and extend the mechanical strength by reorganizing aliphatic backbone chains. Rambabu et al.18 embedded sulfonic acid functionalized fullerene (FF) into a Nafion matrix to prepare a composite membrane. They reported that at 1 wt% FF loading, composite membrane shows an increase in tensile strength by 1.13 fold and in proton conductivity by 1.24 fold with respect to pristine Nafion membrane. Liu et al.19 incorporated multiwalled carbon nanotubes (MWCNTs) that were functionalized with Nafion into a Nafion matrix to prepare a composite membrane. In this work, the mechanical strength and proton conductivity of composite membrane were found to be increased up to a reasonable margin upon incorporating 0.05 wt% Nafion–MWCNTs. These results were attributed to high compatibility between Nafion–MWCNTs and Nafion matrix because Nafion chains on the surface of MWCNTs connect with hydrophilic domains of Nafion matrix and facilitate the dispersion.
Graphene is a single atom thick two-dimensional layer of sp2-hybridized carbon discovered by Geim et al.,20 and is associated with exceptional extended surface, unique π-bond delocalized hexagonal structure, excellent thermomechanical stability and high young's modulus that are beneficial to its wide use in the fields of sensors,21 catalysis,22 adsorption,23 nanoelectronics24 and energy conversion and storage.25–27 For the integration of graphene into PEM, it is essential to functionalize/oxidize sp2-hybridized carbon networks to convert the material as a protonic conductor and electronic insulator.28 The oxygen-related functional groups (e.g. –OH, –O– and –CO2H) over GO were reported to exhibit a conductivity of ∼0.001 S cm−1 at 300 K.29 However, substituting –OH, –O– and –CO2H groups of GO with sulfonic acid containing organic moiety enhances the proton conductivity by many fold owing to a prompt proton conducting behavior of sulfonic acid group. Furthermore, the interfacial hydrogen bonds formed between different kinds of oxygen-related functional groups of sulfonated GO (SGO) and polymer aid in the construction of extended architecture of proton conducting channels, which further facilitate the diffusion of protons.2 Several PEMs were explored with SGO based fillers. As example, Zarrin et al.1 prepared SGO/Nafion composite membrane and demonstrated it to PEMFC operating under high temperature and low humidity. The GO was produced from natural graphite flakes by modified Hummers method and then functionalized using 3-mercaptopropyl trimethoxysilane as sulfonic acid group precursor. They found that maximum power density of SGO/Nafion membrane increases by 3.98 fold over pristine Nafion membrane at 120 °C and 25% RH. Shau et al.17 fabricated the sulfonic acid functionalized graphene (S-graphene)/Nafion composite membrane and applied it in low humidity PEMFC. The functionalization of graphene was performed by sulfonic acid containing aryl radicals to increase the number of sulfonate groups per cluster volume of graphene domain. It was found that peak power density of S-graphene/Nafion membrane increases by 1.36 fold compared to that of pristine Nafion membrane at 70 °C and 20% RH. Beydaghi et al.30 prepared PVA composite membrane that was prepared using SGO and PVA solution. It was found that tensile strength and proton conductivity of SGO/PVA membrane are higher with respect to pristine PVA membrane. The higher tensile strength was due to effectual stress transfer to SGO sheets through interfacial hydrogen bonds. The higher proton conductivity was attributed to chemical interactions between different kinds of oxygen-containing functional groups of SGO and PVA. Although SGO based PEMs are reported by several research groups, the realization of a complete physiochemical, thermomechanical, electrochemical and fuel barrier properties have rarely been investigated.
On the other side, incorporation of metal oxide nanoparticles-anchored SGO sheets into PEM is an ultimate strategy in improving the dispersion of SGO sheets, which facilitates the surface roughness of PEM and compatibility with electrodes. Among the studied metal oxide nanoparticles, Fe3O4 has synthesis flexibility and eco friendliness and is the efficient catalyst for various electrochemical reactions.31,32 Fe3O4 possesses a high density of –OH groups that generate hydrogen bonds with free water existing in membrane thereby slow down the evaporation of water from membrane and facilitate the proton transfer by vehicle mechanism.33 Besides, the interfacial hydrogen bonding between –OH groups of Fe3O4 and sulfonic acid (–SO3H) groups and fluorine atoms (–F) of Nafion enhances thermal and mechanical stabilities of the composite membrane.9 If the Fe3O4 anchored SGO sheets could be effectually intercalated into the Nafion matrix, superior properties with respect to pristine Nafion could be capitalized, which may bring out ameliorated PEMFC performance. Again the existence of Fe3O4 into the polymer matrix is a problem that facilitates the oxidative degradation of PEM when combined with hydroxyl (OH˙) and hydroperoxyl (OOH˙) radicals formed in cathode during PEMFC operation. Recently, Beydaghi et al.9 reported that the addition of 5 wt% Fe3O4–SGO could increase the durability of SPEEK/PVA blend significantly. During 120 h of operation, the fuel cell equipped with Fe3O4–SGO/SPEEK/PVA membrane showed a OCV reduction of 0.65 mV h−1, which is lower by a factor of 1.3 compared to that of fuel cell equipped with SPEEK/PVA membrane (0.85 mV h−1). Thus, addition of Fe3O4 with SGO is a better approach to suppress the effect of oxidative degradation on PEM, because Fe3O4 intercalates between the SGO layers which precludes the direct exposure of Fe3O4 to OH˙ and OOH˙ radicals. The intention of the present study is to investigate the impact of a combination of composite filler (Fe3O4–SGO) and Nafion on performance of PEMFC operating under high temperature and low humidity through its morphological, thermomechanical, electrochemical and fuel barrier properties.
2. Experimental section
2.1. Materials
Natural graphite powder, iron(III) chloride hexahydrate (FeCl3·6H2O, 97%), iron(II) sulphate heptahydrate (FeSO4·7H2O, 99%), sodium nitrate (NaNO3, 99%), sodium nitrite (NaNO2, 97%), sodium borohydride (NaBH4, 98%) and potassium permanganate (KMnO4, 99.3%) were received from Alfa Aesar. Sulfanilic acid (C6H7NO3S, 99%), sulfuric acid (H2SO4, 95%), hydrogen peroxide (H2O2, 32%) and hydrochloric acid (HCl, 35%) were procured from Daejung Chemicals, South Korea. Nafion solution (5%) in lower aliphatic alcohols and ammonium hydroxide (NH3OH, 5 M) were procured from Sigma Aldrich. N,N-Dimethylformamide (DMF) was obtained from Duksan reagents, South Korea. Gas diffusion electrodes (40 wt% Pt on XC-72) were procured form NARA Cell Tech, South Korea.
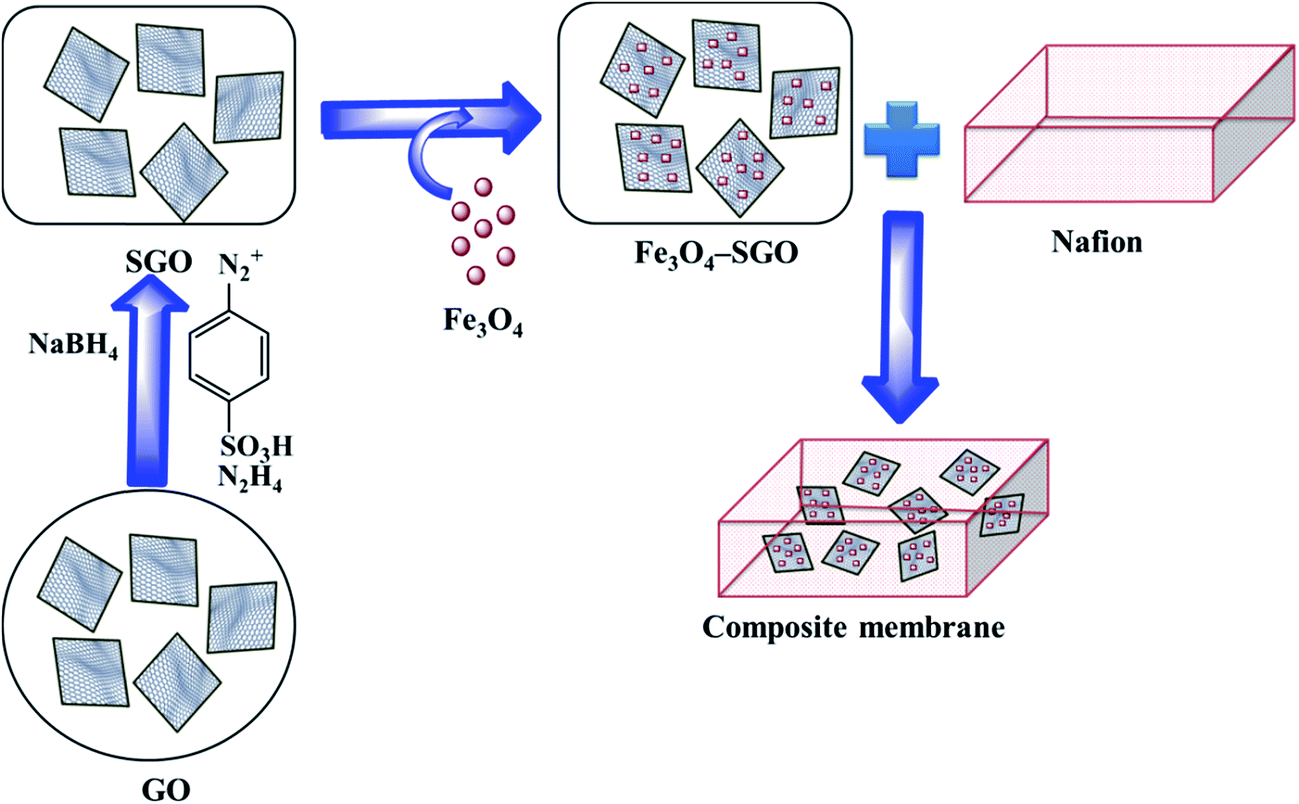
2.2. Synthesis of phenyl sulfonated GO and Fe3O4–SGO composite
First, GO was synthesized from natural graphite powder according to modified Hummers method as described in ref. 34. SGO was then synthesized via a two-step process, similar to a procedure reported in ref. 30. In the first step, partial reduction of GO was performed by NaBH4. A 75 mg of GO was dispersed in 75 mL of deionized (DI) water by ultrasonication for 1 h. Then, 15 mL of 0.1 M NaBH4 was added dropwise into the GO dispersion and stirred at 80 °C for 1 h. The reduced product was collected via centrifugation, washed several times with DI water and dried in oven at 100 °C for 12 h. In the second step, aryl diazonium salt of sulfonic acid was grafted on reduced GO via diazonium coupling. A 75 mg of reduced GO was dispersed in 75 mL of DI water by ultrasonication for 1 h. Separately, 50 mg of sulfanilic acid and 20 mg of NaNO2 were dispersed in 1 M HCl solution by ultrasonication at 0–3 °C. The obtained aryl diazonium salt of sulfonic acid was added into the dispersion of reduced GO with continuous stirring at 0–3 °C. Subsequently, 5 mL of 80% N2H2 was added dropwise and stirred for another 3 h. The resultant product was collected via centrifugation, washed several times with DI water and dried in oven at 100 °C for 12 h.
Fe3O4–SGO composite was prepared via hydrothermal method. A 100 mg of SGO was dispersed in 100 mL of DI water by ultrasonication for 1 h. Separately, desired amount of Fe3O4 nanoparticles (produced by co-precipitation method, similar to a procedure reported in ref. 33) was dispersed in DI water and subsequently added into the SGO suspension. The resultant mixture was further sonicated for 1 h, loaded into the Teflon lined stain less steel autoclave and heated at 180 °C for 12 h. Afterward, the solid product was collected via centrifugation, washed several times with DI water and dried in oven at 100 °C for 12 h.
2.3. Preparation of composite membranes
Nafion composite membranes were prepared via solution casting method. A 500 mg of Nafion (precast membrane at 100 °C for 12 h) was dissolved in 10 mL of DMF. Subsequently, the desired amount of Fe3O4–SGO (to achieve the weight ratios of 0.5, 1.5 or 3%) was impregnated with the above solution to realize a composite membrane. The resultant mixture was sonicated for 1 h followed by mechanical stirring for 12 h. Composite solution was poured on a clean Petri plate and allowed to dry under vacuum at 80 °C for 12 h. Composite membrane thus formed was peeled off from Petri plate and dried at 120 °C for 2 h to remove solvent residues. Nafion (without filler) and GO composite membranes were also prepared to systematically compare their properties with respect to Fe3O4–SGO membrane. The prepared composite membranes such as Nafion/GO (3 wt%), Nafion/Fe3O4–SGO (0.5 wt%), Nafion/Fe3O4–SGO (1.5 wt%) and Nafion/Fe3O4–SGO (3 wt%) has been designated as Nafion/GO-3, Nafion/SGF-0.5, Nafion/SGF-1.5 and Nafion/SGF-3, respectively. The thicknesses of the dry membranes were measured at three random points over the surface using a digital micrometer and were in the range from 50 to 80 μm. Although a high variation observed in thicknesses of dry membranes, the amount of Nafion was controlled in each membrane to be same and this ensures appropriate comparison between various samples. Finally, the membranes were pretreated by boiling in 3% H2O2, 1 M H2SO4 and H2O in sequence each for 1 h. Scheme 1 illustrates the preparation process of Nafion/Fe3O4–SGO composite membrane.
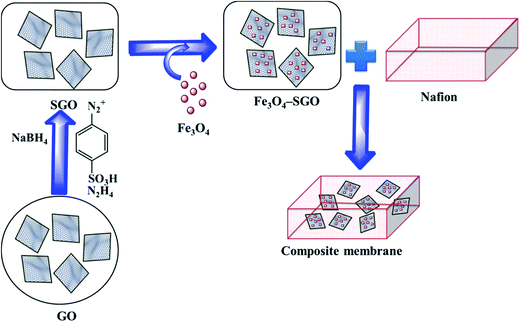 |
| | Scheme 1 Preparation process of Nafion/Fe3O4–SGO composite membrane. | |
3. Characterizations
Surface morphology of GO, SGO and Fe3O4–SGO specimens was observed by transmission electron microscope (TEM) (JEM-2010, JEOL) and field emission scanning electron microscope (FE-SEM) (SUPRA 40VP, ZEISS). Crystalline facets, surface defects, functional groups and thermal stability of GO, SGO and Fe3O4–SGO specimens were investigated by X-ray diffractometer (XRD) (X' pert-MRD equipped with CuKα radiation, Pro Philips), Raman spectrometer (HORIBA-Lab RAM-HR equipped with He–Ne laser beam), Fourier transform infrared spectroscopy (FT-IR) (GX spectrometer equipped with a DTGS detector, PerkinElmer) and thermogravimetric analyzer (TGA) (Q50-TA instruments) respectively. Surface and cross-sectional morphologies of pristine Nafion and composite membranes were observed by using a scanning electron microscope (SEM) (JSM-6400, JEOL). Cross-sectional morphology and corresponding quantitative elemental mappings of composite membrane were performed by energy dispersive X-ray (EDX) spectroscopy coupled with SEM. Topographical difference on pristine Nafion and composite membranes was observed by atomic force microscope (AFM) (multimode-8 model, Bruker). The imaging was performed in contact mode using an ultra-sharp antimony doped silicon tip with a radius of around 20 nm and spring constant of around 40 N m−1. Line profiles were used to investigate the height variation at a particular cross-section on membrane surface. The deviation of actual membrane surface [(x, y)] from an ideal atomically smooth surface (μ) is defined as surface roughness. The surface morphology of the membrane varies from area to area due to the variation in local surface roughness and crumpled surface. The root-mean-square (RMS) roughness (Sq) may give useful information about the surface topography of membrane, and it was calculated using the eqn (1):| | 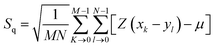 | (1) |
where M and N are respective x and y points in the specimen area, Z (x, y) is the surface and| | 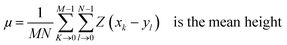 | (2) |
Thermal stability of the membrane specimens was analyzed by TGA (Q50-TA instruments) from 30 to 800 °C at a heating rate of 5 °C min−1 under a nitrogen atmosphere. Thermomechanical stability of the membrane specimens was investigated by using a dynamic mechanical analyzer (DMA) (Q800-TA instruments) from 0 to 200 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere.
4. Measurements
4.1. Water uptake and dimensional stability
Prior dried (100 °C for 5 h) membrane specimens were measured for mass, length and thickness, and then submerged into DI water at 60 °C for 24 h to attain equilibrium. Afterward, the water on the surface of membranes was carefully blotted with tissue paper and mass, length and thickness of membranes were measured again.5,28 The measurement was repeated three times for each membrane using different specimens, and the average value was used for calculation. % of changes in mass, length and thickness were calculated using the eqn (3)–(5):| | 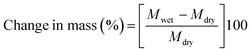 | (3) |
| | 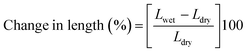 | (4) |
| | 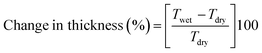 | (5) |
where Mwet, Lwet and Twet are the mass (mg), length (mm) and thickness (μm) of wet membrane specimens, respectively, and Mdry, Ldry and Tdry are the mass, length and thickness of dry membrane specimens, respectively.
4.2. Water contact angle
Wettability of the membrane specimens was measured by static water contact angle method. The measurement was performed at room temperature using a contact angle goniometer (DSA10, Kruss GmbH analyser, Germany). The detailed procedure of contact angle measurement can be found in the ref. 6.
4.3. Ion exchange capacity
Ion exchange capacity (IEC) indicates the number of milliequivalents of ions present in 1 g of a dry membrane.17 The required amount of dry membrane specimen was equilibrated in 0.1 M NaCl for 24 h to exchange H+ with Na+ ions. The released H+ ions were then titrated against 0.01 M solution of NaOH using phenolphthalein as an indicator. By measuring the volume of NaOH consumed, the molar quantity of ionic sites containing H+ ions was determined. The titration was repeated three times for each membrane using different specimens, and the average value was used for calculation. The IEC was calculated using the eqn (6):| |  | (6) |
4.4. Proton conductivity
The proton conductivity of membrane specimens was measured in the longitudinal direction with a four-probe BekkTech cell by alternating current (AC) impedance method.35 A piece of membrane (30 mm × 5 mm) was placed across the Pt probes, and membrane resistance (R, Ω) was measured using a frequency response analyzer (Scitech instrument conjugated with a Keithley-2400 source meter) as a function of temperature under low humidity. The proton conductivity (σ) of membrane specimens was calculated using the eqn (7):| | 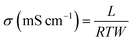 | (7) |
where L is the fixed distance between four Pt probes (i.e., 0.42 cm), R is the resistance (ohm) of the membrane specimen, and T and W are the respective thickness (cm) and width (cm) of the membrane specimen. The above mentioned measurement was repeated at least three times for each membrane using different specimens, and the average resistance value was used for calculation.
4.5. Electrical conductivity
The electrical conductivity of membrane specimens was measured using a four probe method.36 A piece of membrane (30 mm × 5 mm) was placed across the Pt probes, and membrane resistance (R, Ω) was measured using a Keithley Electrometer as a function of applied voltage. The electrical conductivity (σ) of membrane specimens can be calculated using the eqn (8):| | 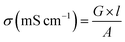 | (8) |
where G is the conductance (Siemens) of the membrane, l is the thickness (cm) of the membrane and A is the active area (cm2) of the membrane.
4.6. Hydrogen (H2) permeability
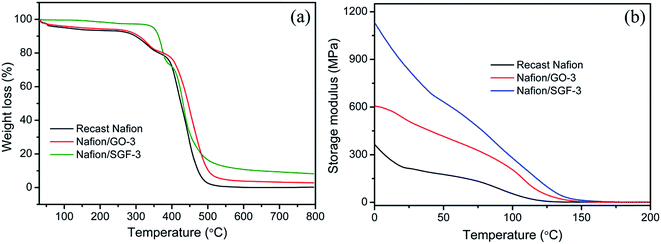
H2-permeability measurements were carried out via traditional constant volume/variable pressure method at 30 °C under anhydrous condition. A detailed method of measuring the H2 permeability can be found in the ref. 37. The measurement was repeated three times for each membrane using different specimens, and the average value was used for calculation. The H2 permeability was calculated using the eqn (9):| | 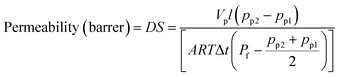 | (9) |
where D is the H2 gas diffusivity coefficient (cm2 S−1), S (cm3 (cm2 s cmHg)−1) is the solubility coefficient, Vp is the constant permeation volume (cm3), l is the film thickness (cm), A is the active membrane surface (cm2), R is the gas universal constant (J mol−1 K−1), T is the temperature (K), Pf is the feed pressure (cmHg) and Δt is the time taken for the pressure to change from pp1 to pp2 (sec).
5. Fabrication of membrane electrode assembly (MEA) and evaluation of PEMFC performance
Commercial gas diffusion electrodes (GDE; NARA Cell Tech, South Korea, thickness = 0.46 mm) with Pt catalyst loading of 0.5 mg cm−1 were adopted to use as the electrodes in MEA fabrication. MEAs with an active electrode area of 2.5 cm2 were obtained by sandwiching membrane between gas diffusion electrodes and subsequent hot pressing under a pressure of 210 kg cm−2 at 110 °C for 3 min.17,38 Each obtained MEAs were coupled with air tight Teflon gaskets and fixed in single cell fixture. PEMFC performance of MEAs was evaluated using Smart II (WonATech, South Korea) fuel cell test station. Flow rate of hydrogen and oxygen gases to their respective anode and cathode were fixed to be 450 and 600 mL min−1, respectively. The relative humidity of PEMFC depends upon the moisture content (mH2O), mass of water vapor (D) and saturated vapor pressure of water (Psat). In order to attain desired relative humidity (i.e., 100% and 25%), the parameters such as gas supply temperature (GST) and dew point humidification temperature (DPHT) were adjusted. After equilibrating the desired humidity level, the cell voltage was measured as a function of current density. The reproducibility of data was ensured by repeating the measurements at least twice with given set of conditions. All MEAs were evaluated in PEMFC atmospheric pressure without applying any back pressure.
6. Results and discussion
6.1. Characterizations of GO, SGO and Fe3O4–SGO
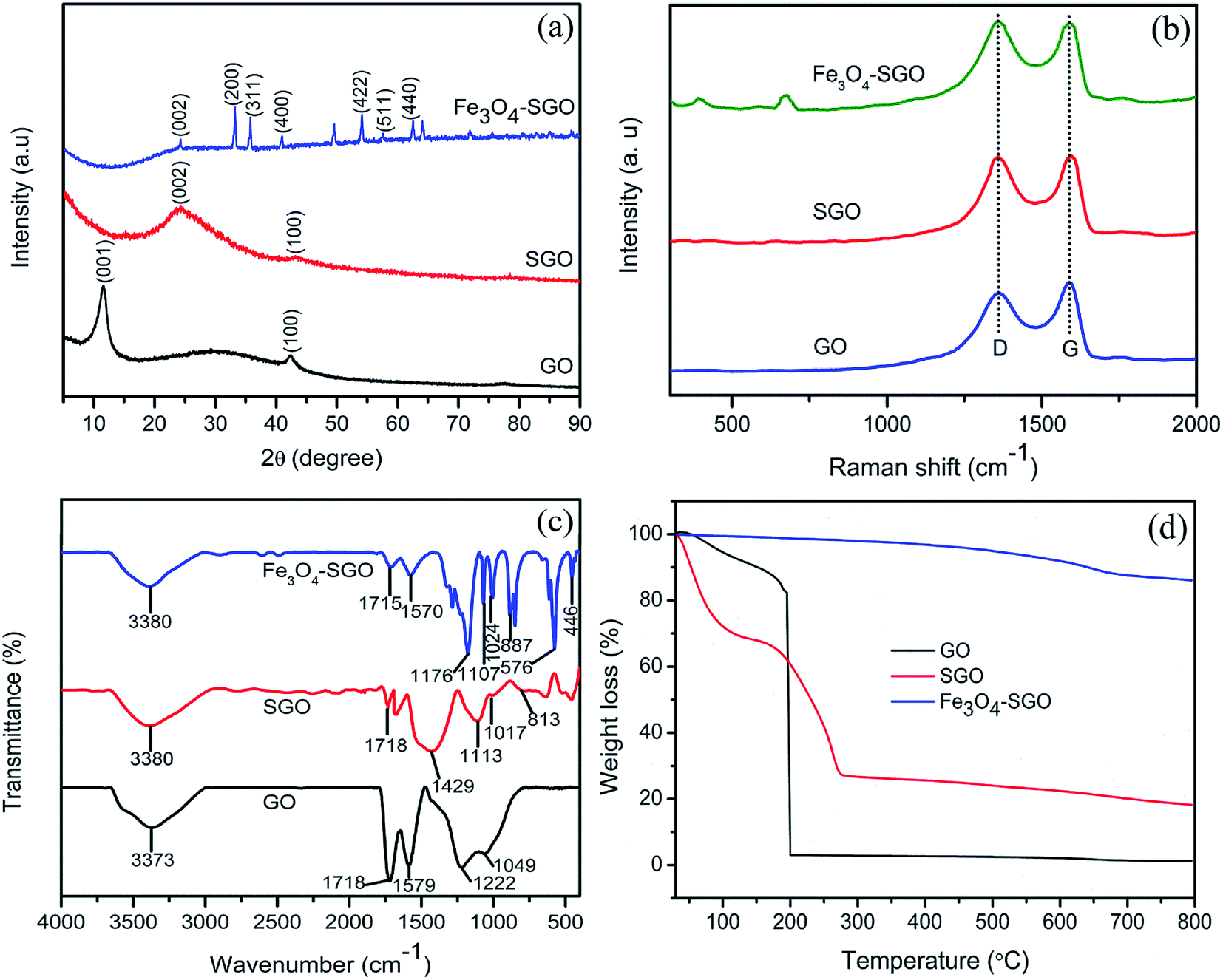
FE-SEM and TEM images on surface morphologies of GO, SGO and Fe3O4–SGO are shown in Fig. 1a–f. GO exhibits the morphology of wavy, multilayered and wrinkled sheets (Fig. 1a and d), due to the intercalations of oxygen-related functionalities. In contrast, flat morphology is observed for SGO (Fig. 1b and e), because of the removal of oxygen-related functionalities via chemical reduction. In the case of Fe3O4–SGO composite, Fe3O4 nanoparticles ranging from 60 to 120 nm are densely anchored over the SGO sheets with homogeneous distribution (Fig. 1c and f), and no Fe3O4 nanoparticles scattered outside of the sheets, representing effectual combination between Fe3O4 and SGO. The electrostatic interactions exerted between –OH group of Fe3O4 and –OH, –O–, –CO2H and –SO3H groups of SGO are responsible for dense anchoring of Fe3O4 over the SGO sheets. The obtained selected area electron diffraction (SAED) pattern (Fig. 1f-inset) ensured the poly-crystalline natured inverse cubic spinel structure of Fe3O4 nanoparticles. To investigate the crystalline structure of GO, SGO and Fe3O4–SGO, the XRD was performed and obtained diffraction patterns are presented in Fig. 2a. The characteristic peak of graphite observed at 26.32° was disappeared and an intense peak observed at 11.56° confirmed the formation of GO, which is ascribed to 001 plane.39 The calculated interplanar distance of GO is 0.75 nm, which is higher than that of graphite and is due to intercalation of oxygen-related functional groups. It can be seen that SGO exhibits the 2θ and interplanar distance of 24.38° and 0.35 nm, respectively. The interplanar distance of SGO is lower than that of GO and is due to the partial restacking of SGO sheets via π–π interactions upon removal of oxygen functionalities.40 In the case of Fe3O4–SGO composite, a broad peak observed at 24.2° is attributed to the 002 reflection plane of SGO, whereas the sharp peaks observed at 33.15, 35.87, 41.01, 54.06, 57.59 and 62.47 are attributed to the 200, 311, 400, 422, 511 and 440 facets of Fe3O4, respectively, which ensure the successful anchoring of Fe3O4 over the surface of SGO. D and G bands are two significant features to characterize the Raman spectra of GO materials, which provide the information on structural defects, degree of disorders and carbon hybridization (Fig. 2b).41,42 The D band at 1359 cm−1 ascribes to the A1g vibration modes of sp3 hybridized disordered carbon lattice, while G band at 1591 cm−1 ascribes to the E2g mode of sp2 hybridized ordered carbon lattice. These bands specify that an intensive oxidation destroys the sp2 character and prompt the defects on carbon lattice. The intensity ratio ID/IG of GO and SGO is 0.87 and 0.9, respectively, which indicate the successful reduction and functionalization of later by –SO3H containing organic moiety. In addition to the abovementioned carbon bands, an A1g mode of Fe3O4 can be found at 669 cm−1, confirming the composite structure of Fe3O4 with SGO sheets of Fe3O4–SGO solid proton conductor. Fig. 2c demonstrates the FT-IR spectra obtained at room temperature for GO, SGO and Fe3O4–SGO. GO oxidation from graphite was verified by the characteristic peaks at 3373, 1718, 1222 and 1049 cm−1, which are assigned to –OH (from intercalated water molecules), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (from carboxylic acid group), C–OH (from carboxylic acid group) and C–O (from epoxy group) stretching, respectively.43–45 The peak appearing at 1579 cm−1 in GO is consistent with graphite, which is related to the stretching vibration of CC from unoxidized graphitic domains.28 Compared to GO, two significant peaks are observed for SGO at 1113 and 1017 cm−1, which are assigned to the respective symmetric and asymmetric stretching modes of OSO in –SO3H groups; the small peak appearing at 813 cm−1 is corresponding to the stretching vibration of p-disubstituted phenyl groups. The apparent reduction in stretching vibration of CO at 1718 cm−1 in FT-IR spectrum of SGO indicate the effective reduction of GO by present method. The presence of Fe3O4 in Fe3O4–SGO composite is confirmed from the bands found at 576 and 887 cm−1, owing to the stretching modes of Fe–O, Fe–O–C, respectively. TGA profile of prepared nanostructures is given in the Fig. 2d. Two significant degradations are observed for GO as follows: (i) from 30 to 100 °C and (ii) from 100 to 203 °C. The former one is due to the removal of moisture mainly physisorbed free water molecules. The later is because of elimination of oxygen-labile functional groups such as carboxyl, epoxy, hydroxyl and so forth. The quantity of oxygen-labile functional groups in GO is about 89.51% according to TGA analysis and the principal reason for excess weight loss at 200 °C is decomposition of carbon atoms of GO together with oxygen during thermal combustion process.42,46 By comparison, an improved thermal stability is observed for SGO with a residual weight of 18.23%, owing to the partial removal of oxygen-labile functional groups via chemical reduction. Notably, the second weight loss in SGO from 150 to 270 °C is 32.91% which verifies the sulfonation of GO to SGO. The removal of oxygen-related functionalities of GO by NaBH4 before sulfonation may lead to incorporate excess number of aryl diazonium salt of sulfanilic acid on GO. The thermal stability of SGO is further improved after anchoring Fe3O4 nanoparticles, as evidenced by higher residual weight of Fe3O4–SGO i.e., 86.03% and is due to the ceramic behavior of the corresponding composite given via Fe3O4 nanoparticles. The hydrolytic stability of Fe3O4 and Fe3O4–SGO composite was measured in 1 M acidic solution at 70 °C to understand the dissolution of Fe3O4; details including figures and explanation, are provided in ESI.†
O (from carboxylic acid group), C–OH (from carboxylic acid group) and C–O (from epoxy group) stretching, respectively.43–45 The peak appearing at 1579 cm−1 in GO is consistent with graphite, which is related to the stretching vibration of CC from unoxidized graphitic domains.28 Compared to GO, two significant peaks are observed for SGO at 1113 and 1017 cm−1, which are assigned to the respective symmetric and asymmetric stretching modes of OSO in –SO3H groups; the small peak appearing at 813 cm−1 is corresponding to the stretching vibration of p-disubstituted phenyl groups. The apparent reduction in stretching vibration of CO at 1718 cm−1 in FT-IR spectrum of SGO indicate the effective reduction of GO by present method. The presence of Fe3O4 in Fe3O4–SGO composite is confirmed from the bands found at 576 and 887 cm−1, owing to the stretching modes of Fe–O, Fe–O–C, respectively. TGA profile of prepared nanostructures is given in the Fig. 2d. Two significant degradations are observed for GO as follows: (i) from 30 to 100 °C and (ii) from 100 to 203 °C. The former one is due to the removal of moisture mainly physisorbed free water molecules. The later is because of elimination of oxygen-labile functional groups such as carboxyl, epoxy, hydroxyl and so forth. The quantity of oxygen-labile functional groups in GO is about 89.51% according to TGA analysis and the principal reason for excess weight loss at 200 °C is decomposition of carbon atoms of GO together with oxygen during thermal combustion process.42,46 By comparison, an improved thermal stability is observed for SGO with a residual weight of 18.23%, owing to the partial removal of oxygen-labile functional groups via chemical reduction. Notably, the second weight loss in SGO from 150 to 270 °C is 32.91% which verifies the sulfonation of GO to SGO. The removal of oxygen-related functionalities of GO by NaBH4 before sulfonation may lead to incorporate excess number of aryl diazonium salt of sulfanilic acid on GO. The thermal stability of SGO is further improved after anchoring Fe3O4 nanoparticles, as evidenced by higher residual weight of Fe3O4–SGO i.e., 86.03% and is due to the ceramic behavior of the corresponding composite given via Fe3O4 nanoparticles. The hydrolytic stability of Fe3O4 and Fe3O4–SGO composite was measured in 1 M acidic solution at 70 °C to understand the dissolution of Fe3O4; details including figures and explanation, are provided in ESI.†
 |
| | Fig. 1 FE-SEM images of (a) GO, (b) SGO and (c) Fe3O4–SGO; TEM images of (d) GO, (e) SGO and (f) Fe3O4–SGO and inset (f) SAED pattern. | |
 |
| | Fig. 2 (a) XRD patterns, (b) Raman spectra, (c) FT-IR spectra and (d) TGA curves of GO, SGO and Fe3O4–SGO. | |
6.2. Characterizations of membranes
6.2.1. Morphological properties.
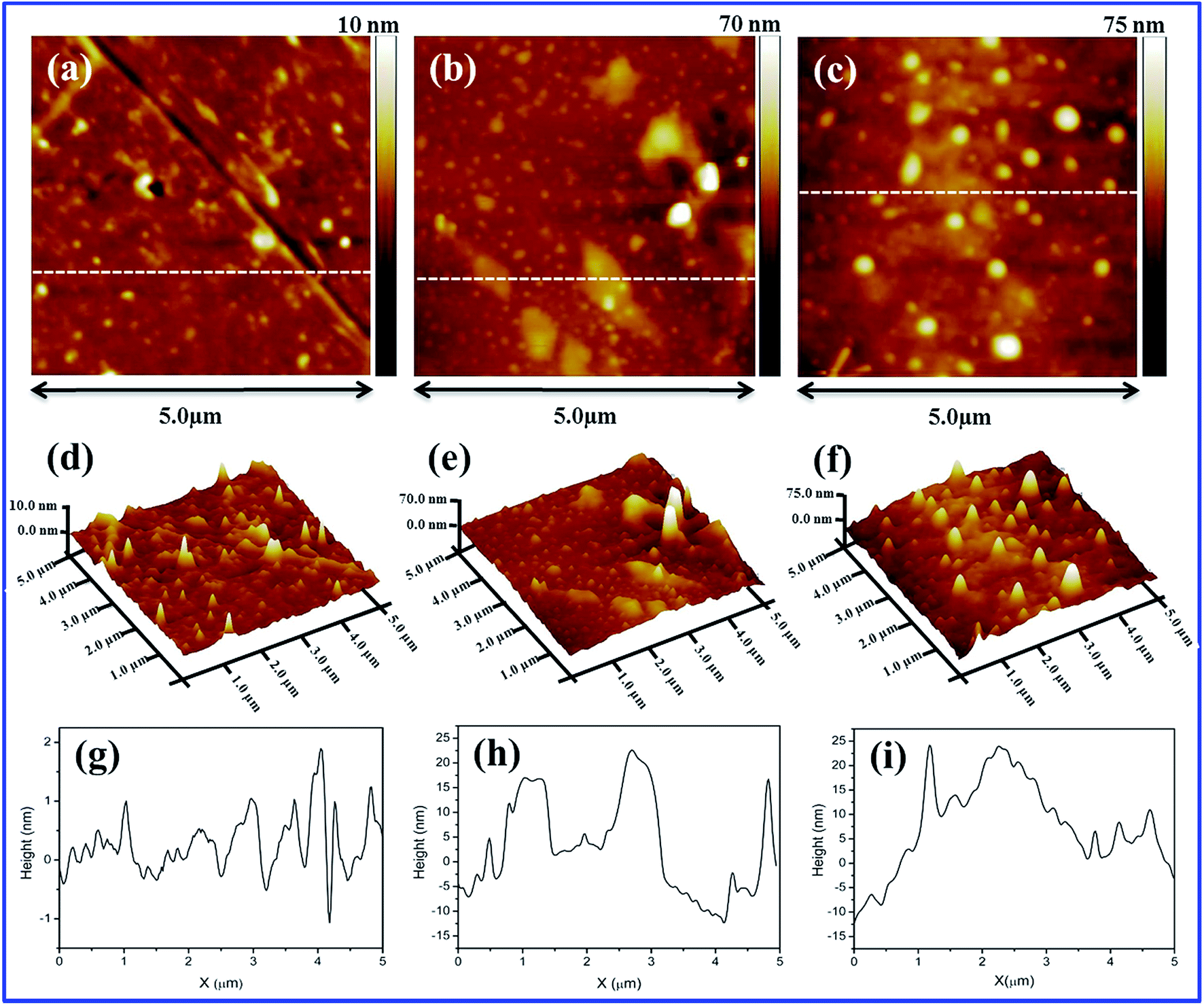
The internal morphology and filler dispersion of the composite membranes are significant factors in determining macroscopic properties such as water uptake, proton conductivity, mechanical strength and gas permeability.28 Surface and cross-sectional SEM images of pristine recast Nafion, Nafion/GO-3 and Nafion/SGF-3 are displayed in Fig. 3a–f. It can be observed that pristine Nafion exhibits a clean and smooth surface (Fig. 3a), while incorporation of GO (Fig. 3b) or Fe3O4–SGO (Fig. 3c) results in surface roughness, which is due to the crumpled behavior of fillers. Compared to pristine Nafion (Fig. 3d), the obvious wrinkles are observed throughout the cross section of Nafion/GO-3 (Fig. 3e) and Nafion/SGF-3 (Fig. 3f) membranes, which ensure the dispersion of fillers. Meanwhile, the Fe3O4–SGO fillers show a more uniform dispersion than GO within Nafion matrix as a result of strong interfacial interactions (hydrogen bonding) between –OH, –O–, –CO2H and –SO3H groups of Fe3O4–SGO and –SO3H groups of Nafion, thereby increasing the mutual compatibility between fillers and Nafion matrix. In such a way, the co-continuous proton conducting channels are generated across the Nafion/SGF-3 membrane, which facilitate the proton conductivity. Scheme S1 (ESI†) shows the possible interactions exerted between Nafion and Fe3O4–SGO in composite membrane. The water contact angle of Nafion/SGF-3 membrane is 87.21° (Fig. 3c-inset), indicating the presence of hydrophilic surfaces due to the presence of polar groups such as –OH, –O– –CO2H and –SO3H. The contact angle of the pristine Nafion membrane was 103.56° (Fig. 3a-inset). However, this value was reduced to 87.21° after incorporating 3 wt% Fe3O4–SGO, indicating an enhancement in hydrophilicity due to an increase in the aforementioned polar groups. The peak found at about 6.3 keV (due to Fe) in the EDX spectrum of Nafion/SGF-3 membrane (Fig. 3h) confirms the incorporation of Fe3O4–SGO in the Nafion matrix. The density and distribution of Fe3O4–SGO in the Nafion matrix were also examined by quantitative energy dispersive elemental mapping. It can be seen that Fe3O4–SGO is uniformly distributed on the entire cross section of Nafion rather than only being located at a particular site (Fig. 4f), which helps to improve the proton conductivity and mechanical stability of composite membranes. 2D and 3D AFM topographies and lines profiles of pristine recast Nafion, Nafion/GO-3 and Nafion/SGF-3 membranes are provided in Fig. 5. The image size of each membrane is 5 μm × 5 μm. Bright and dark regions in the topographies represent the height variations on the membranes. The topography exhibits significant morphological difference on the Nafion membrane before and after incorporating GO or Fe3O4–SGO. Pristine Nafion exhibits a detailed structure with randomly distributed irregular spikes (approximately 100–260 nm width and 1–13 nm height), while the subsidence and protrusions are distributed on the surface of Nafion/GO-3 (approximately 100–960 nm width and 1–90 nm height) and Nafion/SGF-3 (approximately 100–600 nm width and 1–86 nm height) membranes. Detailed structure with spikes might be partially hidden on the surface of composite membranes because the subsidence and protrusions are relatively higher than spikes. The RMS roughness of above specified membranes was calculated by eqn (1). The RMS roughness of pristine Nafion is approximately 1 nm, similar to uniform polymer membrane reported in the literature.17 However, the RMS roughness of Nafion/GO-3 and Nafion/SGF-3 membranes is approximately 10 and 12 nm, respectively. It is noteworthy that extended surface area of GO or Fe3O4–SGO with wrinkled features is responsible for high RMS roughness of composite membranes. This appears to be advantageous toward more water uptake capacity and high compatibility with the electrode during fabrication of membrane electrode assembly.
 |
| | Fig. 3 Surface SEM images of membranes of (a) pristine recast Nafion, (b) Nafion/GO-3 and (c) Nafion/SGF-3; insets of (a), (b) and (c) corresponding water contact angle images; cross-sectional SEM images of membranes of (d) pristine recast Nafion, (e) Nafion/GO-3 and (f) Nafion/SGF-3; EDX spectra of (g) pristine recast Nafion and (h) Nafion/SGF-3. | |
 |
| | Fig. 4 (a) SEM image of selected area of Nafion/SGF-3; corresponding elemental mappings of (b) carbon, (c) oxygen, (d) fluorine, (e) sulfur, (f) iron and (g) overlapping of elements. | |
 |
| | Fig. 5 2D AFM images of membranes of (a) pristine recast Nafion, (b) Nafion/GO-3 and (c) Nafion/SGF-3; 3D AFM images of membranes of (d) pristine recast Nafion, (e) Nafion/GO-3 and (f) Nafion/SGF-3; line profiles of membranes of (g) pristine recast Nafion, (h) Nafion/GO-3 and (i) Nafion/SGF-3. | |
6.2.2. Thermomechanical stabilities.
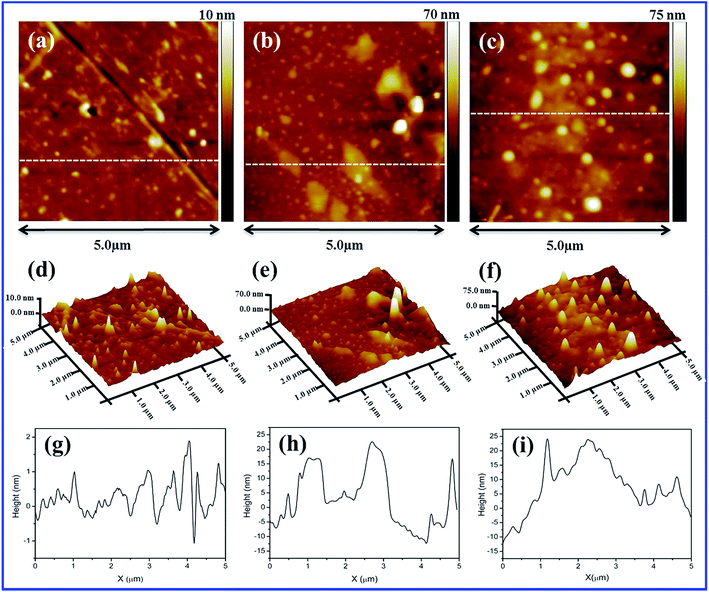
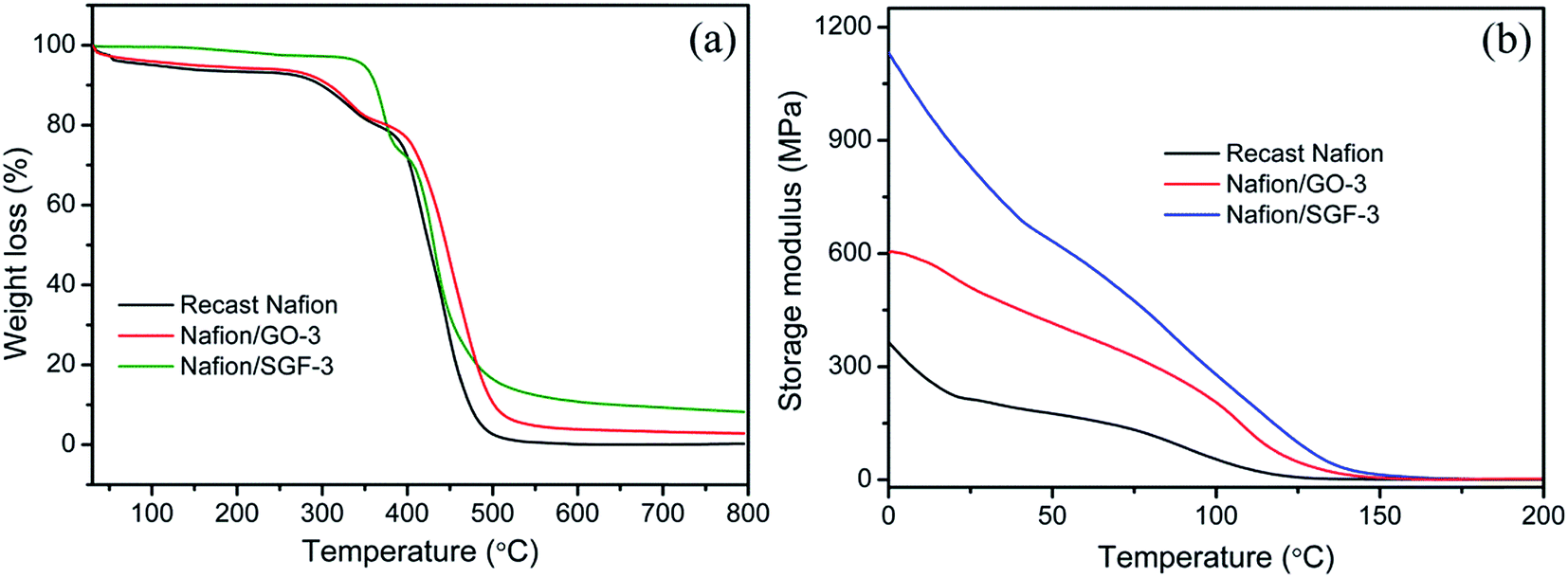
Thermal stability of pristine recast Nafion, Nafion/GO-3 and Nafion/SGF-3 was studied by TGA to explore the effects of filler–polymer interactions, and obtained thermographs are demonstrated in Fig. 6a. Pristine Nafion and composite membranes depicted similar three step weight drops: (i) from 30 to 100 °C that is due to the loss of free water molecules from membranes, (ii) from 240 to 400 °C that is due to the decomposition of –SO3H groups in the side chain of the membranes and (iii) above 400 °C that is because of disintegration of the aliphatic backbones of polymer. It is noteworthy that thermal degradation of composite membranes delays throughout the study and is more pronounced in the temperature range from 400 to 800 °C. Composite membranes showed lower weight drops than pristine Nafion membrane, demonstrating that the addition of GO or Fe3O4–SGO effectively improved the thermal stability of Nafion by prohibiting the movement of aliphatic backbones through interfacial interactions. At 800 °C, the pristine Nafion showed a residual weight of 0.26%, whereas Nafion/GO-3 and Nafion/SGF-3 membranes exhibited the residual weights of 2.89 and 8.24%, respectively. The higher residual weight in composite membranes is a clear manifestation for higher thermal stability of composite membrane containing GO or Fe3O4–SGO. Temperature dependent mechanical properties of pristine recast Nafion and optimized Nafion/GO-3 and Nafion/SGF-3 composite membranes are shown in Fig. 6b. It was found that the maximum value of storage modulus to be 1135 MPa for Nafion/SGF-3 membrane, which is almost 1.87 fold higher than that of Nafion/GO-3 membrane and 3.16 fold higher than that of pristine Nafion membrane. The enhancement in the mechanical strength of Nafion/SGF-3 membrane indicates strong hydrogen bonding interactions due the presence of common –SO3H groups in SGO and Nafion, which strictly restrict the reorganization of polymer chains and toughen the membrane up to a reasonable margin. The Fe3O4 nanoparticles over the SGO sheets help to increase adhesion of fillers with host matrix, which further toughens the membrane. Accordingly, Nafion/SGF-3 composite membrane is extremely beneficial for high temperature and low humidity PEMFC operation.
 |
| | Fig. 6 (a) TGA and (b) DMA curves of pristine recast Nafion and composite membranes. | |
6.2.3. Water uptake, dimensional stability, hydration number and IEC.
Percentage of mass, length and thickness change in membrane after immersing in water at 60 °C for 24 h referred to as the water uptake and dimensional stability as shown in Table 1. It can be seen that pristine Nafion membrane has 26.34% water uptake, whereas Nafion/GO-3 and Nafion/SGF-3 membranes have 31.24 and 35.63% water uptake, respectively. By incorporating GO or Fe3O4–SGO filler, the membrane surface turns to more hydrophilic and is responsible to higher water uptake of corresponding composite membrane. The formation of hydrogen bonds between –OH, –O– and –CO2H groups of high surface fillers and free water molecules help to increase water uptake of Nafion/GO-3 and Nafion/SGF-3 membranes. Then, improvement in water uptake of the GO and Fe3O4–SGO based membranes can increase the proton conductivity of corresponding membranes. Water uptake of Nafion/SGF-X membrane is found to be increased from 26.34 to 35.63% upon increasing the filler from 0 to 3 wt%, however, kinetics of water uptake decrease in 3 wt%, which is probably due to the increasing adhesion at polymer–filler interface. The addition of GO or Fe3O4–SGO also improved the in-plane dimensional stability of membrane during hydration without affecting the water uptake significantly. Pristine Nafion membrane exhibits 17.31% swelling in length direction, whereas the membrane containing optimized wt% GO or Fe3O4–SGO show 11.22 and 11.88% swelling, respectively. It is assumed that during solution casting, the fillers are perpendicularly oriented to the cross section of the membranes, which restricts the relaxation of polymer chains in lateral direction. Then, improvement in in-plane dimensional stability can reduce the delamination of composite membranes from electrodes during PEMFC operation and enhance the cell performance. An increase in swelling in the thickness direction of membrane was also observed upon addition of fillers. This sort of membrane swelling can cause increased pressure between MEA and bipolar plates; however, it can be compensated by compression of the bipolar plates. Collectively, the improvement in water retention capacity without significantly affecting the dimensional stability enables the composite membranes to be well-suited for repeatable PEFC operations. IEC value is considered to correspond to the density of functional groups in the PEMs.47,48Table 1 lists the IEC value of all prepared membranes. IEC for the pristine Nafion membrane was 0.89 meq. g−1, which was lower than the IECs of Nafion/GO-3 (1.03 meq. g−1), and Nafion/SGF-3 (1.36 meq. g−1). The IEC of the Nafion membrane increases with addition of GO or Fe3O4–SGO. This is due to the SGO contained high density of –SO3H groups and Fe3O4 contained high density of –OH groups. The number of water molecules absorbed per unit volume of –SO3H groups is defined as hydration number (λ).36,40,49 This was calculated using the eqn (10):| |  | (10) |
where 18.01 is the molecular weight of water in g mol−1.
Table 1 Ion exchange capacity, water uptake, length change, thickness change, hydration number and proton conductivity values of different composite membranes
| Membrane |
Ion exchange capacity (meq. g−1) |
Water uptake (%) at 60 °C |
Length change (%) at 60 °C |
Thickness change (%) at 60 °C |
Hydration number at 60 °C |
| Recast Nafion |
0.89 |
26.34 ± 2 |
17.31 ± 1 |
6.13 ± 2 |
16.40 |
| Nafion/SGF-0.5 |
0.93 |
30.71 ± 3 |
15.41 ± 2 |
9.18 ± 1 |
18.33 |
| Nafion/SGF-1.5 |
1.12 |
33.36 ± 2 |
14.18 ± 3 |
11.95 ± 2 |
16.51 |
| Nafion/SGF-3 |
1.36 |
35.63 ± 3 |
11.88 ± 2 |
15.79 ± 3 |
14.48 |
| Nafion/GO-3 |
1.03 |
31.24 ± 1 |
11.22 ± 3 |
13.35 ± 2 |
16.79 |
It can be observed that by incorporating 3 wt% GO into the Nafion, the λ value increased from 16.40 to 16.79, which is again due to the high density of functional groups in the composite membranes provided by high surface GO. However, λ value decreased from 16.40 to 14.48 by incorporating 3 wt% Fe3O4–SGO into the Nafion. The decrease in λ of Nafion/SGF-3 membrane can be attributed to the compact structure of membrane due to hydrogen bonding between surface –OH groups of Fe3O4 and –F and –SO3H groups of Nafion.
6.2.4. Proton conductivity and Arrhenius plot.
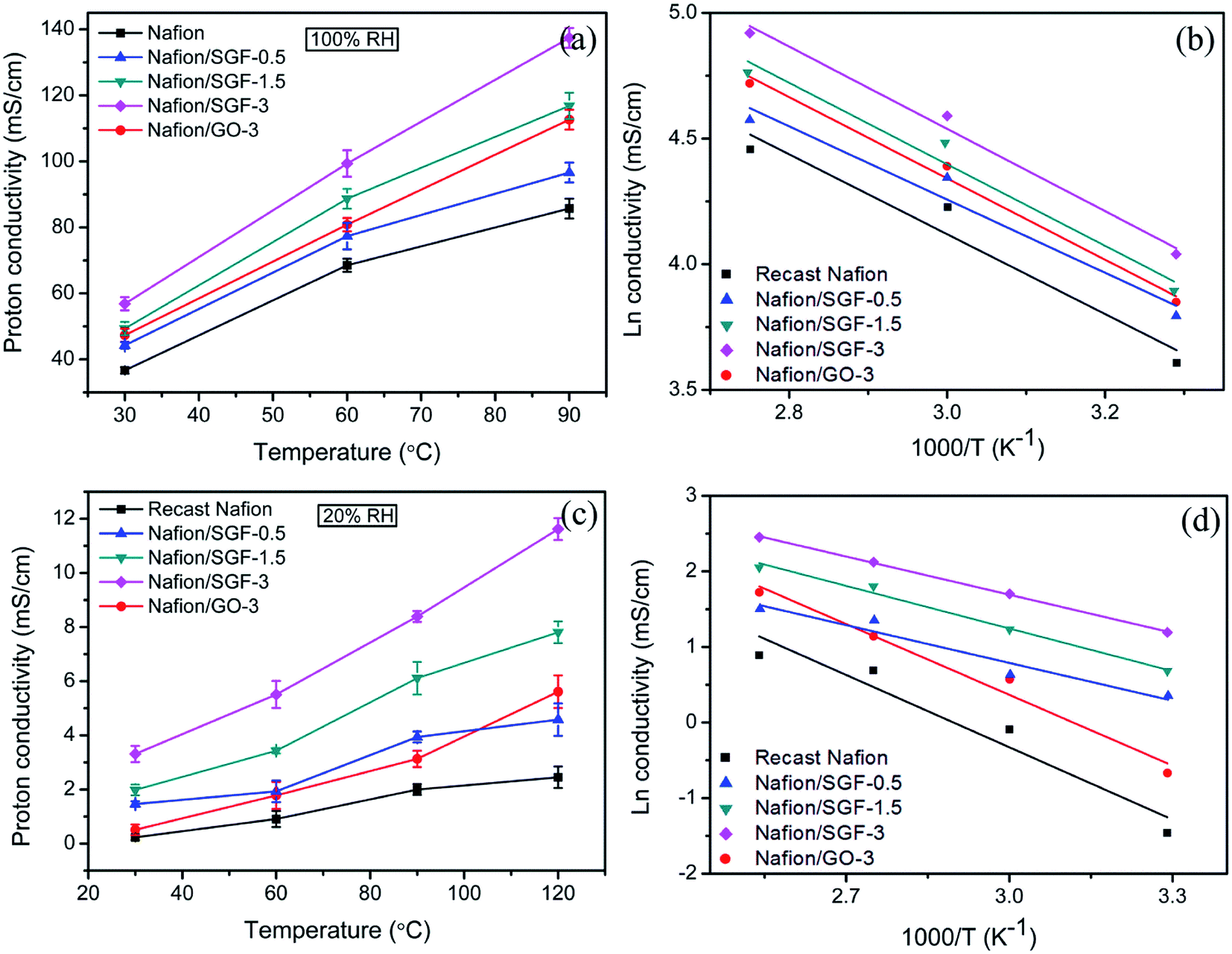
As a key parameter to PEMs, the proton conductivities of membrane specimens were measured at different temperatures under 100% and 20% RH. Fig. 7a shows the all obtained proton conductivities of membrane specimens under 100% RH. It can be observed that pristine Nafion specimen exhibited a proton conductivity of 36.67 mS cm−1 at 30 °C, which increase with temperature and reached a peak value of 85.71 mS cm−1 at 90 °C. The increased proton conductivity obtained is due to the rapid proton movement at high temperature. The transport of protons is relied on the two different mechanisms: (i) vehicular mechanism and (ii) Grotthuss mechanism.50,51 In GO based membrane, the –OH and –CO2H groups can attach to the free water molecules and increase the proton transport of membrane with vehicular mechanism. In Fe3O4–SGO based membrane, the –SO3H groups of SGO increase the bound water in membrane and encourage the proton diffusion from one ionic site (H3O+SO3−) to another ionic site via Grotthuss mechanism, and also hydrogen bonding between the –OH groups of Fe3O4 nanoparticles and free water molecules in membrane encourage the proton conductivity via vehicular mechanism. Since, incorporation of high surface GO or Fe3O4–SGO in Nafion matrix is found to be enhanced the water uptake and IEC up to a reasonable margin, which consequently enhance the proton conductivity of composite membranes. Peak proton conductivities delivered by the Nafion/GO-3 and Nafion/SGF-3 specimens are 112.63 and 137.39 mS cm−1 at 90 °C, which are 1.31 and 1.61 folds higher than that of a pristine Nafion membrane, respectively. On the other side, peak proton conductivity of Nafion/SGF-X specimens is found to be increased from 85.71 to 137.39 mS cm−1 on increasing the Fe3O4–SGO content from 0 to 3 wt%, which may be due to the increase number of –SO3H groups per cluster volume of a domain. Fig. 7c shows all obtained proton conductivities of membrane specimens under 20% RH. These conductivity values are many folds lower in relation to the conductivity values obtained under 100% RH, which demonstrates the critical role of water molecules in proton conduction. At 120 °C, the peak proton conductivity yielded by the pristine Nafion, Nafion/GO-3 and Nafion/SGF-3 specimens is 2.45, 5.61 and 11.62 mS cm−1, respectively. The lower conductivity of Nafion is due to the severe membrane dehydration caused by insufficient volume of bound water molecules, which could be successfully remediated by incorporating the GO or Fe3O4–SGO into the membrane.
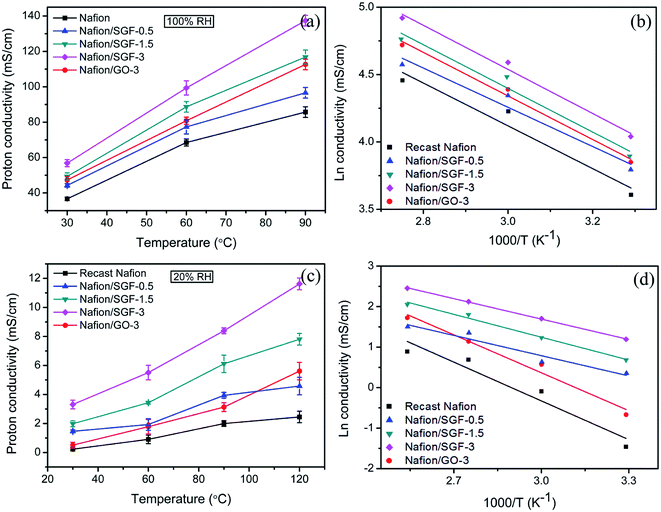 |
| | Fig. 7 Temperature variant proton conductivities of pristine recast Nafion and composite membranes (a) under 100% RH and (c) under 20% RH; corresponding Arrhenius plots of proton conductivities (b) under 100% RH and (d) under 20% RH. | |
The activation energy (Ea) required for the protons to travel through fabricated membranes was estimated using the Arrhenius eqn (11), as reported in the ref. 52 and 53.
| | 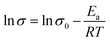 | (11) |
here,
σ and
σ0 are the values of the conductivity and pre-exponential factor, respectively, in mS cm
−1,
Ea is the activation energy required for protons to travel in kJ mol
−1,
R is the gas constant in J mol
−1 K
−1, and
T is the absolute temperature in K. The
Ea of Nafion membrane decreased after incorporating Fe
3O
4–SGO (
Table 2), demonstrating the high density of –SO
3H and other hydrophilic groups were effectually dissociated throughout the Nafion/SGF-X membranes. Considering the
Ea for vehicular mechanism is 16.4 kJ mol
−1 while that for Grotthuss mechanism is between 9.65 and 38.59 kJ mol
−1.
54,55 The results obtained show that both vehicular and Grotthuss mechanisms coexist in the membranes under 100% RH, whereas Grotthuss mechanism dominates more under low humidity, due to lack of free water molecules.
Table 2 Proton conductivity and activation energy values of different composite membranes
| Membrane |
Proton conductivity (mS cm−1) at 60 °C |
Proton conductivity (mS cm−1) at 120 °C |
Activation energy (kJ mol−1) under 100% RH |
Activation energy (kJ mol−1) under 20% RH |
| Recast Nafion |
68.53 ± 2 |
2.45 ± 0.4 |
14.73 |
26.52 |
| Nafion/SGF-0.5 |
77.33 ± 4 |
4.58 ± 0.6 |
13.55 |
25.95 |
| Nafion/SGF-1.5 |
86.66 ± 3 |
7.81 ± 0.4 |
12.05 |
23.21 |
| Nafion/SGF-3 |
99.34 ± 4 |
11.62 ± 0.4 |
10.16 |
21.41 |
| Nafion/GO-3 |
80.82 ± 2 |
5.61 ± 0.6 |
13.28 |
19.22 |
6.2.5. Electrical conductivity.
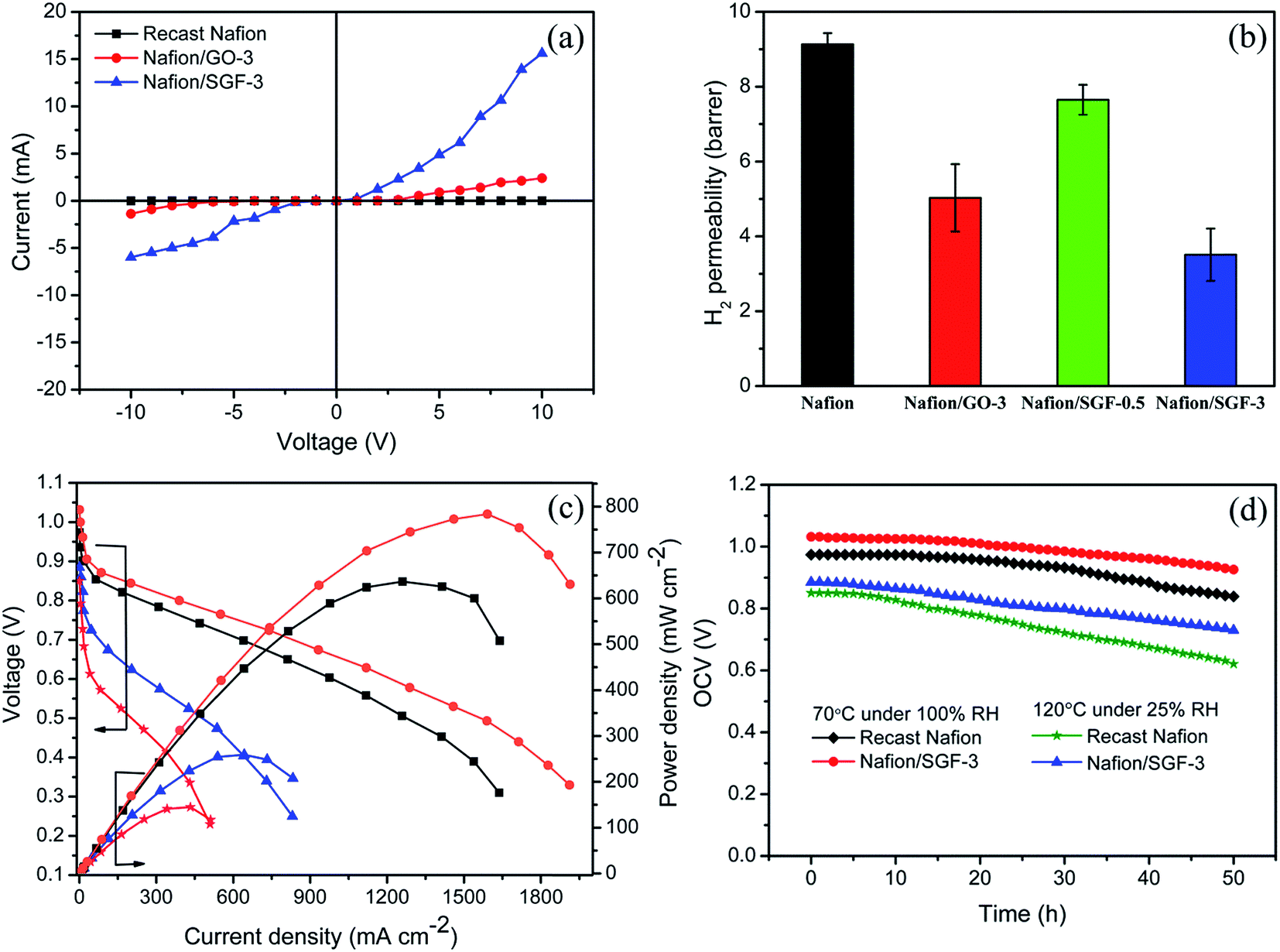
Fig. 8a shows the current–voltage (i–v) characteristics of the pristine recast Nafion, Nafion/GO-3 and Nafion/SGF-3 membranes. Compared with pristine Nafion, there is significant change observed in i–v characteristics of the composite membranes. Nafion membrane reveals negligible currents at all applied voltages from −10 to 10 V, whereas current of the composite membranes increase as increase the voltage, due to the electrically conducting fillers i.e., GO and Fe3O4–SGO. The calculated electrical conductivity for the Nafion/SGF-3 membrane (0.37 mS cm−1) is 3.08 fold higher than that of Nafion/GO-3 (0.12 mS cm−1) and 17.61 fold higher than that of pristine Nafion membrane (0.021 mS cm−1), due to high electrical conductivity and percolation threshold of Fe3O4–SGO.2 However, the obtained electrical conductivity of the Nafion/Fe3O4–SGO membrane is generally less i.e., lesser than the level which can affect the open circuit voltage of membrane during PEMFC operation.
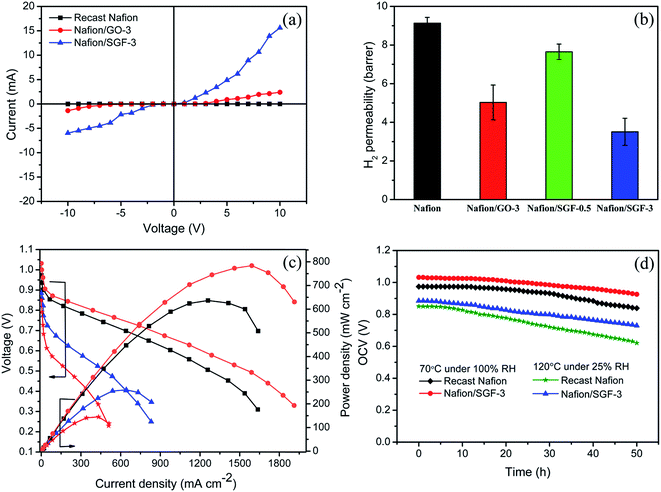 |
| | Fig. 8 (a) Current–voltage (i–v) characteristics of pristine recast Nafion and composite membranes; (b) H2 permeability of pristine recast Nafion and composite membranes; (c) performances of H2/O2 PEMFC at 70 °C under 100% RH with ( ) pristine recast Nafion and ( ) pristine recast Nafion and ( ) Nafion/SGF-3 and at 120 °C under 25% RH with ( ) Nafion/SGF-3 and at 120 °C under 25% RH with ( ) pristine recast Nafion and ( ) pristine recast Nafion and ( ) Nafion/SGF-3; (d) stability test of pristine recast Nafion and Nafion/SGF-3 membrane in fuel cell configuration both at 70 °C under 100% RH and 120 °C under 25% RH. ) Nafion/SGF-3; (d) stability test of pristine recast Nafion and Nafion/SGF-3 membrane in fuel cell configuration both at 70 °C under 100% RH and 120 °C under 25% RH. | |
6.2.6. H2 permeability.
The H2 permeability data for pristine Nafion and composite membranes are shown in Fig. 8b. The higher H2 permeability indicates higher H2 crossover through the membrane. H2 crossover for the Nafion/GO-3 membrane was 9.13 barrer, which was only 52.86% that of pristine Nafion membrane. The reduction in H2 crossover was ascribed to the presence of GO, which made the membrane compact and dense. In the case of Nafion/SGF-3 membrane, the Fe3O4–SGO fillers were tightly wrapped with the aliphatic chains of polymers through interfacial hydrogen bonding, which generate complex structures throughout the cross-section of the membrane. These complex structures exhibit a tortuous path for the diffusion of H2 gas molecules. Therefore, the Nafion/SGF-3 membrane exhibits a further low H2 crossover compared to Nafion/GO-3 membrane.
6.2.7. PEMFC performance.
Fig. 8c shows the polarization and power density curves of PEMFC containing pristine Nafion and Nafion/SGF-3 membranes. The PEMFC performances were measured: (i) at 70 °C under 100% RH and (ii) at 120 °C under 25% RH. At the first condition, the open circuit voltage (OCV) of Nafion/SGF-3 membrane begins from above 1 V, indicating that there is no electronic conductivity effect due to the existence of a small amount of Fe3O4–SGO in Nafion matrix. Peak power density of 636.58 mW cm−2 at a load current density of 1258 mA cm−2 was attained by the PEMFC containing pristine Nafion membrane, whereas the PEMFC containing Nafion/SGF-3 exhibited a peak power density of 783.69 mW cm−2 at a load current density of 1589 mA cm−2. The high performance of Nafion/SGF-3 membrane is primarily due to the high proton conductivity along with enhanced water uptake, attained by high density of –SO3H and –OH groups on Fe3O4–SGO. Also, improvements were found in the ohmic and mass transport regions of the Nafion/SGF-3 membrane when compared to the pristine Nafion, demonstrating that this membrane has high efficacy for the free flow of protons and supply of ions to the cathode reaction sites. At the second condition (nearer dry condition), the PEMFC containing pristine Nafion and Nafion/SGF-3 yielded peak power densities of 144.89 and 258.82 mW cm−2 at the load current densities of 431.36 and 640.73 mA cm−2, respectively. In pristine Nafion case, due to the limited availability of water in membrane, the electro-osmotic drag of water from the anode to cathode and inadequate back diffusion of water from the cathode to anode cause the MEA to dehydrate. MEA dehydration results in an increase of ohmic resistance, leading to reduced cell performance. By contrast, the Nafion/SGF-3 membrane hold a greater amount of water due to the presence of Fe3O4–SGO, which keep the membrane sufficiently wet under low humidity and enable the corresponding PEMFC to sustain without losing the performance excessively. Table S1 (ESI†) summarized the various Nafion based membrane performance results reported in literatures, for comparison with present study.1,4,17,56–66
Accordingly, the present study provides a novel Nafion composite membrane with Fe3O4–SGO filler particles which helps to improve proton conductivity and reduce H2 gas crossover. However, O2 gas crossover from the cathode to the anode through PEM is a crucial drawback that leads to generation of H2O2 and consequently membrane degradation. For the determination of O2 gas crossover of prepared membranes, OCV of the studied membranes-equipped PEMFC was examined for 50 h at 70 °C under 100% RH (Fig. 8d). After 50 h of operation, the OCV of the pristine Nafion and Nafion/SGF-3 membrane were found to decrease from 0.97 to 0.83 V and 1.03 to 0.92 V, respectively. The lower OCV reduction rate of composite membrane indicates its good barrier property against O2 gas diffusion from the cathode to anode, which can be attributed to the presence of Fe3O4–SGO. To address effect of high temperature operation, OCV of the membranes-equipped PEMFC was examined at 120 °C under 25% RH. By comparison, the accelerated OCV degradations are observed for both pristine Nafion and Nafion/SGF-3 membranes, which represent the rapid chemical degradation of membranes during high temperature operation.
7. Conclusion
Nafion composite membranes, incorporating different wt% of Fe3O4–SGO, were successfully fabricated via a facile solution casting method. In the composite membranes, the number of –SO3H groups per cluster volume domain was increased by SGO, and the hydrogen bonding networks between –SO3H groups were extended by Fe3O4. Because of the high density of –SO3H, –CO2H and –OH groups on Fe3O4–SGO, the water uptake, ion exchange capacity and proton conductivity of the composite membrane significantly increased. The incorporation of Fe3O4–GO restructured the aliphatic chains of Nafion through hydrogen bonding, which strengthened the membrane mechanically and thermally by 3.16 and 31.6 fold, respectively. By the combined efforts of high surface area and strong-acid functionalities of Fe3O4–GO, composite membrane harvested number of protons and ameliorates the PEMFC performance at high temperature and low humidity condition compared to the PEMFC containing pristine Nafion membrane.
Conflicts of interest
The all authors declare that they have no conflict of interest.
Acknowledgements
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20164030201070). This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (NRF-2017R1A2B4005230).
References
- H. Zarrin, D. Higgins, Y. Jun, Z. Chen and M. Fowler, J. Phys. Chem. C, 2011, 115, 20774–20781 CrossRef CAS.
- M. Vinothkannan, A. R. Kim, G. Gnanakumar, J. M. Yoon and D. J. Yoo, RSC Adv., 2017, 7, 39034–39048 RSC.
- K. D. Papadimitriou, F. Paloukis, S. G. Neophytides and J. K. Kallitsis, Macromolecules, 2011, 44, 4942–4951 CrossRef CAS.
- K. Ketpang, K. Lee and S. Shanmugam, ACS Appl. Mater. Interfaces, 2014, 6, 16734–16744 CrossRef CAS.
- A. M. Baker, L. Wang, W. B. Johnson, A. K. Prasad and S. G. Advani, J. Phys. Chem. C, 2014, 118, 26796–27802 CrossRef CAS.
- S. Neelakandan, N. K. Jacob, P. Kanagaraj, R. M. Sabarathinam, A. Muthumeenal and A. Nagendran, RSC Adv., 2016, 6, 51599–51608 RSC.
- S. Zhou, S. D. Hai and D. Kim, Fuel Cells, 2012, 12, 589–598 CrossRef CAS.
- S. G. Peera, S. Meenakshi, K. H. Gopi, S. D. Bhat, P. Sridhar and S. Pitchumani, RSC Adv., 2013, 3, 14048–14056 RSC.
- H. Beydaghi, M. Javanbakht, A. Bagheri, P. Salarizadeh, H. G. Zahmatkesh, S. Kashefi and E. Kowsari, RSC Adv., 2015, 5, 74054–74064 RSC.
- V. Parthiban, S. Akula, S. G. Peera, N. Islam and A. K. Sahu, Energy Fuels, 2016, 30, 725–734 CrossRef CAS.
- H. Zhang, T. Zhang, J. Wang, F. Pei, Y. He and J. Liu, Fuel Cells, 2013, 13, 1155–1165 CrossRef CAS.
- V. Tricoli and F. Nannetti, Electrochim. Acta, 2003, 48, 2625–2633 CrossRef CAS.
- E. Chalkova, C. Wang, S. Komarneni, J. K. Lee, M. V. Fedkin and S. N. Lvov, ECS Trans., 2009, 25, 1141–1150 CAS.
- P. Kalappa and J. H. Lee, Polym. Int., 2007, 56, 371–375 CrossRef CAS.
- Q. Yang, J. Liu, J. Yang, M. P. Kapoor, S. Inagaki and C. Li, J. Catal., 2004, 228, 265–272 CrossRef CAS.
- A. R. Kim, M. Vinothkannan and D. J. Yoo, Composites, Part B, 2017, 130, 103–118 CrossRef CAS.
- A. K. Sahu, K. Ketpang, S. Shanmugam, O. Kwon, S. Lee and H. Kim, J. Phys. Chem. C, 2016, 120, 15855–15866 CrossRef.
- G. Rambabu, N. Nagaraju and S. D. Bhat, Chem. Eng. J., 2016, 306, 43–52 CrossRef CAS.
- Y. L. Liu, Y. H. Su, C. M. Chang, Suryani, D. M. Wang and J. Y. Lai, J. Mater. Chem., 2010, 20, 4409–4416 RSC.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS.
- W. Chen, L. Duan, L. Wang and D. Zhu, Environ. Sci. Technol., 2008, 42, 6862–6868 CrossRef CAS.
- D. H. Kim, J. H. Ahn, W. M. Choi, H. S. Kim, T. H. Kim, J. Song, Y. Y. Huang, Z. Liu, C. Lu and J. A. Rogers, Science, 2008, 320, 507–511 CrossRef CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS.
- X. Wang, L. Zhi and K. Mullen, Nano Lett., 2008, 8, 323–326 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H. S. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS.
- M. Vinothkannan, A. R. Kim, K. S. Nahm and D. J. Yoo, RSC Adv., 2016, 6, 108851–108863 RSC.
- M. R. Karim, K. Hatakeyama, T. Matsui, H. Takehira, T. Taniguchi, M. Koinuma, Y. Matsumoto, T. Akutagawa, T. Nakamura, S. Noro, T. Yamada, H. Kitagawa and S. Hayami, J. Am. Chem. Soc., 2013, 135, 8097–8100 CrossRef CAS.
- H. Beydaghi, M. Javanbakht and E. Kowsari, Ind. Eng. Chem. Res., 2014, 53, 16621–16632 CrossRef CAS.
- G. Gnana kumar, C. J. Kirubaharan, D. J. Yoo and A. R. Kim, Int. J. Hydrogen Energy, 2016, 41, 13208–13219 CrossRef CAS.
- J. Salamon, Y. Sathishkumar, K. Ramachandran, Y. S. Lee, D. J. Yoo, A. R. Kim and G. G. Kumar, Biosens. Bioelectron., 2015, 64, 269–276 CrossRef CAS.
- H. Beydaghi and M. Javanbakht, Ind. Eng. Chem. Res., 2015, 54, 7028–7037 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- K. H. Lee, J. Y. Chu, A. R. Kim, K. S. Nahm, C. J. Kim and D. J. Yoo, J. Membr. Sci., 2013, 434, 35–43 CrossRef CAS.
- S. Gahlot and V. Kulshrestha, ACS Appl. Mater. Interfaces, 2015, 7, 264–272 CrossRef CAS.
- L. Zhao, Y. Li, H. Zhang, W. Wu, J. Liu and J. Wang, J. Power Sources, 2015, 286, 445–457 CrossRef CAS.
- A. R. Kim, M. Vinothkannan and D. J. Yoo, Int. J. Hydrogen Energy, 2017, 42, 4349–4365 CrossRef CAS.
- A. Ghosh, S. Basu and A. Verma, Fuel Cells, 2013, 13, 355–363 CrossRef CAS.
- S. Gahlot, P. P. Sharma, V. Kulshrestha and P. K. Jha, ACS Appl. Mater. Interfaces, 2014, 6, 5595–5601 CrossRef CAS.
- J. Liu, D. Takeshi, K. Sasaki and S. M. Lyth, Fuel Cells, 2014, 14, 728–734 CrossRef CAS.
- M. Vinothkannan, C. Karthikeyan, G. Gnana kumar, A. R. Kim and D. J. Yoo, Spectrochim. Acta, Part A, 2015, 136, 256–264 CrossRef CAS.
- N. Jiang, Z. Xiu, Z. Xie, H. Li, G. Zhao, W. Wang, Y. Wu and X. Hao, New J. Chem., 2014, 38, 4312–4320 RSC.
- X. Liu, N. Wen, X. Wang and Y. Zheng, Nanomater. Nanotechnol., 2015, 5, 14–20 CrossRef.
- X. Li, J. Feng, Y. Du, J. Bai, H. Fan, H. Zhang, Y. Peng and F. Li, J. Mater. Chem. A, 2015, 3, 5535–5546 RSC.
- B. Dehghanzad, M. K. R. Aghjeh, O. Rafeie, A. Tavakoli and A. J. Oskooie, RSC Adv., 2016, 6, 3578–3585 RSC.
- A. Muthumeenal, S. Neelakandan, D. Rana, T. Matsuura, P. Kanagaraj and A. Nagendran, Fuel Cells, 2014, 14, 853–861 CrossRef CAS.
- S. P. Tung and B. J. Hwang, Fuel Cells, 2007, 7, 32–39 CrossRef CAS.
- S. Gahlot, H. Gupta, P. K. Jha and V. Kulshrestha, ACS Omega, 2017, 2, 5831–5839 CrossRef CAS.
- X. H. He, Y. Zheng, H. L. Yao, Y. W. Chen and D. F. Chen, Fuel Cells, 2014, 14, 26–34 CrossRef CAS.
- A. Bagheri, M. Javanbakht, H. Beydaghi, P. Salarizadeh, A. Shabanikia and H. S. Amoli, RSC Adv., 2016, 6, 39500–39510 RSC.
- Z. Jiang, X. Zhao, Y. Fu and A. Manthiram, J. Mater. Chem., 2012, 22, 24862–24869 RSC.
- K. Oh, B. Son, J. Sanetuntikul and S. Shanmugam, J. Membr. Sci., 2017, 541, 386–392 CrossRef CAS.
- Y. Jin, S. Qiao, J. C. D. Costa, B. J. Wood, B. P. Ladewig and G. Q. Lu, Adv. Funct. Mater., 2007, 17, 3304–3311 CrossRef CAS.
- K. D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- K. Feng, B. Tang and P. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 1481–1488 CrossRef CAS.
- X. He, G. He, A. Zhao, F. Wang, X. Mao, Y. Yin, L. Cao, B. Zhang, H. Wu and Z. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 27676–27687 CrossRef CAS.
- A. K. Mishra, N. H. Kim, D. Jung and J. H. Lee, J. Membr. Sci., 2014, 458, 128–135 CrossRef CAS.
- R. Kumar, C. Xu and K. Scott, RSC Adv., 2012, 2, 8777–8782 RSC.
- D. C. Lee, H. N. Yang, S. H. Park, K. W. Park and W. J. Kim, J. Membr. Sci., 2015, 474, 254–262 CrossRef CAS.
- S. Sasikala, S. V. Selvaganesh, A. K. Sahu, A. Carbone and E. Passalacqua, J. Membr. Sci., 2016, 499, 503–514 CrossRef CAS.
- K. Ketpang, B. Son, D. Lee and S. Shanmugam, J. Membr. Sci., 2015, 488, 154–165 CrossRef CAS.
- W. Zhengbang, T. Haolin and P. Mu, J. Membr. Sci., 2011, 369, 250–257 CrossRef.
- V. Ramani, H. R. Kunz and J. M. Fenton, J. Membr. Sci., 2004, 232, 31–44 CrossRef CAS.
- Y. Kim, K. Ketpang, S. Jaritphun, J. S. Park and S. Shanmugam, J. Mater. Chem., 2015, 3, 8148–8155 RSC.
- K. Ketpang, S. Shanmugam, C. Suwanboon, N. Chanunpanich and D. Lee, J. Membr. Sci., 2015, 493, 285–298 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12768e |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c and
Dong Jin
Yoo
c and
Dong Jin
Yoo
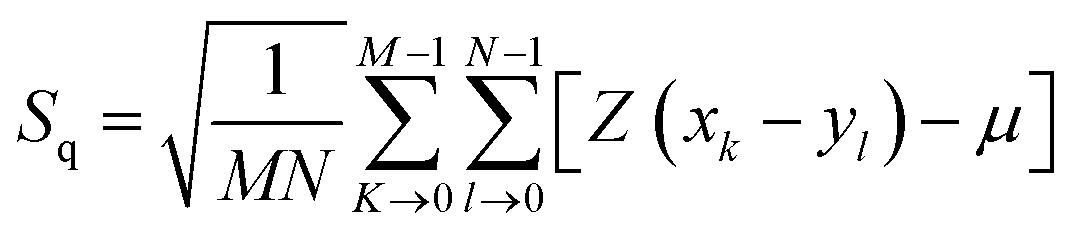
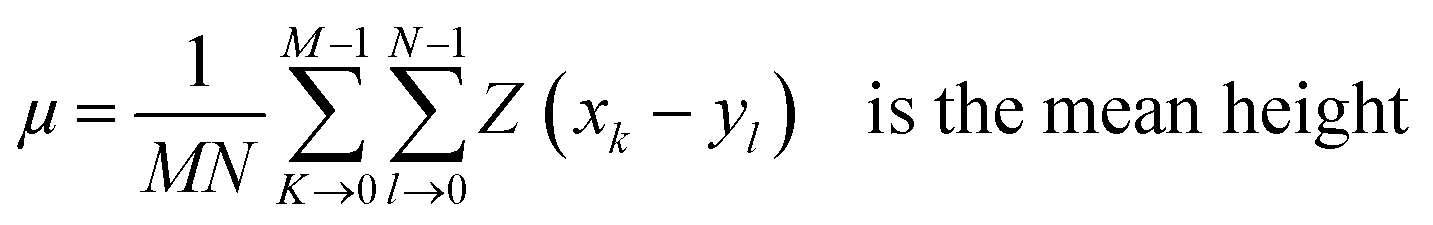
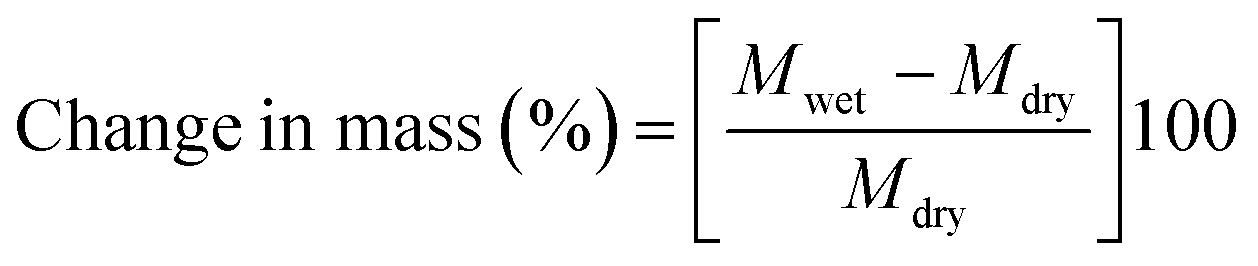
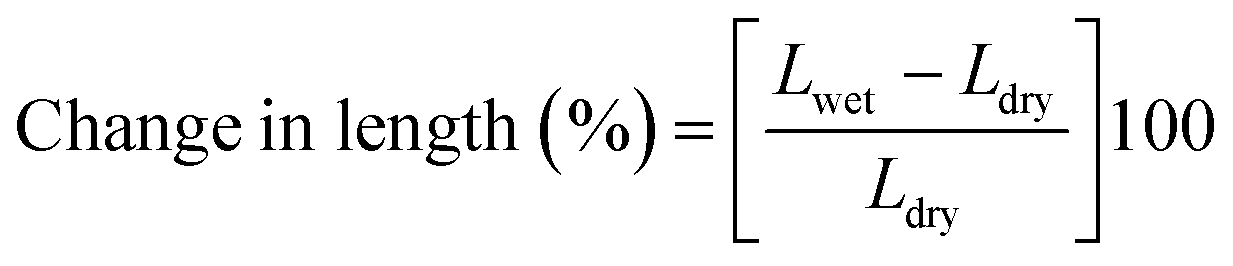
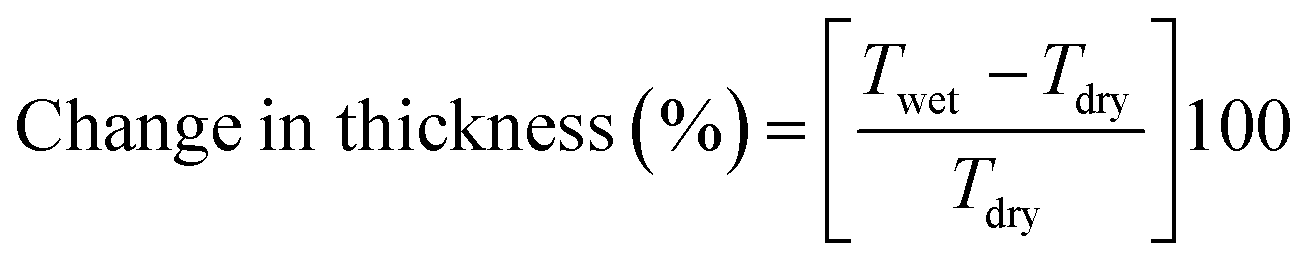
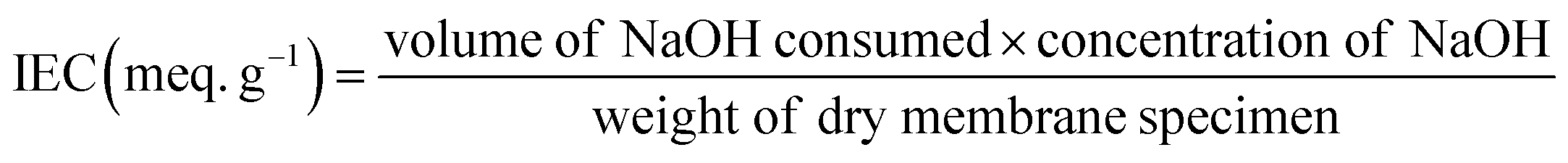
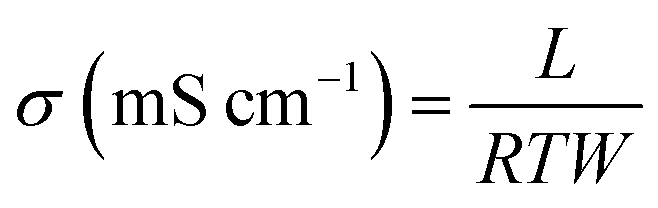
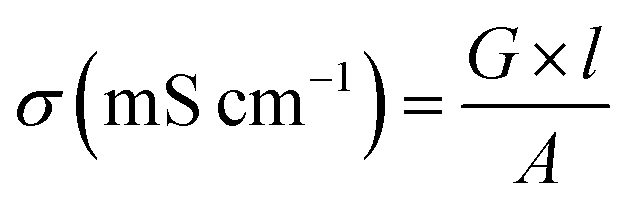
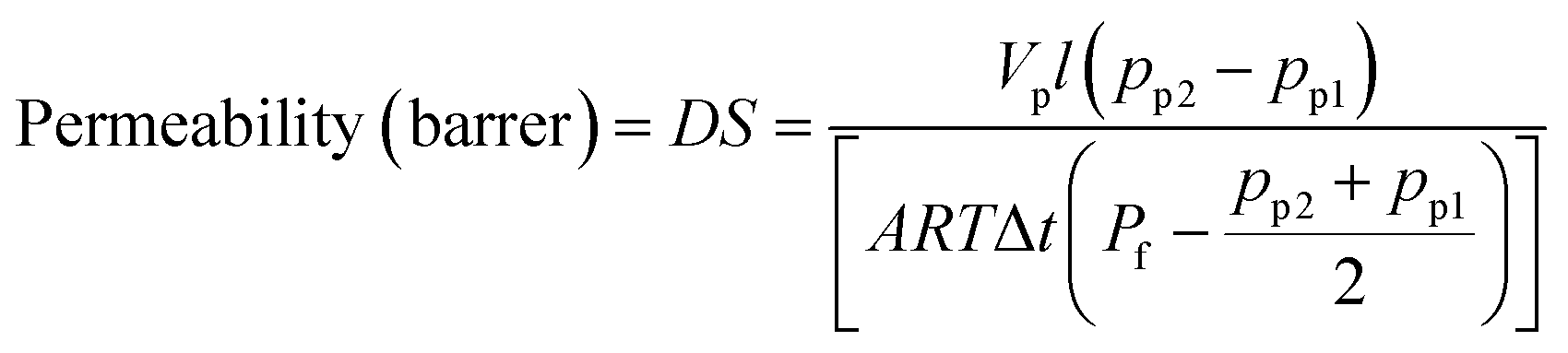



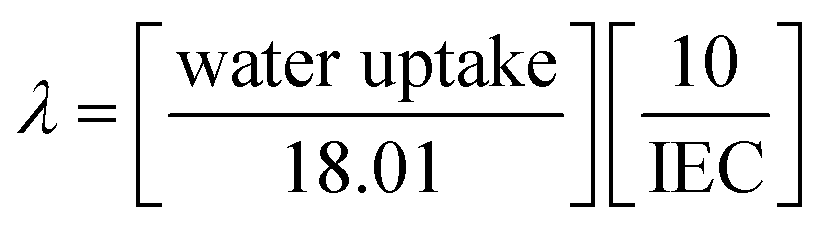
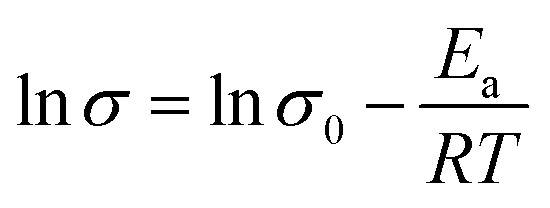
 ) pristine recast Nafion and (
) pristine recast Nafion and ( ) Nafion/SGF-3 and at 120 °C under 25% RH with (
) Nafion/SGF-3 and at 120 °C under 25% RH with ( ) pristine recast Nafion and (
) pristine recast Nafion and ( ) Nafion/SGF-3; (d) stability test of pristine recast Nafion and Nafion/SGF-3 membrane in fuel cell configuration both at 70 °C under 100% RH and 120 °C under 25% RH.
) Nafion/SGF-3; (d) stability test of pristine recast Nafion and Nafion/SGF-3 membrane in fuel cell configuration both at 70 °C under 100% RH and 120 °C under 25% RH.