
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogen-free guanidinium-based perovskite solar cell with enhanced stability†
Narayan Chandra Deb Nath,
Kicheon Yoo and
Jae-Joon Lee *
*
Department of Energy & Materials Engineering, Research Center for Photoenergy Harvesting & Conversion Technology (phct), Dongguk University, Seoul, 100-715, Republic of Korea. E-mail: jjlee@dongguk.edu; Tel: +82-2-2260-4979
First published on 14th May 2018
Abstract
Herein, we report a new halogen-free and excellent stable perovskite, GAPb(SCN)3, which was prepared from the symmetric guanidinium cation (GA+) with nearly zero dipole moment, and pseudohalogen (SCN−), for the fabrication of stable perovskite solar cells. GAPb(SCN)3 exhibits an orthorhombic crystal phase, with optical band gap of 1.43 eV. The orthorhombic crystal phase shifted to a cubic phase, due to the formation of PbS beyond 150 °C. GAPb(SCN)3 itself showed excellent stability upon being exposed to ambient conditions for 30 days, without degradation of its optical or crystallographic properties. This superior stability could be attributable to the strong electrostatic interaction between SCN− and Pb2+, and hydrogen bonding between SCN− and GA+. Even though it has a suitable band gap, it exhibited a significantly lower efficiency of ca. 0.11%. The very low performance could be attributable to the significantly low light absorption coefficient, and large non-radiative recombination of photo-induced charges via an oxidized form of S at the TiO2/perovskite interface.
1. Introduction
Perovskites (MAPbX3, X = Cl, Br, or I and MA = CH3NH3) have distinguished themselves from other photovoltaic materials due to their unique features, which include a strong light absorption coefficient (1.5 × 104 cm−1 at 550 nm), tunable band gap, ambipolar charge transport, long carrier lifetime, and low-cost solution-processability.1,2 The remarkable increase in the efficiency of perovskite-based solid-state solar cells (PSCs) from 3.8% to over 21% in 5 years has attracted much attention in the solar cell community.3–8Despite the success in their photovoltaic performance, PSCs still face several critical challenges, such as device stability under ambient conditions.9 In particular, bare MAPbX3 itself cannot survive in atmospheric moisture, resulting in the instability of PSCs under ambient conditions.10 Therefore, PSCs have to be fabricated mostly in an inert atmosphere. Although this seriously limits the application of the devices, an effective solution to this issue still remains a major obstacle to overcome for the commercialization of PSCs.11–13
While a water-resistive coating, prepared by employing hydrophobic poly(methylmethacrylate) on HTM, has been used to improve the stability of PSCs, the intrinsic vulnerability of perovskites to moisture remains unchanged.13 A layered hybrid perovskite, formed by incorporating phenylethylammonium (C6H5(CH2)2NH3+) in MAPbI3 perovskite, has been shown to exhibit better moisture stability.12 In addition, mixed halide perovskites, MAPbI3−xBrx, formed by replacing 10–15 mol% iodide (I−) with Br−, has also been shown to possess better moisture stability, which was attributed to the stronger interaction between Br− and MA+.6 Similar observations have been reported for the mixed perovskite MAPbI3−x(SCN)x, which involves the partial replacement of I− with pseudohalide (thiocyanate, SCN−).14–16 This anion has similar properties to I−, such as a similar ionic radius. The incorporation of SCN− groups into the sensitizer or redox couple of a dye-sensitized solar cell (DSSC) has been shown to be a useful strategy to enhance the cell performance.17,18 PSCs based on MAPbI3−x(SCN)x exhibit better intrinsic stability in humid air than conventional MAPbI3 perovskite.14,15,19 The strong interaction between SCN− and Pb2+ contributes to improving the humid air stability of the perovskites.19
Meanwhile, some studies have examined the structure and electronic properties of GAPbI3 perovskite (where GA = C(NH2)3), which contains the larger GA cation with nearly zero dipole moment, and have suggested that the high thermodynamic stability and favorable band gap of GAPbI3 could make it a potential and stable candidate for future photovoltaic applications.20–22 Recently, the perovskite MA1−xGAxPbI3, which is based on a combination of GA/MA cations, was reported to exhibit an average PCE of over 19%, with excellent device stability.23 The introduction of GA increased the number of interactions (e.g., hydrogen bonding) in the MAPbI3 crystal, which seemed to be the ultimate cause behind the superior stability.
In this study, we have developed a new perovskite material, GAPb(SCN)3, prepared from symmetric GA+ and pseudohalogen (SCN−) instead of halide, for the fabrication of stable PSCs. Fig. 1 shows that GAPb(SCN)3 exhibits an orthorhombic crystal phase with the space group Pbcn. GAPb(SCN)3 showed an excellent intrinsic stability against air for 30 days, without degradation of the optical and crystallographic properties. The excellent intrinsic stability was attributed to the strong electrostatic interaction between SCN− and Pb2+, and hydrogen bonding interactions between SCN+ and GA+ (Fig. 1), even though the cell efficiency with GAPb(SCN)3 was ca. 0.11% PCE.
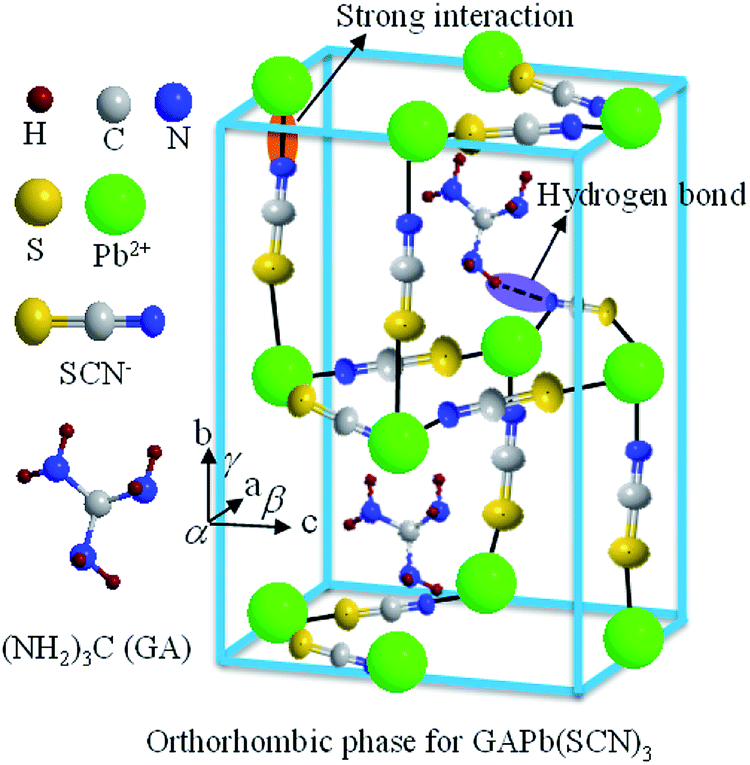 | ||
| Fig. 1 Schematic of the orthorhombic crystal phase of GAPb(SCN)3 perovskite, along with the possible interactions inside the crystal lattice. | ||
2. Experimental section
2.1. Materials
All reagents and solvents were purchased from Sigma-Aldrich, unless otherwise mentioned.2.2. Preparation of perovskite
The perovskite was prepared by either a one- or two-step method. For the one-step method, the precursor solution (A) was prepared by mixing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of lead thiocyanate (Pb(SCN)2) and guanidine thiocyanate (GASCN) in N,N-dimethylformamide (DMF). The perovskite films were prepared by spin-coating the precursor A on SnO2:F layers (FTO) (15 mm × 20 mm, 8/sq, Pilkington Co. Ltd) at 2000 rpm for 30 s, followed by heating at different temperatures (140–200 °C). For the two-step method, Pb(SCN)2 was first dissolved in dimethyl sulfoxide (DMSO) at a concentration of 500 mg mL−1. Then, the solution was spin coated on FTO at 2000 rpm for 30 s, and heated at 100 °C in air for 1 h, to obtain a uniform film of Pb(SCN)2. Then, a solution of 10 mg mL−1 GASCN in isopropanol (IPA), was dropped on top of the Pb(SCN)2 film, and maintained for 30 s, followed by spin-coating at 3000 rpm for 25 s. Next, the films were heated at different temperatures (140 to 200 °C). All the solutions were filtered through a 0.45 μm nylon filter to obtain a clear solution.
:1 molar ratio of lead thiocyanate (Pb(SCN)2) and guanidine thiocyanate (GASCN) in N,N-dimethylformamide (DMF). The perovskite films were prepared by spin-coating the precursor A on SnO2:F layers (FTO) (15 mm × 20 mm, 8/sq, Pilkington Co. Ltd) at 2000 rpm for 30 s, followed by heating at different temperatures (140–200 °C). For the two-step method, Pb(SCN)2 was first dissolved in dimethyl sulfoxide (DMSO) at a concentration of 500 mg mL−1. Then, the solution was spin coated on FTO at 2000 rpm for 30 s, and heated at 100 °C in air for 1 h, to obtain a uniform film of Pb(SCN)2. Then, a solution of 10 mg mL−1 GASCN in isopropanol (IPA), was dropped on top of the Pb(SCN)2 film, and maintained for 30 s, followed by spin-coating at 3000 rpm for 25 s. Next, the films were heated at different temperatures (140 to 200 °C). All the solutions were filtered through a 0.45 μm nylon filter to obtain a clear solution.
2.3. Preparation of perovskite solar cells
The preparation of TiO2 electrodes is described in the ESI. Then, the perovskite layers were prepared on the TiO2 films using an optimized one-step method only. In brief, the perovskite precursor A was dropped on mp-TiO2 film, and kept for 30 s for better penetration, followed by spin-coating at 2000 rpm for 30 s. The resulting perovskite films were annealed at 150 °C in air for 30 min. Then, spiro-MeOTAD solution, comprising 56 mg 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-MeOTAD), 27 mg of 4-tert-butyl pyridine, and 6 mg of bis(trifluoromethane)sulfonimide lithium salt (Li-TFSI) in 1 mL of chlorobenzene, was spin-coated on the perovskite layer at 2500 rpm for 20 s, for the preparation of the HTM layer. Finally, a 160 nm-thick Au electrode was deposited, via thermal evaporation.2.4. Characterization
The UV-Vis absorption spectra of all electrodes were measured using UV-Visible spectrophotometry (Scinco, S-3100, Korea). A surface profiler (Alpha-Step IQ) was used to measure the thickness of the perovskite layer for the calculation of the molar absorption coefficient and optical band gap of the perovskite. Photoluminescence (PL) spectra were obtained using fluorescence spectrophotometry (F-4500, Hitachi) operating at 400 V, with an excitation slit of 1 nm, and an emission slit of 10 nm. For the PL measurements, a thin film of GAPb(SCN)3 perovskite was placed in a sample holder, and the excitation wavelength was fixed at 520 nm for all samples. X-ray diffraction (XRD) measurements of the samples were carried out to observe the crystalline phase using a powder diffractometer (Rigaku D/Max-2500) in Bragg–Bretano geometry under CuKα radiation. The surface morphology was determined using field-emission scanning electron microscopy (FESEM, JEOL, JSM-6700F) equipped with an energy-dispersive X-ray (EDX) detector. The J–V characteristics of the solar cells were measured using a photovoltaic power meter (Polaronix K101/LAB20, McScience, Korea) and a solar simulator (Polaronix K201, McScience) equipped with a 200 W Xenon lamp. The power of the simulated light was calibrated to 100 mW cm−2 (1 sun) using a standard mono-Si solar cell (PVM-396, PV Measurements Inc.), which was certified by the National Renewable Energy Laboratory (NREL). X-ray photoelectron spectroscopy (XPS) analysis was performed using a spectrometer (PHI 5000 VersaProbe II, Japan) equipped with an Al Kα monochromator X-ray source running at 20 kV, a hemispherical electron energy analyzer, and a multichannel detector. The fitting and background subtraction of XPS curves were accomplished using Casa XPS version 2.3.16 software. Ultraviolet photoemission spectroscopy (UPS) measurements were performed using a spectrometer (PHI 5000 VersaProbe II, Japan) with an ultrahigh vacuum of 3 × 10−6 Pa.3. Results and discussion
The GA-cation and SCN−-based iodine-free perovskite, GAPb(SCN)3, was first synthesized to investigate its photoelectrochemical properties for solar cell applications. Fig. 2 shows the surface morphology for different GAPb(SCN)3 films, prepared using a one-step method at different temperatures (150 to 200 °C) for 30 min. Regular-shaped morphologies started to form on FTO at 150 °C. The morphology for the one-step method appeared as two-dimensional (2-D) rhombic, while for the two-step it was 3-D orthorhombic (see Fig. S1 in the ESI†). According to the crystal responses in the XRD spectra (Fig. 3 and Fig. S2†), both exhibited the orthorhombic crystal phase of GAPb(SCN)3 perovskite with the space group of Pnma. Lattice parameters of a = 6.78, b = 5.21, and c = 13.76 (Å) were obtained from the XRD spectra. These were consistent with the orthorhombic crystal phase of the GA+-based perovskite, GAPbI3.21 They were also consistent with that for MAPbI3−x(SCN)x, as the partial incorporation of SCN− groups resulted in a slight tilt in the crystal lattice of the conventional perovskite, MAPbI3, leading to a pseudo-orthorhombic crystal phase.19 Additional peaks, which appear at 13.1 and 27.37°, correspond to the hexagonal phase (H).24 The ratio of S to Pb was found to be ∼3 (EDX spectra, Fig. S3†), supporting the formation of perovskite. The tolerance factor (TF) of ca. 1.04 for GAPb(SCN)3, calculated according to Goldschmidt's equation, TF = (rA + r0)/√2(rB + r0), where, rA is the radius of organic cation (GA+ = 3 Å), rB the radius of metal cation (Pb2+ = 1.33 Å), and r0 the radius of anion (SCN− = 2.20 Å),25 also suggests the formation of a perovskite material.26 Further, the N1s XPS analysis confirmed the formation of GAPb(SCN)3, as described in Fig. 4a.23 | ||
| Fig. 2 SEM images of GAPb(SCN)3 perovskite samples, which were prepared using a one-step method, and activated at different temperatures for 30 min (insets show the photographs of the compounds on FTO). | ||
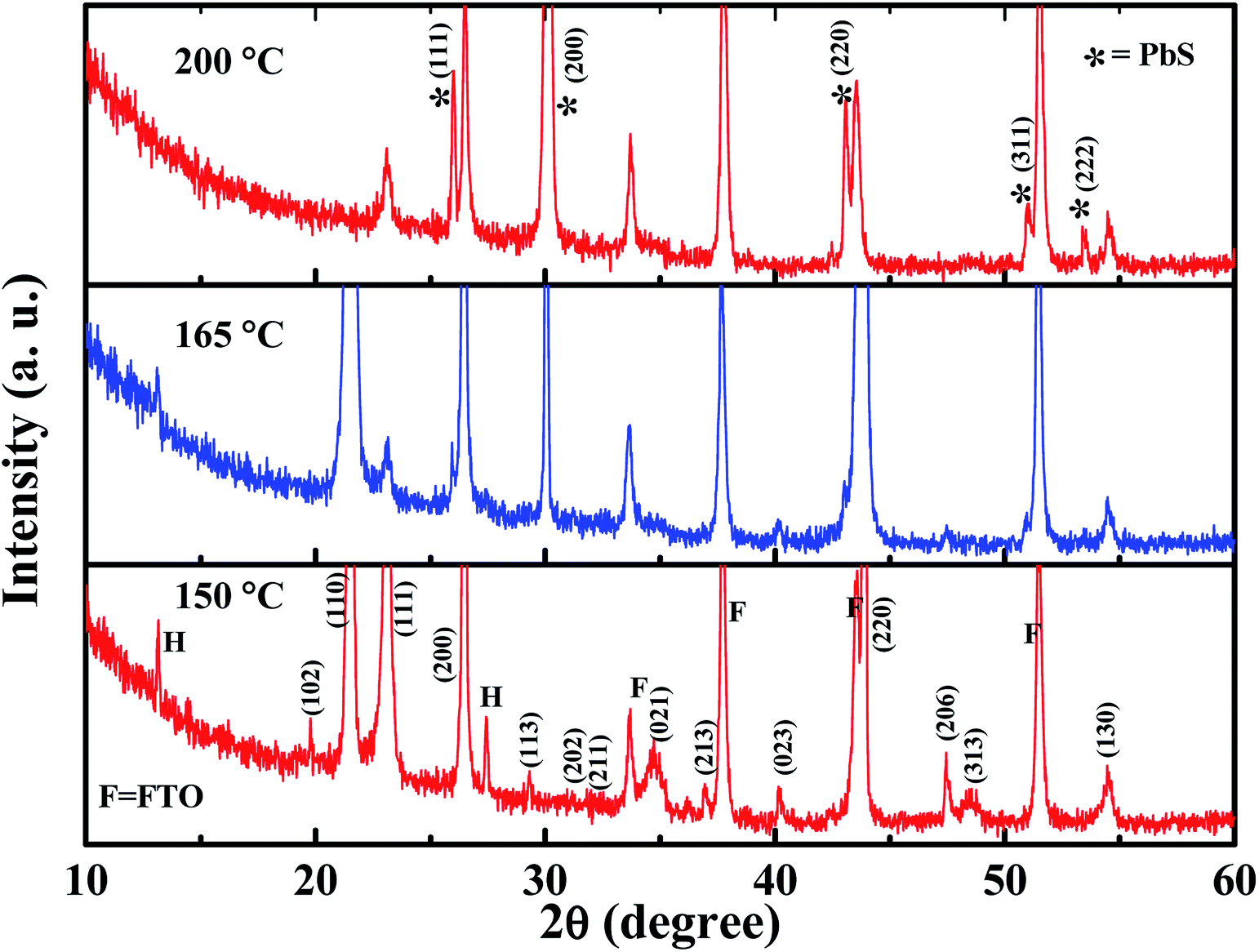 | ||
| Fig. 3 XRD spectra of GAPb(SCN)3 perovskites prepared using the one-step method, and activated from 150 to 200 °C for 30 min. | ||
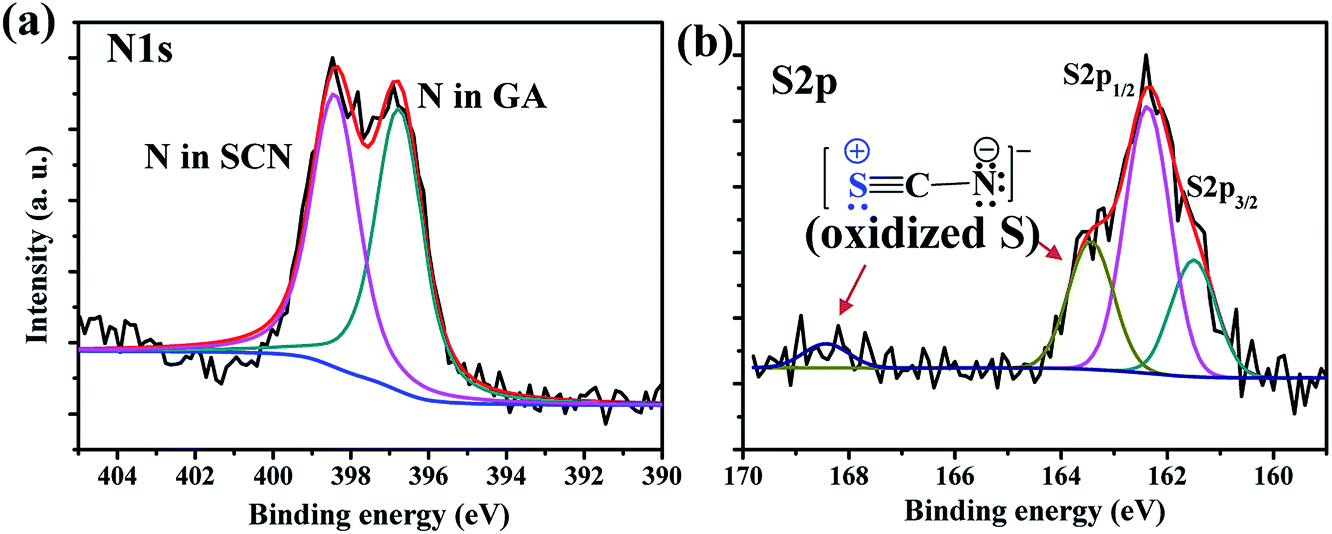 | ||
| Fig. 4 (a) N1s and (b) S2p XPS spectra of GAPb(SCN)3 perovskite films, which were prepared by the one-step method, and activated at 150 °C for 30 min. | ||
The films activated at 140 °C showed irregular-shaped morphologies (Fig. S4a and b†). Essentially, the films contained the starting material, Pb(SCN)2, which was confirmed by the XRD spectra (Fig. S4c†). An additional cubic crystal (Fig. 2 and Fig. S1†) started to form at above 150 °C. The XRD spectrum of the sample activated at 165 °C highlights a deviation from the orthorhombic phase (Fig. 3 and Fig. S2†). Interestingly, all the crystals became cubic at 200 °C. For the samples activated at 200 °C, the XRD spectra (Fig. 3 and Fig. S2†) exhibit a totally different crystallinity. Peaks are present at 25.7, 30.2, 43, 50.9, 53.2 and 52.5°, corresponding to the cubic crystal phase of PbS.27,28 Further, the formation of PbS is supported by the EDX spectra as having the S/Pb (elemental) ratio of ∼1 (Fig. S3†). This is attributed to the degradation of SCN− to sulfur (S) and CN− at above 150 °C, which is similar to the formation of a small amount of S impurities observed during the preparation of MAPbI3−x(SCN)x.14,19 Then, S reacted with Pb2+ at high temperature to form PbS with a cubic crystal phase. The whole phenomenon was further confirmed by the synthesis of pure and powder forms of PbS from perovskite precursor A at 200 °C using an anti-solvent technique. The precursor A was added into octanol (as the anti-solvent), and the mixture with the ratio of 55:45 (octanol/precursor A) was heated at 200 °C, followed by constant stirring for 30 min. This technique nucleated large pure cubic crystals of PbS, which were characterized by SEM, XRD, and EDX (Fig. S5†). The XRD spectrum was consistent with that for PbS reported elsewhere.27
Further, a Lewis acid–base adduct approach was used to prepare a compact and homogeneous GAPb(SCN)3 film (see ESI† for preparation), which has been previously used to prepare high-quality MAPbI3 perovskite films using DMSO as a Lewis base.29 Recently, TU was used as Lewis base for the preparation of better quality FAPbI3 films, as S-donors (TU) are stronger than O-donors (DMSO).30 In the present study, TBP and TU were used as Lewis bases. The formation of PbS cubic crystals was favored even at 150 °C for TU, as it acted as sulfur source (Fig. S6†). However, TBP exhibited a two-dimensional (2-D) rhombic structure, which also had the similar orthorhombic crystal phase of GAPb(SCN)3 perovskite (Fig. S7†). However, it contained few cubic crystals, even at 150 °C.
Fig. 5a shows the light absorption spectrum of GAPb(SCN)3, prepared using the one-step method. It exhibits an absorption maximum at about 550 nm, with the onset absorption edge of ca. 800 nm. On the other hand, GAPb(SCN)3 prepared using the two-step method showed a significant low absorption in the wavelength region between 400 and 700 nm, indicating that it is a worse absorber for solar cell applications (Fig. S8†). Hereafter, we characterized only the GAPb(SCN)3 perovskite prepared using the one-step method for further applications. The direct optical band gap was determined by plotting the square of the Kubelka–Munk function (α × photon energy (hν)) vs. hν (Fig. 5b), where α is the absorption coefficient (determined by the equation α = 2.303 × A (absorbance)/d (film thickness in nm)), h is the plank constant, and ν is the photon frequency.31 This direct optical band gap was found to be about ca. 1.43 eV, which is in accordance with previously reported theoretical values for GAPbI3.21 This indicates that the replacement of I− with SCN− did not affect the optical band gap much. It has also been reported that the partial replacement of I− with SCN− in the perovskite MAPb(SCN)2I did not affect its optical band gap.14 Then, the valence band (VB) energy of GAPb(SCN)3 was estimated to be ca. −5.61 eV at the vacuum level (Fig. 5c), which is slightly lower than that for the conventional perovskite, MAPbI3, reported elsewhere.32 From the observed optical band gap, a conduction band (CB) energy of ca. −4.17 eV was estimated, which is slightly higher than that of TiO2. However, its band positions were well aligned for charge separation alone with n-type TiO2 and p-type HTM of spiro-MeOTAD (Fig. 5d).
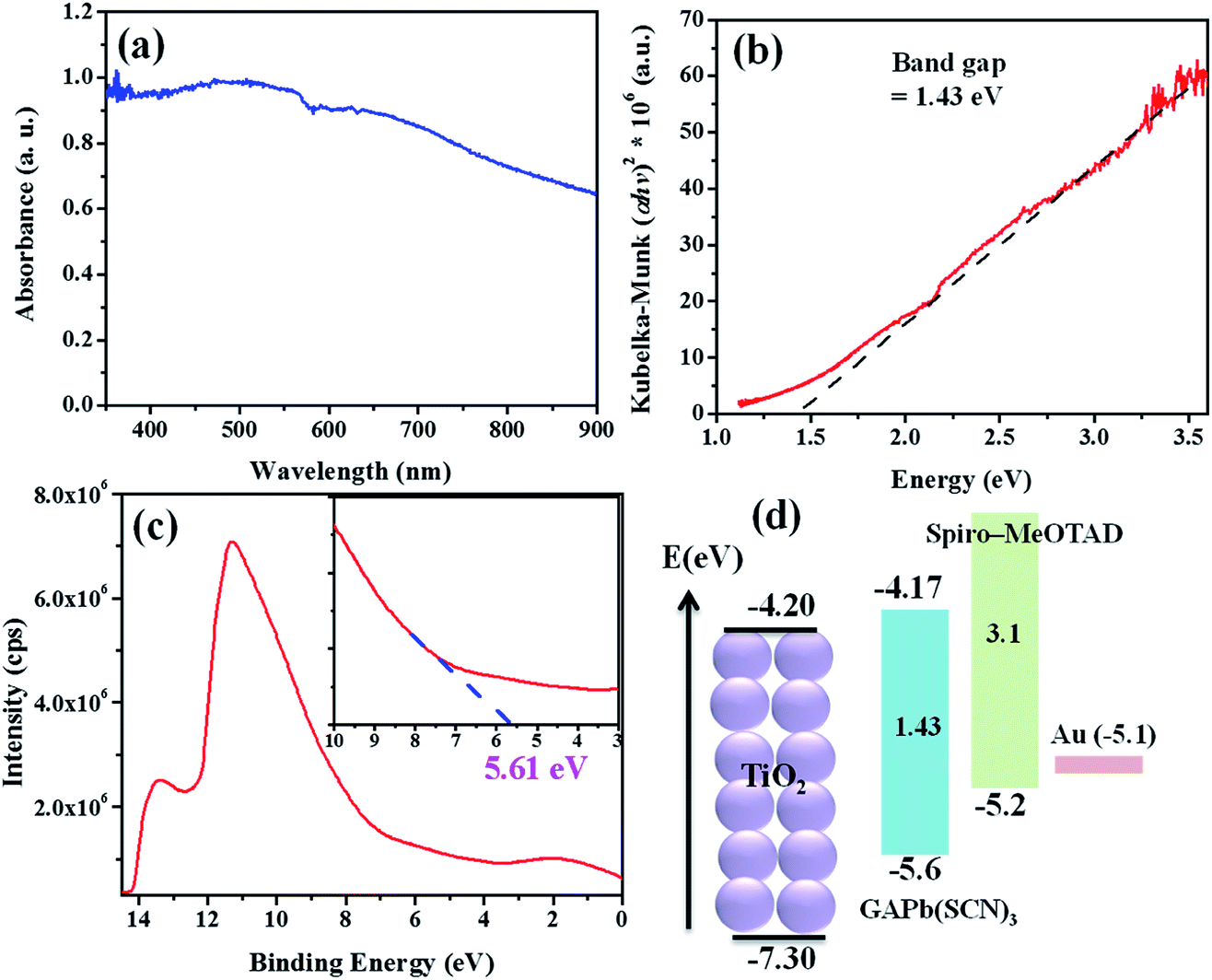 | ||
| Fig. 5 (a) UV-Vis spectra, (b) Kubelka–Munk plot, (c) UPS, and (d) energy levels for GAPb(SCN)3 perovskites prepared using the one-step method, and activated at 150 °C for 30 min. | ||
As mentioned above, the stability of perovskite under ambient conditions is a very concerning issue that limits its outdoor application. We therefore studied the stability of GAPb(SCN)3 upon exposure to air for 30 days. Fig. 6 shows the photographs, UV-Vis spectra, and XRD spectra of GAPb(SCN)3 upon exposure to air for 30 days. The photographs clearly indicate that the color of the perovskite itself did not degrade (Fig. 6a). The identical light absorption pattern in the UV-Visible spectrum suggests that the perovskite retained all of its optical properties, e.g. absorption co-efficient and optical band gap (Fig. 6b).14 The identical XRD spectrum confirms the high stability of the orthorhombic crystals of GAPb(SCN)3 perovskite after exposure in air for 30 days (Fig. 6c). Therefore, it is obvious that the stability of GAPb(SCN)3 itself is superior to that of the conventional perovskite, MAPbI3.14 This is consistent with the recent report on the perovskite MA1−xGAxPbI3, which possesses a combination of GA and MA cations, in which the incorporation of GA increases the number of interactions (e.g., hydrogen bonding) in the MAPbI3 crystal, inducing superior stability.23 The excellent stability can be attributed to the strong electrostatic interaction between SCN− and Pb2+, and hydrogen bonding between SCN− and GA+, described in Fig. 1. This is also consistent with the strong interaction existing in MAPbI3−x(SCN)x perovskite.19
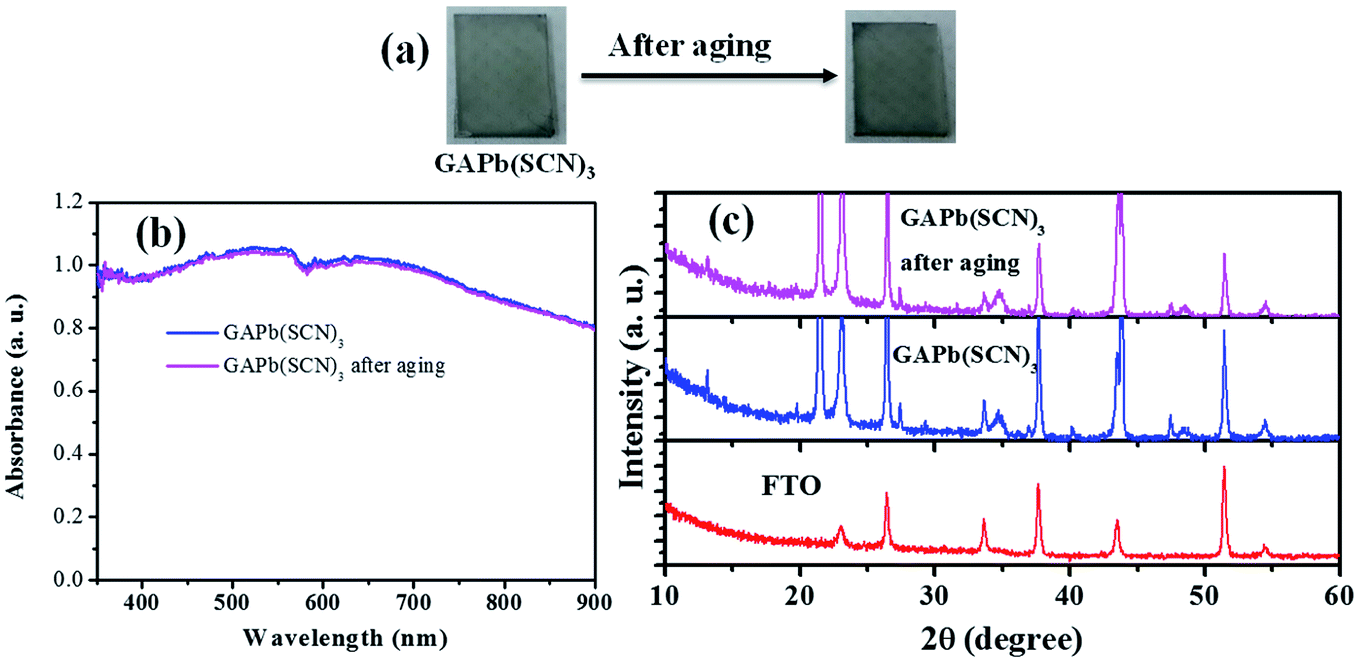 | ||
| Fig. 6 (a) Photographs, (b) UV-Vis spectra, and (c) XRD spectra of GAPb(SCN)3 perovskite, along with aging in air for 30 days. | ||
Then, we constructed a solar device with the GAPb(SCN)3 perovskite using spiro-MeOTAD as the HTM layer, and Au as counter electrode, as shown in Fig. 7a. The current–voltage (J–V) curve
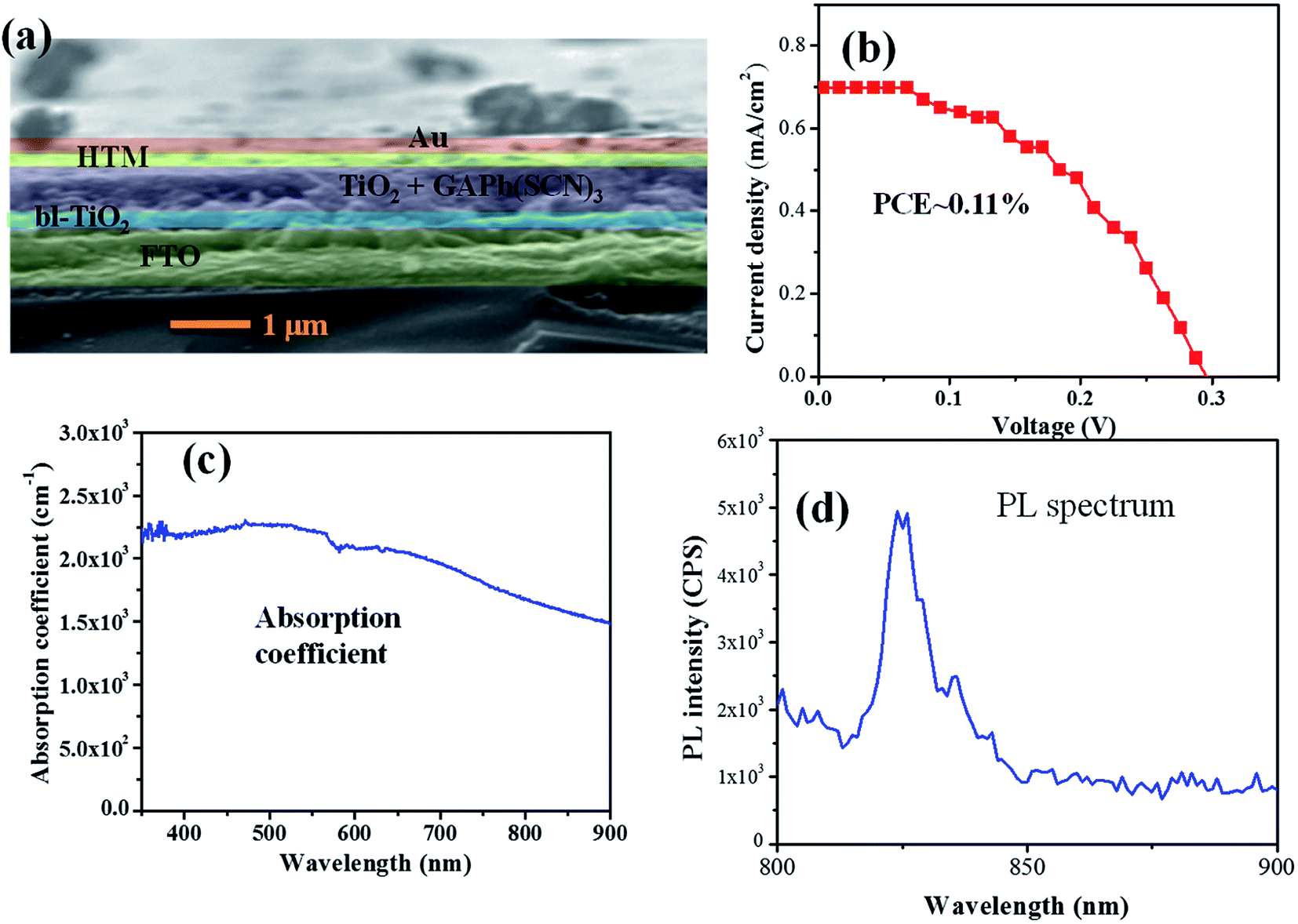 | ||
| Fig. 7 (a) Device cross-sectional SEM image, (b) device current–voltage characteristic, (c) UV-Vis absorption coefficient variation, and (d) photoluminescence (PL) spectrum (excitation wavelength = 520 nm) of GAPb(SCN)3 perovskite films, prepared using the one-step method, and activated at 150 °C for 30 min. | ||

| Perovskite | Voc (V) | Jsc (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| GAPb(SCN)3 | 0.30 | 0.70 | 55.78 | 0.11 |
| GAPb(SCN)2.6I0.4 | 0.46 | 1.23 | 57.71 | 0.33 |
| GAPb(SCN)2.2I0.8 | 0.69 | 1.79 | 53.89 | 0.67 |
| GAPb(SCN)1.8I1.2 | 0.66 | 3.68 | 51.18 | 1.25 |
Among the three suggested intermediates, the formation of 3 was favored, because N is more electronegative than S. The XPS spectrum of S2p showed extra peaks, appearing at higher energies of about ∼163.44 and 168.43 eV (Fig. 4b). These peaks could be attributable to the formation of the oxidized form of S in the intermediate 3, which is consistent with the XPS results of oxidized S reported elsewhere.35 The formation of trap states, formed within the band gap of GAPb(SCN)3, facilitated the recombination of photo-induced charges, as well as significantly decreasing the Voc of the solar device.36,37 The recombination behavior was consistent with the high trap density formed by Cu2+ reduction for Cu-based perovskite, which corresponds to a very low PCE.38
We also prepared hybrid perovskites, GAPb(SCN)3−yIy (y = doping level of iodide (I−) ranging from 0.4 to 1.2), by replacing the SCN− with I−, to reduce the recombination of photo-induced charges associated with SCN−. Rod-like shapes were obviously formed at the doping level of y = 0.4 (Fig. 8a). In addition to the rod-shaped structures, the nucleation of round crystals was observed at y = 0.8, which almost disappeared at the doping level of 1.2. The hybrid perovskites showed an additional absorption edge near 470 nm, compared with that for pure GAPb(SCN)3 (Fig. 8b). The hybrid perovskite (GAPb(SCN)2.2I0.8) exhibited a highest PCE of 1.25% (Fig. 8c and Table 1), which might be associated with higher light absorption and reduced trap states due to the oxidized form of S, compared with those for pure GAPb(SCN)3.
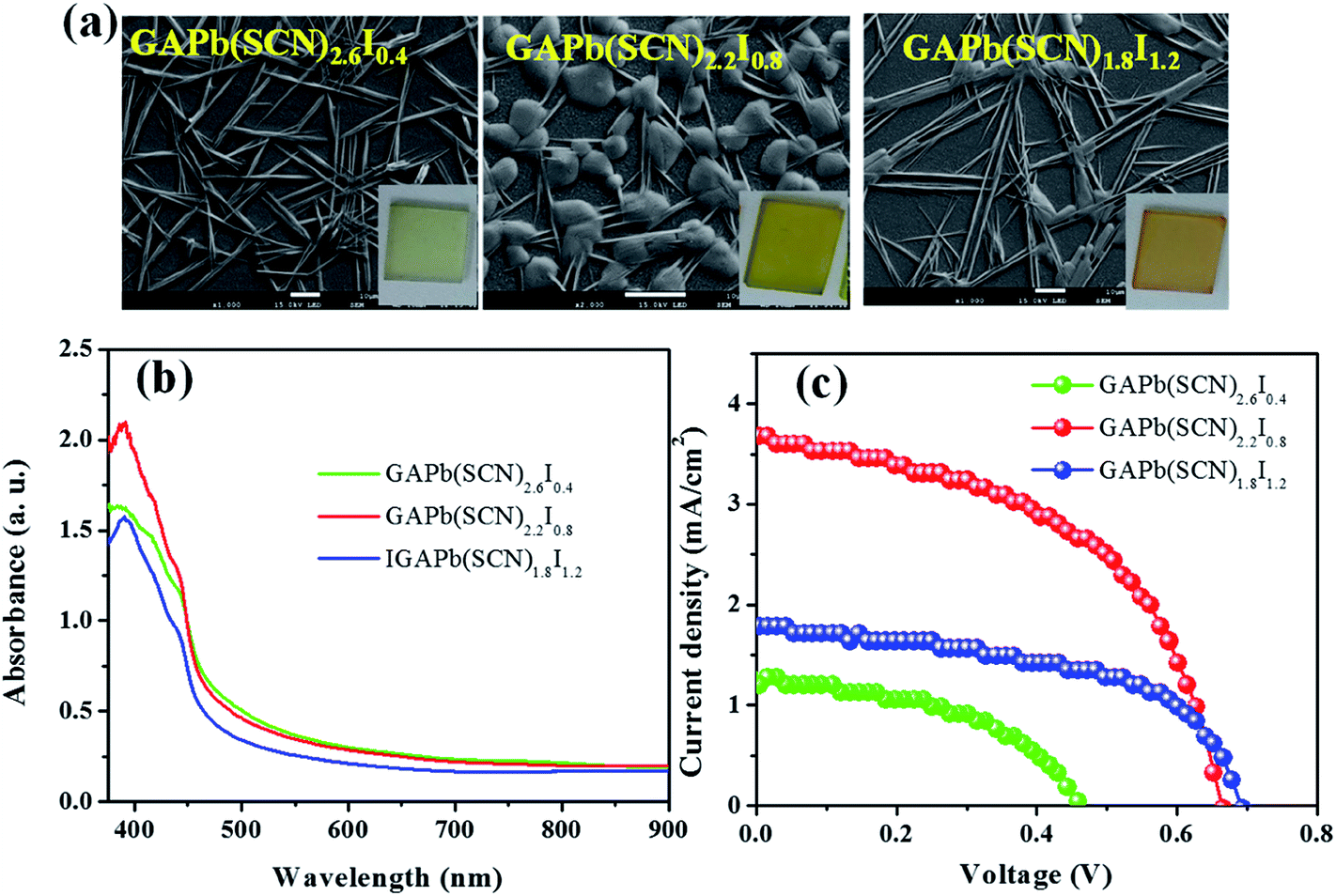 | ||
| Fig. 8 (a) SEM images with photographs, (b) UV-Vis spectra, and (c) current–voltage (J–V) characteristic parameters for hybrid GAyPb(SCN)3−yIy perovskites (y = doping level), which were prepared using the one-step method, and activated at 150 °C for 30 min. | ||
4. Conclusions
In this report, we prepared a new halogen-free and excellent stable perovskite, GAPb(SCN)3 from symmetric GA+ and pseudohalogen (SCN−) for stable PSCs, which exhibited an orthorhombic crystal phase with a moderate optical band gap of 1.43 eV. The pure GAPb(SCN)3 perovskite could be formed at around 150 °C, while a cubic PbS was formed beyond 150 °C. At temperatures above 150 °C, the SCN− ion degraded to S, which eventually reacted with Pb2+ to form PbS. The GAPb(SCN)3 perovskite showed excellent intrinsically stability upon exposure to ambient conditions for 30 days, without degradation to its optical or crystallographic properties. The orthorhombic crystal phase of GAPb(SCN)3 was found to be thermodynamically stable, with a strong electrostatic interaction between SCN− and Pb2+, and hydrogen bonding between SCN+ and GA+. Even though it showed a suitable band gap, the device containing GAPb(SCN)3 exhibited a low PCE of ca. 0.11%, due to the significantly low light absorption coefficient of the perovskite, and high non-radiative recombination of photo-induced charges, caused by the formation of large trap states at the TiO2/perovskite interface. However, a PCE of up to 1.25% was achieved by reducing the non-radiative recombination at the interface with the partial replacement of SCN− with I− in the perovskite. The PCE could be further improved by increasing the light absorption in the visible region via the partial introduction of additional cations, e.g., FA or MA in the GAPb(SCN)3 perovskite, which is currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2015M1A2A2054996, NRF-2016R1A2B2012061). It was also supported by the Technology Development Program to Solve Climate Changes of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2016M1A2A2940912). This work was also supported by the Dongguk University Research Fund of 2016.References
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef.
- H. S. Jung and N.-G. Park, Small, 2015, 11, 10–25 CrossRef PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef PubMed.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Energy Environ. Sci., 2016, 9, 1989–1997 Search PubMed.
- T. Leijtens, G. E. Eperon, S. Pathak, A. Abate, M. M. Lee and H. J. Snaith, Nat. Commun., 2013, 4, 2885 Search PubMed.
- M. Gratzel, Nat. Mater., 2014, 13, 838–842 CrossRef PubMed.
- J. H. Kim, S. T. Williams, N. Cho, C.-C. Chueh and A. K. Y. Jen, Adv. Energy Mater., 2015, 5, 1401229 CrossRef.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., 2014, 126, 11414–11417 CrossRef.
- S. N. Habisreutinger, T. Leijtens, G. E. Eperon, S. D. Stranks, R. J. Nicholas and H. J. Snaith, Nano Lett., 2014, 14, 5561–5568 CrossRef PubMed.
- Q. Jiang, D. Rebollar, J. Gong, E. L. Piacentino, C. Zheng and T. Xu, Angew. Chem., Int. Ed., 2015, 54, 7617–7620 CrossRef PubMed.
- Y. Chen, B. Li, W. Huang, D. Gao and Z. Liang, Chem. Commun., 2015, 51, 11997–11999 RSC.
- Q. Tai, P. You, H. Sang, Z. Liu, C. Hu, H. L. W. Chan and F. Yan, Nat. Commun., 2016, 7, 11105 CrossRef PubMed.
- N. C. D. Nath, I. S. Jung, P.-J. Park and J.-J. Lee, Electrochim. Acta, 2015, 161, 95–99 CrossRef.
- H. Greijer Agrell, J. Lindgren and A. Hagfeldt, Sol. Energy, 2003, 75, 169–180 CrossRef.
- Q. Tai, P. You, H. Sang, Z. Liu, C. Hu, H. L. W. Chan and F. Yan, Nat. Commun., 2016, 7, 11105 CrossRef PubMed.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari, M. K. Nazeeruddin, M. Grätzel and F. De Angelis, Nano Lett., 2014, 14, 3608–3616 CrossRef PubMed.
- G. Giorgi, J.-I. Fujisawa, H. Segawa and K. Yamashita, J. Phys. Chem. C, 2015, 119, 4694–4701 Search PubMed.
- H.-S. Kim, I.-H. Jang, N. Ahn, M. Choi, A. Guerrero, J. Bisquert and N.-G. Park, J. Phys. Chem. Lett., 2015, 6, 4633–4639 CrossRef PubMed.
- A. D. Jodlowski, C. Roldán-Carmona, G. Grancini, M. Salado, M. Ralaiarisoa, S. Ahmad, N. Koch, L. Camacho, G. de Miguel and M. K. Nazeeruddin, Nat. Energy, 2017, 2, 972–979 CrossRef.
- L. Dimesso, A. Quintilla, Y. M. Kim, U. Lemmer and W. Jaegermann, Mater. Sci. Eng., B, 2016, 204, 27–33 CrossRef.
- Y. Iwadate, K. Kawamura, K. Igarashi and J. Mochinaga, J. Phys. Chem., 1982, 86, 5205–5208 CrossRef.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2015, 6, 3430–3433 RSC.
- J. D. Patel, F. Mighri, A. Ajji and S. d. Elkoun, Mater. Sci. Appl., 2012, 03(02), 4 Search PubMed.
- S. S. Rao, I. K. Durga, C. V. Tulasi-Varma, D. Punnoose, L. J. Cheol and H.-J. Kim, New J. Chem., 2015, 39, 7379–7388 RSC.
- N. Ahn, D.-Y. Son, I.-H. Jang, S. M. Kang, M. Choi and N.-G. Park, J. Am. Chem. Soc., 2015, 137, 8696–8699 CrossRef PubMed.
- J.-W. Lee, H.-S. Kim and N.-G. Park, Acc. Chem. Res., 2016, 49, 311–319 CrossRef PubMed.
- X. Lu, Z. Zhuang, Q. Peng and Y. Li, Chem. Commun., 2011, 47, 3141–3143 RSC.
- N. J. Jeon, J. Lee, J. H. Noh, M. K. Nazeeruddin, M. Grätzel and S. I. Seok, J. Am. Chem. Soc., 2013, 135, 19087–19090 CrossRef PubMed.
- S. Mastroianni, F. D. Heinz, J. H. Im, W. Veurman, M. Padilla, M. C. Schubert, U. Wurfel, M. Gratzel, N. G. Park and A. Hinsch, Nanoscale, 2015, 7, 19653–19662 RSC.
- Y. Li, W. Yan, Y. Li, S. Wang, W. Wang, Z. Bian, L. Xiao and Q. Gong, Sci. Rep., 2015, 5, 14485 CrossRef PubMed.
- L. Qie, W. Chen, X. Xiong, C. Hu, F. Zou, P. Hu and Y. Huang, Adv. Sci., 2015, 2, 1–6 Search PubMed.
- N. C. D. Nath, S. Sarker, A. J. Saleh Ahammad and J.-J. Lee, Phys. Chem. Chem. Phys., 2012, 14, 4333–4338 RSC.
- G. I. Lee, N. C. D. Nath, S. Sarker, W. H. Shin, A. J. S. Ahammad, J. K. Kang and J.-J. Lee, Phys. Chem. Chem. Phys., 2012, 14, 5255–5259 RSC.
- D. Cortecchia, H. A. Dewi, J. Yin, A. Bruno, S. Chen, T. Baikie, P. P. Boix, M. Grätzel, S. Mhaisalkar, C. Soci and N. Mathews, Inorg. Chem., 2016, 55, 1044–1052 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00639c |
| This journal is © The Royal Society of Chemistry 2018 |