
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copolymerization of CO2 and epoxides mediated by zinc organyls†
Christoph Wulf,
Ulrike Doering and
Thomas Werner *
*
Leibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: Thomas.Werner@catalysis.de
First published on 18th January 2018
Abstract
Herein we report the copolymerization of CHO with CO2 in the presence of various zinc compounds R2Zn (R = Et, Bu, iPr, Cy and Ph). Several zinc organyls proved to be efficient catalysts for this reaction in the absence of water and co-catalyst. Notably, readily available Bu2Zn reached a TON up to 269 and an initial TOF up to 91 h−1. The effect of various parameters on the reaction outcome has been investigated. Poly(ether)carbonates with molecular weights up to 79.3 kg mol−1 and a CO2 content of up to 97% were obtained. Under standard reaction conditions (100 °C, 2.0 MPa, 16 h) the influence of commonly employed co-catalysts such as PPNCl and TBAB has been investigated in the presence of Et2Zn (0.5 mol%). The reaction of other epoxides (e.g. propylene and styrene oxide) under these conditions led to no significant conversion or to the formation of the respective cyclic carbonate as the main product.
1. Introduction
CO2 is the by-product from combustion of fossil resources and chemical processes and its increasing concentration in the atmosphere is linked to global climate change.1 Thus, the utilization of the greenhouse gas CO2 as a C1-building block2–4 has attracted much attention due to its low cost, availability and the potential to substitute fossil fuel based feedstocks. Reductive transformations of CO2 to produce basic chemicals, e.g. formic acid5 or methanol require stoichiometric amounts of reductants such as silanes, boranes or hydrogen.6 In contrast the addition of CO2 to strained rings, such as oxetanes or epoxides, is a non-reductive process.7 The catalytic coupling of CO2 with epoxides 1 to generate cyclic carbonates 2 or polycarbonates 3 is an atom economic reaction (Scheme 1). The thermodynamically favored product of this reaction is the cyclic carbonate 2.8 Lower reaction temperatures and suitable catalysts allow kinetic control, thus the polycarbonates might be favored. | ||
| Scheme 1 Reaction of epoxides and CO2. | ||
Over the past two decades significant efforts have been made in industry and academia to develop efficient catalysts for the selective formation of either cyclic carbonates9–12 or polycarbonates from epoxides and CO2.13–18
Polycarbonates produced by this reaction are used even on industrial scale e.g. for the production of polyurethanes.19–23 Current industrial processes for the production of polycarbonates are based on the condensation of diols with highly toxic phosgene. In 1969 Inoue and co-workers were the first to report the synthesis of poly(propylene carbonate) from CO2 and propylene oxide utilizing partially hydrolyzed Et2Zn to initiate the polymerization.24,25 Since this pioneering work many catalysts have been developed for the copolymerization of CO2 with epoxides.13–18 Especially zinc based catalyst systems were shown to be efficient. A variety of initiating systems based on different zinc species alone26,27 as well as in combination with additives e.g. ZnO and diprotic activators (e.g. glutaric acid)28 have been reported.29–31 Moreover, transition metal complexes based on zinc proved to be highly efficient and selective. In this context, Darensbourg et al. developed zinc phenoxide catalysts, which exhibit high turnover capabilities for the copolymerization of cyclohexene oxide (CHO) and CO2.32–34 Coates and co-workers developed zinc-β-diiminate-complexes for the synthesis of monodispersed, highly alternating copolymers with high molecular weight.35–38 The group of Williams demonstrated a zinc-based macrocyclic bimetallic catalyst, which showed remarkable activity even at atmospheric pressure of carbon dioxide.39–41 More recently, Rieger et al.42–44 reported dinuclear zinc-β-diiminate complexes and Dinjus and co-workers45 complexes based on the N4–N,N-bis(2-pyridinecarboxamide)-1,2-benzene chelating ligand for the copolymerization of CO2 and CHO.
Notably, most of these systems require halogen containing compounds, such as ammonium and phosphonium salts which are commonly employed co-catalyst for the synthesis of cyclic- as well as polycarbonates from epoxides and CO2. We are generally interested in the reaction between epoxides and CO2.46–54 Most recently we reported a zinc based binary catalytic system for the synthesis of cyclic carbonates.55 Herein we report the efficient copolymerization of CHO and CO2 in the presence of organozinc compounds in catalytic amounts under co-catalyst and halogen-free conditions.
2. Experimental
2.1 Material
The zinc organyls ZnR2 were obtained in solution from commercial sources as follows and used as received: Et2Zn (c = 1.1 M in toluene) from Sigma Aldrich, Bu2Zn (c = 1.0 M in heptane) from Acros, iPr2Zn (c = 1.0 M in toluene) from Sigma Aldrich, Cy2Zn (c = 0.4 M in diethyl ether) from Sigma Aldrich and Ph2Zn as solid from Strem. Epoxides were obtained from commercial sources as follows and distilled over CaH2 prior the use: cyclohexene oxide from Acros, propylene oxide from Acros, styrene oxide from Acros, 4-methyl-1,2-cyclohexene oxide from Alfa Aesar, (+)-limonene-1,2-epoxide and butylene oxide from Sigma Aldrich and 2-(3,4-epoxycyclohexyl)-ethyl-trimethoxy-silane and 2-(3,4-epoxycyclohexyl)-ethyl-triethoxy-silane from abcr. All solvents were obtained dried from Acros over molecular sieves.2.2 Measurements
1H and 13C spectra were recorded with a Bruker 300 Fourier (300 MHz), Bruker AV 300 (300 MHz) and Bruker AV 400 (400 MHz). Shifts δ are stated in ppm. The spectra were calibrated to the rest signal of the applied solvent. CDCl3: 1H δ = 7.27 ppm, 13C δ = 77.00 ppm.The experiments were carried out under increased pressure in a Multiple Reactor System 5000 and Compact Micro Reactor 5000 from Parr. The molar masses and dispersities were analyzed employing size exclusion chromatography (SEC) 1100 GPC from Agilent Technologies with a refraction index detector at 40 °C. The measurements were performed at a constant temperature of 40 °C using three columns with a polyester copolymer network as stationary phase (PSS GRAM 1000 Å, 5 μm particle size, 8.0 × 300 mm; PSS GRAM 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Å, 5 μm particle size, 8.0 × 300 mm; PSS GRAM 1000000 Å). For calibration polystyrene standards from Polymer Standards Service (PSS) were used. Unstabilized THF (HPLC grade) was applied as the mobile phase with a flow rate of 1 mL min−1. For this purpose around 10 mg of the sample were dissolved in 1 mL THF. Ethylene glycol was used as reference peak. For the recording and the evaluation of the measurement the software PSS WINGPC 6® UniChrom from PSS was used.
000 Å, 5 μm particle size, 8.0 × 300 mm; PSS GRAM 1000000 Å). For calibration polystyrene standards from Polymer Standards Service (PSS) were used. Unstabilized THF (HPLC grade) was applied as the mobile phase with a flow rate of 1 mL min−1. For this purpose around 10 mg of the sample were dissolved in 1 mL THF. Ethylene glycol was used as reference peak. For the recording and the evaluation of the measurement the software PSS WINGPC 6® UniChrom from PSS was used.
2.3 General procedures for the copolymerization with zinc organyls
In a 45 cm3 stainless-steel autoclave a solution of R2Zn (0.25 mmol, 0.5 mol%) was added dropwise to a solution of cyclohexene oxide (4.91 g, 50 mmol) in 2 mL toluene. The reactor was sealed and charged with 0.5–5.0 MPa CO2 at 60–100 °C. The reaction mixture was stirred for 1–48 h. Subsequently, the reactor was cooled to ≤20 °C in an ice bath and CO2 was released slowly. After the removal of all volatile components in a vacuum the polymer was solved in 20 mL of CH2Cl2 and precipitated with 50 mL of a solution of MeOH and 5% HCl. The precipitate was filtered off and dried to yield a polymer as a colorless solid.2.4 General procedure for the copolymerization with Et2Zn
In a 45 cm3 stainless-steel autoclave Et2Zn (0.25 mmol, 0.23 mL, 15 wt% in toluene, 0.5 mol%) was added dropwise to a solution of cyclohexene oxide (4.91 g, 50 mmol) in 2 mL toluene. The reactor was sealed and charged with 2.0 MPa CO2 at 100 °C. The reaction mixture was stirred for 16 h. Subsequently, the reactor was cooled to ≤20 °C in an ice bath and CO2 was released slowly. After the removal of all volatile components in vacuum the polymer was solved in 20 mL of CH2Cl2 and precipitated with 50 mL of a solution of MeOH and 5% HCl. The precipitate was filtered off and dried to yield 5.28 g polymer (66%) as a colorless solid.3. Results and discussion
Initially we studied different readily available organozinc compounds as catalysts under standard reaction conditions (0.5 mol% R2Zn, 100 °C, 2.0 MPa, 16 h, Table 1). In the absence of R2Zn neither the formation of polycarbonate nor cyclohexene carbonate (CHC) was observed (entry 1). In the presence of Et2Zn (0.5 mol%) the formation of a polycarbonate with a carbonate content of >85% was obtained as the main product (entry 2). Interestingly, this was achieved in the absence of a co-catalyst and without the addition of water. Even though the use of Bu2Zn led to a product with higher molecular weight the incorporation of CO2 decreased significantly <80% (entry 3). iPr2Zn and Cy2Zn gave similar results in respect to the TON and polycarbonate content while the Mn of the product almost doubled in the presence of Cy2Zn (entry 4 and 5). The polymer with the lowest Mn of 11.7 kg mol−1 and a polycarbonate to polyether ratio of 81:19 was obtained with Ph2Zn (entry 6). As expected the reduction of the amount of Et2Zn from 0.5 mol% to 0.25 mol% led to a significant increase of the Mn from 10.5 to 43.1 kg mol−1 respectively (entry 2 vs. 7). Even though the CO2-content also increased to >99%, the yield dropped below 10%. Similar trends were observed when 0.25 mol% of R2Zn (R = Bu, iPr, Cy and Ph) were employed.47,56 Notably, if the reaction was carried out under argon instead of CO2 a high molecular polyether (Mn = 76.4 kg mol−1) was obtained in 16% yield (entry 8). Even though the highest yields were obtained in toluene other solvents such as CH2Cl2 and THF are also suitable.47,56 For further investigations we did MALDI-TOF experiments for the polymer obtained with Ph2Zn (Table 1, entry 6). The spectrum shows the alternating nature of the polycarbonate with repeating units for the monomer of 142 g mol−1 (Fig. S1†).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R2Zn | TON | Yb,c/% | PC:PE ratiod |
Mn/kg mol−1e | Đe | Tg/°C |
| a Reaction conditions: 50 mmol CHO, 0.5 mol% R2Zn, 2 mL toluene, p(CO2) = 2.0 MPa, T = 100 °C, t = 16 h.b Isolated yield of the polymer after precipitation from CH2Cl2 with 5 mol% HCl in MeOH.c Yield of CHC determined by 1H NMR from the reaction mixture in parenthesis.d Determined by comparison of the integrals arising from the methine protons in the 1H NMR spectra from the polycarbonate (PC) and polyether (PE) unit.e Determined by SEC in THF, calibrated with polystyrene standards.f 0.25 mol% Et2Zn.g Reaction was carried out under inert atmosphere (argon). | |||||||
| 1 | — | — | 0 (0) | — | — | — | — |
| 2 | Et2Zn | 145 | 63 (7) | 87:13 |
10.5 | 5.0 | 89.5 |
| 3 | Bu2Zn | 154 | 60 (10) | 77:23 |
20.8 | 3.6 | 98.1 |
| 4 | iPr2Zn | 166 | 64 (9) | 87:13 |
12.1 | 3.0 | — |
| 5 | Cy2Zn | 162 | 66 (11) | 90:10 |
22.2 | 2.6 | — |
| 6 | Ph2Zn | 107 | 46 (7) | 81:19 |
11.7 | 4.4 | 81.6 |
| 7f | Et2Zn | 65 | 6 (<1) | >99:<1 |
43.1 | 4.6 | 83.1 |
| 8g | Et2Zn | 31 | 16 (0) | <1:>99 |
76.4 | 2.2 | — |

Subsequently, we studied the influence of various reaction parameters (p(CO2), T, t) for all of the tested zinc compounds since all of them showed similar activity and selectivity as well as promising results in respect to the polymer properties such as amount of incorporated CO2 and Mn.
First the influence of the CO2-pressure on the copolymerization of CHO and CO2 was investigated (Table 2). For all of the tested zinc compounds R2Zn an increase of the pressure to 5.0 MPa led to higher turnover numbers and increased isolated polymer yields compared to 2.0 MPa (Table 2, entries 1–5 vs. Table 1, entries 2–6).
| Entry | R2Zn | p(CO2)/MPa | TON | Yb,c/% | PC:PE ratiod |
Mn/kg mol−1e | Đe |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 50 mmol CHO, 0.5 mol% R2Zn, 2 mL toluene, p(CO2) = 0.5–5.0 MPa, T = 100 °C, t = 16 h.b Isolated yield of the polymer after precipitation from CH2Cl2 with 5 mol% HCl in MeOH.c Yield of CHC determined by 1H NMR from the reaction mixture in parenthesis.d Determined by comparison of the integrals arising from the methine protons in the 1H NMR spectra from the PC and PE unit.e Determined by SEC in THF, calibrated with polystyrene standards. | |||||||
| 1 | Et2Zn | 5 | 167 | 67 (5) | 94:6 |
16.8 | 5.5 |
| 2 | Bu2Zn | 5 | 197 | 65 (7) | 68:32 |
24.9 | 3.1 |
| 3 | iPr2Zn | 5 | 174 | 87 (4) | 90:10 |
11.4 | 5.7 |
| 4 | Cy2Zn | 5 | 170 | 51 (7) | 97:3 |
21.8 | 3.0 |
| 5 | Ph2Zn | 5 | 137 | 59 (8) | 87:13 |
22.8 | 3.2 |
| 6 | Et2Zn | 0.5 | 70 | 20 (3) | 81:19 |
9.9 | 6.3 |
| 7 | Bu2Zn | 0.5 | 79 | 18 (4) | 29:71 |
11.6 | 4.6 |
| 8 | iPr2Zn | 0.5 | 100 | 33 (5) | 68:32 |
7.7 | 5.3 |
| 9 | Cy2Zn | 0.5 | 54 | 18 (4) | 81:19 |
4.3 | 4.4 |
| 10 | Ph2Zn | 0.5 | 57 | 15 (2) | 29:71 |
6.9 | 3.8 |
Interestingly, in the presence of Et2Zn the Mn increased from 10.5. to 16.8 kg mol−1 while for Ph2Zn the Mn increased to 22.8 kg mol−1 (Table 2, entries 1 and 5 vs. Table 1, entries 2 and 6). At a lower CO2 pressure of 0.5 MPa the turnover number, polymer yield and polycarbonate content decreased for all R2Zn compounds (Table 2, entries 6–10 vs. Table 1, entries 2–6). Except for Et2Zn the Mn dropped significantly.
Since the polycarbonate is the kinetic product of the reaction of CO2 and CHO, we subsequently studied the influence of the reaction temperature.8 Thus the reaction was carried out at 60 °C in the presence of the different organozinc compounds (Table 3). As expected in all cases the formation of the cyclic carbonate which is the thermodynamic product decreased significantly for the different ZnR2 while the turnover number decreased slightly in all cases (Table 3 vs. Table 1, entries 2–6). Notably, for Et2Zn the polycarbonate content dropped from 94% to 16% while the Mn increased from 10.5 to 79.3 kg mol−1 (Table 3, entry 1 vs. Table 1, entry 2). In the presence of the other zinc compounds ZnR2 (R = Bu, iPr, Cy or Ph) polymers obtained at 60 °C showed similar CO2 content compared the products which were obtained at 100 °C (Table 3, entries 2–5 vs. Table 1, entries 3–6). Interestingly, for iPr2Zn and Ph2Zn the Mn at lower temperature was about three times higher compared to 100 °C (Table 3, entries 3 and 5 vs. Table 1, entries 4 and 6). The observation that with decreasing reaction temperature the Mn increases is in accordance with work previously reported by Soga and co-workers.56 They investigated the effect of the temperature on the copolymerization of propylene oxide and CO2 using an alumina-supported diethylzinc catalyst. In general the observed dispersities of the products were higher compared to the polymers obtained at 100 °C (Table 3, entries 1–5 vs. Table 1, entries 2–6).
| Entry | R2Zn | T/°C | TON | Yb,c/% | PC:PE ratiod |
Mn/kg mol−1e | Đe |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 50 mmol CHO, 0.5 mol% R2Zn, 2 mL toluene, p(CO2) = 2.0 MPa, T = 60 °C, t = 16 h.b Isolated yield of the polymer after precipitation from CH2Cl2 with 5 mol% HCl in MeOH.c Yield of CHC determined by 1H NMR from the reaction mixture in parenthesis.d Determined by comparison of the integrals arising from the methine protons in the 1H NMR spectra from the PC and PE unit.e Determined by SEC in THF, calibrated with polystyrene standards. | |||||||
| 1 | Et2Zn | 60 | 113 | 52 (0) | 16:84 |
79.3 | 4.4 |
| 2 | Bu2Zn | 60 | 123 | 53 (0) | 71:29 |
23.0 | 10.3 |
| 3 | iPr2Zn | 60 | 115 | 45 (0) | 84:16 |
37.5 | 7.3 |
| 4 | Cy2Zn | 60 | 80 | 52 (0) | 94:6 |
27.9 | 7.4 |
| 5 | Ph2Zn | 60 | 91 | 37 (≤1) | 68:32 |
30.2 | 7.3 |
Consequently we investigated the influence of the reaction time on the copolymerization for three different dialkyl zinc compounds (R2Zn, R = Et, Bu, iPr). Bu2Zn showed the highest initial activity with 45% conversion of the starting material after 1 h (Fig. 1).47,56 In the presence of Et2Zn and iPr2Zn the conversion of CHO was 31% and 25% after 1 h, respectively. Notably, after 4 h more than 50% of the starting material was converted. Subsequently the reaction rate decreased in all cases which resulted in a maximum conversion of only 86% for iPr2Zn after 16 h. The exponential formation of the polycarbonate was accompanied by a linear increase in the cyclic carbonate content of the reaction mixture which is in accordance with previously reported work by Górecki and Kuran.47,57 Notably, the CHC-content in all cases did not exceed 10% after 16 h.
 | ||
| Fig. 1 Influence of the dialkyl zinc compounds (ZnR2, R = Et, Bu, iPr) on the conversion of CHO (copolymerization of CHO and CO2, observed CHC ≤10%) in dependence on the reaction time. Reaction conditions: 50 mmol CHO, 0.5 mol% R2Zn, 2 mL toluene, p(CO2) = 2.0 MPa, T = 100 °C, t = 1–16 h. The data points are the results for separate batches. | ||
In the presence of Et2Zn the Mn of the obtained polycarbonate showed a linear increase from 8.9 to 28.3 kg mol−1 during the first 8 h (Fig. 2). Subsequently the Mn decreased to 12.2 kg mol−1 after 16 h. This might be addressed to transesterification reactions as suggested by Vandenberg and Tian.58 This result is comparable to the molecular weights obtained by Williams et al. using mixed salts prepared from Et2Zn and acetic acid as a catalyst.59
 | ||
| Fig. 2 Influence of the dialkyl zinc compound Et2Zn on the Mn of CHO (copolymerization of CHO and CO2, observed CHC ≤10%) in dependence on the reaction time. Reaction conditions: 50 mmol CHO, 0.5 mol% R2Zn, 2 mL toluene, p(CO2) = 2.0 MPa, T = 100 °C, t = 1–16 h. The data points are the results for separate batches. | ||
Notably, no clear trend was observed for the other zinc compounds (R2Zn, R = Bu and iPr).47,56
However, in all cases the CO2 content of the obtained polymer was approximately constant (85%) over time. In further investigations, the influence of different co-catalysts was tested (Table 4). The copolymerization was performed with Et2Zn (0.5 mol%) under standard reaction conditions (2.0 MPa, 100 °C, 16 h) in the presence of commonly used co-catalysts.60–66 Notably, in all cases the utilization of a co-catalyst led only to the formation of the cyclic carbonate and/or low molecular weight products/oligomers. Tetra-n-butylammonium bromide (TBAB) and bis(triphenylphosphine)iminium chloride (PPNCl) are known to catalyze the addition of CO2 to epoxides yielding cyclic carbonates.67,68 Both co-catalyst provided similar results mainly producing the cyclic carbonate in 63% and 82% yield respectively (entry 1 and 2). Known as catalysts for ring-opening-polymerization (ROP) of cyclic carbonates, 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) and triazabicyclodecene (TBD) were used as co-catalyst to decrease the cyclic side product.64,69 In the presence of DBU the formation of an oligomer (Mn = 510 g mol−1) with a polycarbonate content of 55% was observed (entry 3). A similar result was obtained with TBD as the co-catalyst (entry 4) while 4-dimethylaminopyridine (DMAP) showed no significant conversion (entry 5). It is generally discussed that TBD hinders the depolymerization of the polycarbonate through deprotonation of the in situ formed hydroxyl end-group.70,71
|
|||||
|---|---|---|---|---|---|
| Entry | Co-catalyst | TON | Yb,c/% | PC:PE ratiod |
Mn/mol−1e |
| a Reaction conditions: 50 mmol CHO, 0.5 mol% R2Zn, 0.5 mol% co-catalyst, 2 mL toluene, p(CO2) = 2.0 MPa, T = 100 °C, t = 16 h.b Yield of polymer determined by 1H NMR from the reaction mixture.c Yield of CHC determined by 1H NMR from the reaction mixture in parenthesis.d Determined by comparison of the integrals arising from the methine protons in the 1H NMR spectra from the PC and PE unit.e Determined by SEC in THF, calibrated with polystyrene standards. | |||||
| 1 | TBAB | 141 | 7 (63) | 0:100 |
270 |
| 2 | PPNCl | 172 | 3 (82) | 0:100 |
260 |
| 3 | DBU | 66 | 18 (14) | 55:45 |
510 |
| 4 | TBD | 61 | 18 (12) | 71:29 |
590 |
| 5 | DMAP | 16 | 8 (<1) | 65:35 |
490 |

Additionally, we studied the conversion of various other epoxides with CO2. In the presence of Et2Zn (0.5 mol%) under our standard reaction conditions (2.0 MPa, T = 100 °C, t = 16 h, Table 5). For the simple propylene oxide a conversion of 8% and low weight polymer with high polyether content was reached (entry 1). This is a lower polymer yield then previous reported by Inoue where 5.0–6.0 MPa CO2-pressure and the Et2Zn/H2O system was used.24,25 For the conversion of butylene oxide only 7% of the oligomeric ether and 5% of the respective cyclic carbonate were observed (entry 2). The reaction of styrene oxide led to higher TON of 59 and 20% yield of an oligomer with a Mn < 500 g mol−1 (entry 3) which is higher than the obtained yield of 2% by the Et2Zn/H2O system by Endo and co-workers. Epichlorohydrin showed a poor yield of the corresponding cyclic carbonate of only 2% (entry 4) which is lower than the result of Endo and co-workers.24 Notably, other cyclohexane based epoxides could not be converted to the corresponding polycarbonates (entries 5–8).
| Entry | Monomer | Xepoxide/% | TON | Yb,c/% | Mn/g mol−1d |
|---|---|---|---|---|---|
| a Reaction conditions: 0.5 mol% Et2Zn, 2 mL toluene, p(CO2) = 2.0 MPa, T = 100 °C, t = 16 h.b Yield of polymer determined by 1H NMR from the reaction mixture.c Yield of CHC determined by 1H NMR from the reaction mixture in parenthesis.d Determined by SEC in THF, calibrated with polystyrene standards.e 50 mmol epoxide.f 25 mmol epoxide.g 17 mmol epoxide.h 14 mmol epoxide.i 11 mmol epoxide.j Only Polyether was observed. | |||||
| 1e |  |
8 | 15 | 7j (<1) | <500 |
| 2f |  |
12 | 25 | 7j (5) | <500 |
| 3e |  |
28 | 59 | 20j (9) | <500 |
| 4f | 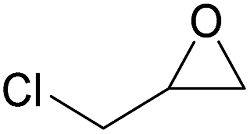 |
2 | 1 | 0 (2) | — |
| 5f |  |
<1 | — | — | — |
| 6g |  |
<1 | — | — | — |
| 7h |  |
<1 | — | — | — |
| 8i |  |
27 | 57 | 27j (0) | <500 |
In the case of limonene oxide this might be addressed to steric hindrance which was observed before by Coates71 and Anwander72 (entry 5). The alkoxy silyl functionalized substrates showed no conversion which might be addressed to a reaction of these groups with the catalyst.72,73 The methyl substituted cyclohexene oxide was partially converted to an oligomeric ether (entry 8).
4. Conclusions
Zinc organyls (R2Zn, R = Et, Bu, iPr, Cy and Ph) efficiently mediate the copolymerization of CO2 and CHO. Under the standard reaction conditions (100 °C, 2.0 MPa) an initial TOF of up to 91 h−1 (for Bu2Zn) and TONs up to 269 after 16 h were achieved. Polycarbonates with molecular weights up to 79.3 kg mol−1 and a CO2 content up to 97% were obtained. The effect of various parameters on the reaction outcome has been investigated. An increase of the pressure to 5.0 MPa led to higher turnover numbers and increased isolated polymer yields compared to 2.0 MPa. However, at this pressure also the highest dispersities were observed. Higher molecular weight products could be isolated at lower reaction temperature of 60 °C. Several commonly employed readily available co-catalysts were studied in combination with Et2Zn. However, the co-catalysts facilitated rather the formation of the cyclic carbonate than the production of the polycarbonate. Moreover different epoxides were tested in the copolymerization with CO2, unfortunately only low conversions and/or the formation of the respective cyclic carbonates were observed as the major product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank the Federal Ministry of Research and Education (BMBF) for financial support (Chemische Prozesse und stoffliche Nutzung von CO2: Technologien für Nachhaltigkeit und Klimaschutz, grant 033RC1004A).References
- IPCC, Climate Change 2013: The Physical Science Basis, in Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K.Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013, ch. SPM, pp. 1–30, DOI:10.1017/CBO9781107415324.004.
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281–7305 CAS.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 1–15 Search PubMed.
- G. A. Filonenko, D. Smykowski, B. M. Szyja, G. Li, J. Szczygieł, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2015, 5, 1145–1154 CrossRef CAS.
- S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. V. Stein, U. Englert, M. Holscher, J. Klankermayer and W. Leitner, Chem. Sci., 2015, 6, 693–704 RSC.
- C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS PubMed.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- A. C. Kathalikkattil, R. Babu, J. Tharun, R. Roshan and D.-W. Park, Catal. Surv. Asia, 2015, 19, 223–235 CrossRef CAS.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- D. J. Darensbourg, S. J. Lewis, J. L. Rodgers and J. C. Yarbrough, Inorg. Chem., 2003, 42, 581–589 CrossRef CAS PubMed.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 CAS.
- M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- M. Luo, Y. Li, Y.-Y. Zhang and X.-H. Zhang, Polymer, 2016, 82, 406–431 CrossRef CAS.
- Y. Qin, X. Sheng, S. Liu, G. Ren, X. Wang and F. Wang, J. CO2 Util., 2015, 11, 3–9 CrossRef CAS.
- M. A. Subhani, B. Köhler, C. Gürtler, W. Leitner and T. E. Müller, Angew. Chem., Int. Ed., 2016, 55, 5591–5596 CrossRef CAS PubMed.
- T. E. Müller, C. Gürtler, A. Kermagoret, Y. Dienes, I. Busygin, B. Köhler and W. Leitner, DE102010043409A1, 2014.
- T. E. Müller, C. Gürtler, M. Wohak, J. Hofman, A. M. Subhani, W. Leitner, I. Peckermann and A. Aurel, EP2703425A1, 2014.
- T. E. Müller, C. Gürtler, A. M. Subhani and W. Leitner, EP2845873A1, 2015.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part C: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- S. Inoue, H. Koinuma and T. Tsuruta, Makromol. Chem., 1969, 130, 210–220 CrossRef CAS.
- K. Soga, K. Uenishi, S. Hosoda and S. Ikeda, Makromol. Chem., 1977, 178, 893–897 CrossRef CAS.
- K. L. Orchard, J. E. Harris, A. J. P. White, M. S. P. Shaffer and C. K. Williams, Organometallics, 2011, 30, 2223–2229 CrossRef CAS.
- M. Ree, J. Y. Bae, J. H. Jung and T. J. Shin, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1863–1876 CrossRef CAS.
- W. Kuran, Prog. Polym. Sci., 1998, 23, 919–992 CrossRef CAS.
- M. S. Super and E. J. Beckman, Trends Polym. Sci., 1997, 5, 236–240 CAS.
- H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS.
- D. J. Darensbourg and M. W. Holtcamp, Macromolecules, 1995, 28, 7577–7579 CrossRef CAS.
- D. J. Darensbourg, J. R. Wildeson, J. C. Yarbrough and J. H. Reibenspies, J. Am. Chem. Soc., 2000, 122, 12487–12496 CrossRef CAS.
- D. J. Darensbourg, M. W. Holtcamp, G. E. Struck, M. S. Zimmer, S. A. Niezgoda, P. Rainey, J. B. Robertson, J. D. Draper and J. H. Reibenspies, J. Am. Chem. Soc., 1999, 121, 107–116 CrossRef CAS.
- M. Cheng, N. A. Darling, E. B. Lobkovsky and G. W. Coates, Chem. Commun., 2000, 2007–2008 RSC.
- M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738–8749 CrossRef CAS PubMed.
- S. D. Allen, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 14284–14285 CrossRef CAS PubMed.
- L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS PubMed.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535–9542 CrossRef CAS PubMed.
- M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A.-K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef CAS PubMed.
- M. Reiter, A. Kronast, S. Kissling and B. Rieger, ACS Macro Lett., 2016, 5, 419–423 CrossRef CAS.
- S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579–4582 RSC.
- M. Adolph, T. A. Zevaco, C. Altesleben, S. Staudt and E. Dinjus, J. Mol. Catal. A: Chem., 2015, 400, 104–110 CrossRef CAS.
- T. Werner, N. Tenhumberg and H. Büttner, ChemCatChem, 2014, 6, 3493–3500 CrossRef CAS.
- H. Büttner, K. Lau, A. Spannenberg and T. Werner, ChemCatChem, 2015, 7, 459–467 CrossRef.
- H. Büttner, J. Steinbauer and T. Werner, ChemSusChem, 2015, 8, 2655–2669 CrossRef PubMed.
- C. Kohrt and T. Werner, ChemSusChem, 2015, 8, 2031–2034 CrossRef CAS PubMed.
- W. Desens and T. Werner, Adv. Synth. Catal., 2016, 358, 622–630 CrossRef CAS.
- W. Desens, C. Kohrt, M. Frank and T. Werner, ChemSusChem, 2015, 8, 3815–3822 CrossRef CAS PubMed.
- H. Büttner, J. Steinbauer, C. Wulf, M. Dindaroglu, H.-G. Schmalz and T. Werner, ChemSusChem, 2017, 10, 1076–1079 CrossRef PubMed.
- H. Büttner, C. Grimmer, J. Steinbauer and T. Werner, ACS Sustainable Chem. Eng., 2016, 4, 4805–4814 CrossRef.
- N. Tenhumberg, H. Büttner, B. Schäffner, D. Kruse, M. Blumenstein and T. Werner, Green Chem., 2016, 18, 3775–3788 RSC.
- W. Desens, C. Kohrt, A. Spannenberg and T. Werner, Org. Chem. Front., 2016, 3, 156–164 RSC.
- K. Soga, K. Hyakkoku, K. Izumi and S. Ikeda, J Polymer Sci. Polymer Chem. Ed., 1978, 16, 2383–2392 CrossRef CAS.
- P. Górecki and W. Kuran, J Polymer Sci. Polymer Lett. Ed., 1985, 23, 299–304 CrossRef.
- E. J. Vandenberg and D. Tian, Macromolecules, 1999, 32, 3613–3619 CrossRef CAS.
- K. L. Orchard, J. E. Harris, A. J. P. White, M. S. P. Shaffer and C. K. Williams, Organometallics, 2011, 30, 2223–2229 CrossRef CAS.
- M. Reiter, P. T. Altenbuchner, S. Kissling, E. Herdtweck and B. Rieger, Eur. J. Inorg. Chem., 2015, 2015, 1766–1774 CrossRef CAS.
- M. Mandal, D. Chakraborty and V. Ramkumar, RSC Adv., 2015, 5, 28536–28553 RSC.
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- B. Han, L. Zhang, S. J. Kyran, B. Liu, Z. Duan and D. J. Darensbourg, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1938–1944 CrossRef CAS.
- A. K. Diallo, E. Kirillov, M. Slawinski, J. M. Brusson, S. M. Guillaume and J. F. Carpentier, Polym. Chem., 2015, 6, 1961–1971 RSC.
- Y.-Y. Zhang, X.-H. Zhang, R.-J. Wei, B.-Y. Du, Z.-Q. Fan and G.-R. Qi, RSC Adv., 2014, 4, 36183–36188 RSC.
- D. J. Darensbourg, in Adv. Inorg. Chem., ed. A. Michele and E. Rudi van, Academic Press, 2014, vol. 66, pp. 1–23 Search PubMed.
- F. D. Monica, S. V. C. Vummaleti, A. Buonerba, A. D. Nisi, M. Monari, S. Milione, A. Grassi, L. Cavallo and C. Capacchione, Adv. Synth. Catal., 2016, 358, 3231–3243 CrossRef CAS.
- A. Sibaouih, P. Ryan, M. Leskela, B. Rieger and T. Repo, Appl. Catal., A, 2009, 365, 194–198 CrossRef CAS.
- D. J. Darensbourg, W. C. Chung, A. D. Yeung and M. Luna, Macromolecules, 2015, 48, 1679–1687 CrossRef CAS.
- C. Li, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2015, 67, 449–458 CrossRef CAS.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, W. C. Ellis and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 1215–1218 CrossRef CAS PubMed.
- E. Le Roux, A. De Mallmann, N. Merle, M. Taoufik and R. Anwander, Organometallics, 2015, 34, 5146–5154 CrossRef CAS.
- K. Su, T. D. Tilley and M. J. Sailor, J. Am. Chem. Soc., 1996, 118, 3459–3468 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12535f |
| This journal is © The Royal Society of Chemistry 2018 |