
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceD-Glucosamine production from chitosan hydrolyzation over a glucose-derived solid acid catalyst
Hongkui Zhanga,
Yuting Lua,
Yuanhao Wang *a,
Xingrong Zhang*b and
Tingyu Wangc
*a,
Xingrong Zhang*b and
Tingyu Wangc
aFaculty of Light Industry and Chemical Engineering, Dalian Polytechnic University, Dalian 116023, China. E-mail: yuanhaowang@126.com
bState Key Laboratory of Mineral Processing, Beijing General Research Institute of Mining and Metallurgy, Beijing 102600, China. E-mail: nichao6666@126.com
cCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215006, China
First published on 1st February 2018
Abstract
A glucose-based solid acid catalyst (GSA) was synthesized by hydrothermal carbonization and its physicochemical properties were explored by various characterization techniques including IR, TG and SEM. In addition, its catalytic performance towards D-glucosamine formation from the hydrolysis of chitosan was extensively investigated to determine the effects of reaction parameters, such as reaction temperature, time and mass ratio of catalyst and reactants. The experimental results revealed that the yield of targeted product D-glucosamine could reach as high as 98.1% under optimal conditions (temperature: 110 °C; time: 6 h). After six catalytic cycles, no evident deactivation was observed, suggesting the satisfactory stability of the investigated solid acid catalyst. This might provide insight on the development of suitable catalyst systems for D-glucosamine formation to replace homogeneous catalysts.
Introduction
Chitosan is a partial deacetylation product of chitin which consists of β-(1-4)-2-amino-D-glucose and β-(1-4)-2-acetamido-D-glucose linked via 1,4-β-glucosidic bonds. It is the second most abundant biopolymer on earth coming from marine invertebrates, insects, fungi1 and bacteria etc.; obtained after treatment with 40% concentrated sodium hydroxide solution2 and produced on a scale of billions of tons annually.3 To date, chitosan has received more and more attention as a functional biopolymer for various applications from pharmaceuticals to commodity chemicals because of its non-toxicity, biodegradability, biocompatibility,4 non-antigenicity, antibacterial properties5,6 and pilot-scale production. Its physicochemical properties greatly rely on the structure and the molecular weight. Therefore, chitosan generally presents special physiological properties, such as anti-tumor, anti-thrombotic and anti-cancer, if its molecular weight is reduced to a certain extent.7 Therefore, many methods including acid hydrolysis,8,9 formic acid,10 nitrous acid hydrolysis,11 phosphoric acid,12 thermal degradation,13 oxidative–reductive degradation14 and enzymatic degradation15,16 have been developed to degrade chitosan. In order to achieve the promising degradable effect,17 a combination of methods, such as the combination of acetic acid and sonication,18 H2O2 and gamma irradiation,19 H2O2 and microwave,20 acetic acid and microwave,21 mechanical-force-assisted with H2SO4 (ref. 22) and solid-state mechano-chemical method23 have been employed. Ning Yan had investigated the degradation performance of acid to chitosan in different solvents, such as the system of aprotic polar solvents with water using an acid catalyst24 and ethylene glycol (EG) under the catalysis of sulfuric acid.25 After the degradation of chitosan by different hydrolysis methods, chitosan with low molecular weight, chitosan oligermers or D-glucosamine can be obtained, and they are widely used in food, medicine, cosmetics and material science fields.Among these hydrolysis products, D-glucosamine is considered as one of the useful medicaments because of its biological, pharmaceutical and nutrimental effects.15 D-Glucosamine can also be used in wound healing, bone regeneration and as an antibacterial agent in dentistry.26 Apart from that, many materials in pharmaceutical, food and special materials processing industries27–29 might be synthesized from D-glucosamine, because it contains active amino groups. Based on these various applications, many hydrolysis methods, such as enzymatic degradation by Penicillium decumbens CFRNT15 (ref. 30) or cocktail,31 oxidative degradation in concentrated nitrous acid32 or hydrogen peroxide33 hydrolysis with concentrated HCl or HCl–H3PO4 and acid hydrolysis in ionic liquids34 have been designed to produce D-glucosamine from chitosan. The degradation process of chitosan by expensive enzymes occurred under mild conditions, but possessed several drawbacks, such as the slow reaction rate, difficulty to handle,35–37 quick deactivation of the enzymes38 and the formation of other byproducts.32 At present, degradation of chitosan to synthesize D-glucosamine by a strong liquid acid, is the most common and effective method, but the degradation process normally requires excess acid loading, complex reactors and produce effluent disposal problems.39 In the presence of the excess acid, further degradation of D-glucosamine had taken place, leading to less yield.40,41
In addition to the above mentioned method, chitosan hydrolysis by a carbon-based solid acid may be a promising method to produce D-glucosamine. On the contrary to the homogeneous liquid acid, the heterogeneous solid acid catalyst could be easily separated from the reaction system, thereby reusable for the next catalytic cycle. Hence, hydrolysis of other polysaccharides by carbon-based solid acid catalyst had already been deeply investigated regarding its satisfactory performance. Ayumu Onda et al.42 had reported the selective hydrolysis of cellulose into glucose using sulfonated active carbon. Nata et al.43 had prepared a highly sulfonated solid acid catalyst for the cornstarch hydrolysis. It had been demonstrated that the carbon-based solid acid catalyst can adsorb oligosaccharides as well as enzymes, resulting in the efficient catalytic hydrolysis.44,45 Daizo Yamaguchi44 had reported that an amorphous carbon material with –SO3H, –COOH and –OH groups present the high activity and selectivity for the hydrolysis of cellulose, and the functions of different groups had been discussed.
In the present study, a carbon-based solid acid catalyst bearing –SO3H, –COOH and –OH groups was synthesized by a hydrothermal method using renewable glucose as the precursor and used as an environment-friendly and recyclable catalyst for D-glucosamine production from chitosan hydrolysis. The influences of experimental parameters were studied to identify the optimal reaction conditions. Additionally, several characterization techniques, such as IR, TG and SEM were carried out to determine the physicochemical features of the as-prepared catalyst in order to establish the structure–activity relationship.
Experimental
Materials
D-Glucose, sulfuric acid, citric acid, acrylic acid, sodium hydroxide and other chemical regents were purchased from Sigma (St. Louis, USA) and used without further purification. Chitosan (800–2000 cP) with a DD value 45–97% was purchased from Sigma (St. Louis, USA), and used directly with no pretreatment.Catalyst preparation
Glucose-derived solid acid catalyst was prepared by one-pot hydrothermal carbonization, as described in previous literature.36 Briefly, a mixture of glucose (5.0 g), citric acid (2.5 g), hydroxyethylsulfonic acid (1.5 g) and deionized water (40.0 mL) was placed in a Teflon-lined stainless steel autoclave (100 mL), and maintained at 180 °C for 4 h. After cooling down to room temperature, the precipitate was thoroughly washed with hot deionized water and dried at 80 °C for 8 h in a vacuum oven. The achieved catalyst is labelled as GSA in the following section.The acid density (mmol g−1) of the investigated catalyst was determined by the neutralization titration method.46 Typically, catalyst (0.2 g) was added to an aqueous solution of NaCl (0.1 mol L−1, 20.0 mL). The mixture was sonicated for 1 h and then magnetically stirred for 24 h at room temperature. After centrifugal separation, the obtained filtrate (5.0 mL) was titrated with aqueous NaOH (0.05 mol L−1) with phenolphthalein as the indicator.
Catalyst characterization
FT-IR spectra of the GSA catalyst were recorded and qualitatively analyzed using (FT/IR-430, JASCO, Japan). Samples were prepared by grinding into fine powders with KBr. The spectrum was recorded in the range of 400–4000 cm−1.Thermo-gravimetric analysis (TGA) measurement was conducted on a thermal analyzer TGA/SDTA 851 (Perkin-Elmer, USA). Samples (20 mg) were placed in corundum crucibles non-hermetically closed with lids, and heated under argon from room temperature to 400 °C at a heating rate of 5 °C min−1.
Field emission scanning electron microscopy (FE-SEM) images of the samples were obtained by scanning electron microscope (JEOL, JSM-6500 LV).
NH3-TPD technique was used to estimate the acidity of the GSA catalyst. Prior to NH3 adsorption, the catalyst was outgassed at 300 °C for 1 h. After cooling down to room temperature, NH3 adsorption was carried out under a flow of NH3/He (10 vol%, 50 mL min−1) and the temperature was maintained for 30 min. Then, the flow was switched to He (50 mL min−1) so as to remove the weakly adsorbed NH3. NH3-TPD was conducted in the He flow (50 mL min−1) by increasing the temperature to 800 °C with a heating rate of 5 °C min−1, while the desorbed NH3 molecules were detected using online mass spectrometry (Inficon quadrupole).
Hydrolysis of chitosan
Performance of the catalyst was investigated by chitosan hydrolysis reaction as illustrated in Scheme 1. Chitosan was completely dissolved in a hydrochloric acid solution under magnetic stirring and then transferred to a Teflon-lined stainless steel autoclave, as well as the appropriate GSA sample. The role of hydrochloride was to protonate the amine groups of chitosan, avoiding the combination of amine groups with solid acid sites. After the reaction, the solution was cooled down to room temperature and was filtered. Formed D-glucosamine was quantitatively determined using HPLC (Watese2695) equipped with UV and refractive index (RI) detectors and a Shodex SUGAR SC1011 column (8 × 300 mm). The column was operated at 70 °C and the operating temperature of detectors was set at 35 °C. The mixture solution of NaOAc and HAc was used as the mobile phase at a flow rate of 0.6 mL min−1. The injection volume was 10 μL of filtrate (25 mm). The yield of D-glucosamine was calculated according to:| D-Glucosamine yield (%) = 100B[161DD + 203(1 − DD)]/m0 | (1) |
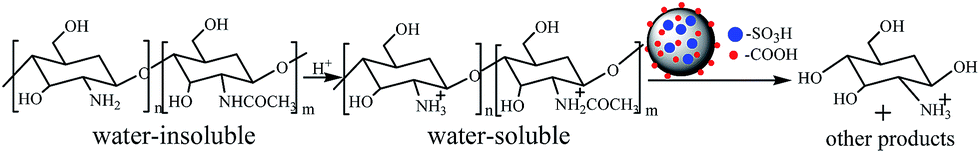 | ||
| Scheme 1 The hydrolysis of chitosan over GSA catalyst. | ||
Catalyst recyclability
The recyclability of GSA catalyst for the hydrolysis of chitosan was investigated to estimate its catalytic stability. The reactions were repeated under identical experimental conditions as mentioned above. After each test, the aged catalyst was filtered, washed with hot deionized water and dried at 80 °C overnight for the next run.Results and discussion
Physicochemical properties of GSA catalyst
The FT-IR spectrum of the solid acid is shown in Fig. 1(a). An obvious band due to the O–H stretching mode of the –COOH groups was noticed at 3420 cm−1. The bands at 1614 cm−1 and 1385 cm−1 were assigned to aromatic-like C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching mode in polyaromatic compound. The band at 1709 cm−1 corresponds to the CO stretching mode of the –COOH group.47 The band at 1210 cm−1 was attributed to the sulfonate absorption of C4–SO3H.43,48 The vibration bands at 1035 cm−1 (–SO2– symmetric stretching) evidenced the presence of –SO3H group.49
C stretching mode in polyaromatic compound. The band at 1709 cm−1 corresponds to the CO stretching mode of the –COOH group.47 The band at 1210 cm−1 was attributed to the sulfonate absorption of C4–SO3H.43,48 The vibration bands at 1035 cm−1 (–SO2– symmetric stretching) evidenced the presence of –SO3H group.49
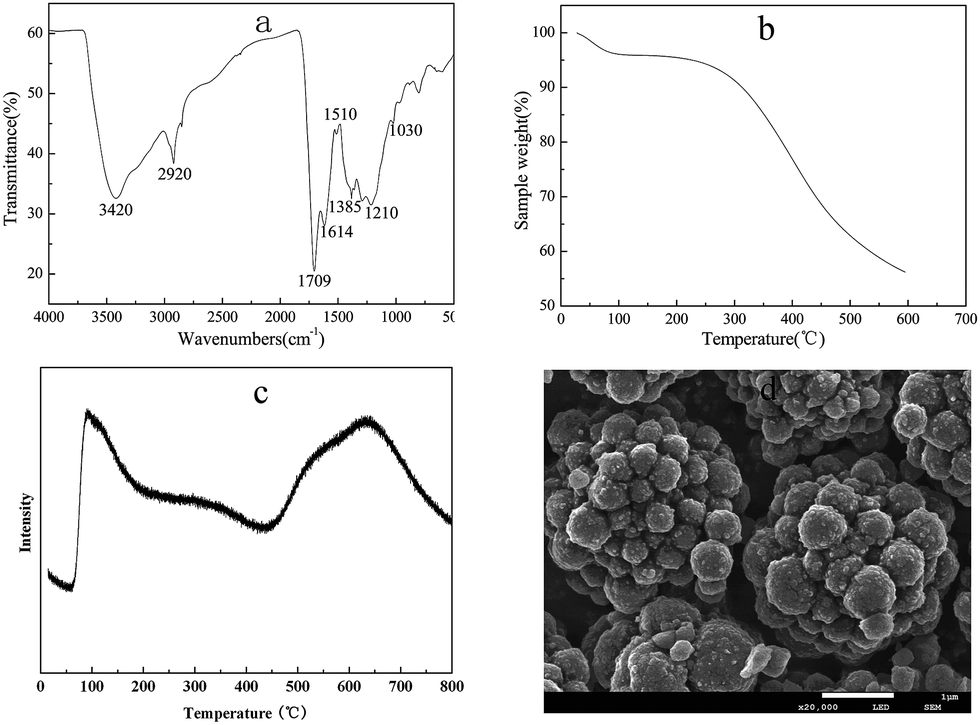 | ||
| Fig. 1 FT-IR spectrum (a), TGA curve (b), NH3-TPD (c) and SEM images (d) of the as-prepared GSA catalyst. | ||
TGA curve of GSA sample is also presented in Fig. 1(b). The observed weight loss (ca. 5%) below 100 °C is normally caused by the evaporation of adsorbed water on the catalyst surface.50 It can be seen from Fig. 2(b) that only a 5% weight loss occurred with further increase in the temperature up to 250 °C, suggesting a good thermal stability of the GSA catalyst. It could be effectively used below 250 °C. In this work, reactions for the chitosan hydrolysis were conducted below 120 °C. Therefore, the as-prepared GSA material was suitable for this reaction.
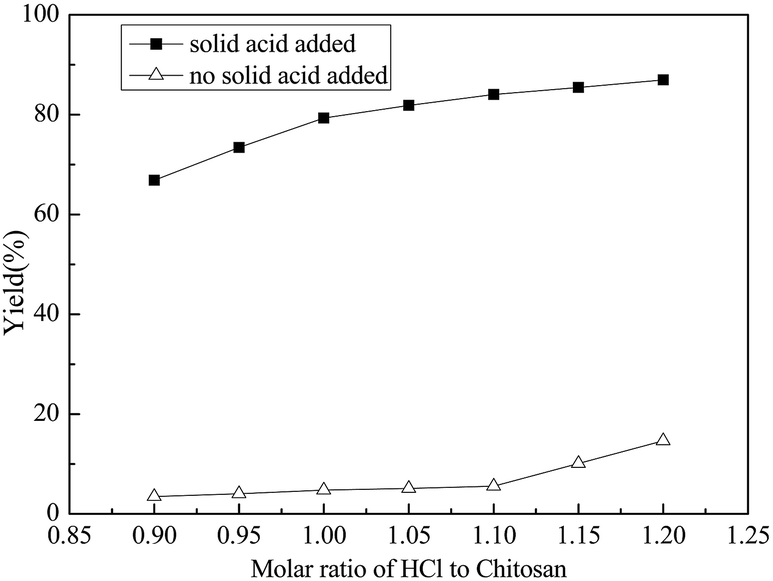 | ||
| Fig. 2 Effect of the molar ratio of hydrochloric acid to chitosan on the yield of glucosamine (chitosan: 1.0 g, mass of GSA: 0.2 g, reaction time: 4 h, reaction temperature: 100 °C). | ||
The acid density of the synthesized carbon-based solid acid was 1.9 mmol g−1, which was determined by the neutralization titration method. Fig. 1(c) shows the NH3-TPD profile of solid acid catalysts. The peaks of NH3-desorption indicate the existence of acid sites with various strengths, and it was associated with the acidity strength. Normally, the higher desorption temperature corresponds to the stronger acid sites. The first peak, which was in the range of 100–200 °C maximized at 110 °C, had been assigned for the hydrogen-bound ammonia (weak).49 The acid site on the GSA catalyst surface was denoted as a medium strong acid site, based on the peak at 340 °C. The third peak at 650 °C corresponds to very strong acid sites. Considering the acid sites of the prepared GSA catalyst, the very strong acid sites were assigned to the –SO3H groups,50,51 whereas the weak and medium strong acid sites were assigned to the –OH groups and the –COOH groups on the catalyst surface.
As observed from SEM image (Fig. 1(d)), the solid acid particles produced by hydrothermal carbonization possessed spherical shapes with diameters in the range of 200–500 nm. This result is consistent with the reports of Nata et al.43 The presence of acrylic acid may promote glucose dehydration and polymerization, leading to more particle formation. Furthermore, acrylic acid might stabilize the newly formed carbonaceous particles and prevent their further aggregation in hydrothermal carbonization process, in order to obtain small particle sizes.
Influence of hydrochloric acid concentration on the hydrolysis reaction
Hydrochloric acid providing H+ ions to protonate the amine groups of chitosan mainly possessed two merits. On one hand, chitosan could be completely dissolved in water if the amine groups were balanced by the adequate hydrogen ions.18 On the other hand, the sufficient protonation of the amine groups might avoid the neutralization reaction between the amine groups and the solid acid. However, excess hydrochloric acid may lead to the degradation of the targeted D-glucosamine.Based on the above concern, the influence of the molar ratio of hydrochloric acid and chitosan on the hydrolysis reaction was studied and the results are shown in Fig. 2. When the ratio was less than 1.1, the yield of D-glucosamine was less than 5.0%. This might be because the H+ ions provided by HCl mainly protonated the amine groups and the hydrolysis of chitosan was hindered. Moreover, the unprotonated amine groups of chitosan may have combined with acid sites in GSA sample, which might be partially related to the less catalytic behaviour. However, further increase of the amount of hydrochloric acid resulted in the increase of D-glucosamine yield. It was assumed that when an excess of H+ ions are supplied by HCl, the amount of solid acid consumed by the amino groups will be less. As a result, the yield of D-glucosamine increased gradually, with the same amount of solid acid. When the proportion of hydrochloric acid and chitosan exceeded 1.1, the amine groups were sufficiently protonated by an adequate number of hydrogen ions. The H+ ions produced by HCl could lead to the hydrolysis of chitosan and eliminate the drawbacks that were mentioned in the introduction. Thus, 1.10 is the most appropriate molar ratio of hydrochloric acid to chitosan.
Influence of time, temperature and catalyst mass on the yield of D-glucosamine
The effect of hydrolysis time on the D-glucosamine yield was studied (chitosan: 1.0 g, mass of GSA: 0.2 g, reaction temperature: 100 °C, nHCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nchitosan: 1.1) and the results are shown in Fig. 3. The yield of D-glucosamine increased from 68.7% to 91.2% with time up to 6 h, and slightly decreased to 88.6% with further increase of time to 8 h. It is generally accepted that a suitable extension of the reaction time is favourable for a solid acid to be in contact with the chitosan molecules and subsequently break the glycosidic bonds. However, further increase of time may lead to further degradation of the targeted product52 and form other undesirable species such as 5-hydroxymethyl furfural.53
:nchitosan: 1.1) and the results are shown in Fig. 3. The yield of D-glucosamine increased from 68.7% to 91.2% with time up to 6 h, and slightly decreased to 88.6% with further increase of time to 8 h. It is generally accepted that a suitable extension of the reaction time is favourable for a solid acid to be in contact with the chitosan molecules and subsequently break the glycosidic bonds. However, further increase of time may lead to further degradation of the targeted product52 and form other undesirable species such as 5-hydroxymethyl furfural.53
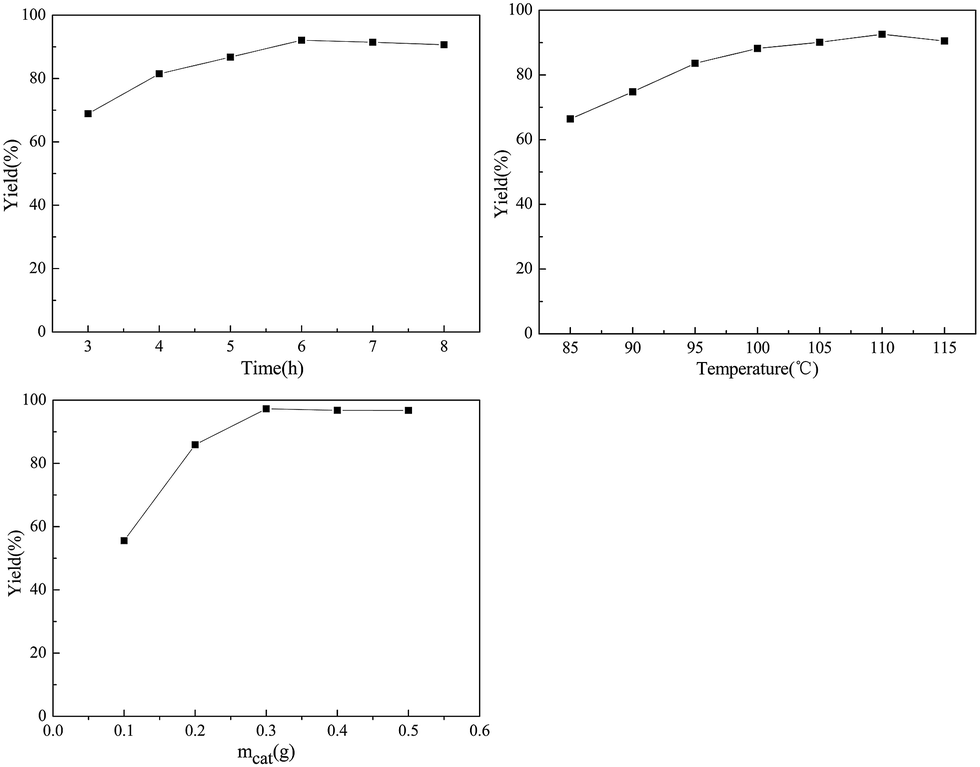 | ||
| Fig. 3 Effect of time, temperature and catalyst mass (mcat) on the yield of D-glucosamine. | ||
The reaction temperature effect on the hydrolysis of chitosan was also studied (chitosan: 1.0 g, solid acid amount: 0.2 g, reaction time: 6.0 h, nHCl:nchitosan: 1.1). It was observed that the D-glucosamine yield progressively increased with temperature and the maximum value (92.3%) was achieved at 110 °C. A slight decline was observed upon heating to 115 °C. It revealed that the reaction temperature greatly affects the yield of D-glucosamine.36 Based on the above results, the optimum reaction temperature might have to be maintained at 110 °C.
After probing the effects of reaction time and temperature, further studies were conducted to evaluate the role of the catalyst's mass (chitosan: 1.0 g, reaction time: 6.0 h, temperature: 110 °C nHCl:nchitosan: 1.1). The yield of D-glucosamine significantly increased with the increase of the catalyst's mass from 0.1 to 0.3 g. When the weight of the catalyst was 0.3 g, D-glucosamine yield could reach as high as 91.3% at 110 °C, after a 6 h reaction. With the addition of more sample, a slight decline of D-glucosamine yield took place. The hydrolysis of chitosan with the GSA catalyst is a heterogeneous system. It was assumed that the hydrogen bonds among –OH groups in the GSA catalyst caused the adsorption of oligosaccharides, and the –SO3H groups efficiently attacked the adsorbed oligosaccharides.44 If the amount of the GSA catalyst is less, the few number of hydroxyl groups can not completely adsorb the chitosan molecules on the surface of the catalyst. Consequently, the –SO3H or the –COOH groups can not efficiently attack the 1,4-β-glucosidic bonds of chitosan. Thus, the optimum amount of solid acid was set at 0.3 g.
Effect of DD of chitosan on D-glucosamine production
Chitosan is a partially deacetylated product of chitin, and the D-glucosamine unit rely on the degree of deacetylation (DD). The yield of D-glucosamine with different DD of chitosan was investigated and the results are shown in Fig. 4. It was apparent that the D-glucosamine yield was closely related with the DD of chitosan. Under optimal reaction conditions, the yield of D-glucosamine increased from 40.0% to 98.1% with the increase of the DD of chitosan from 0.45 to 0.97. The result indicates that the GSA catalyst can cause the fracture of the chitosan, but the deacetylation of the chitosan is very weak.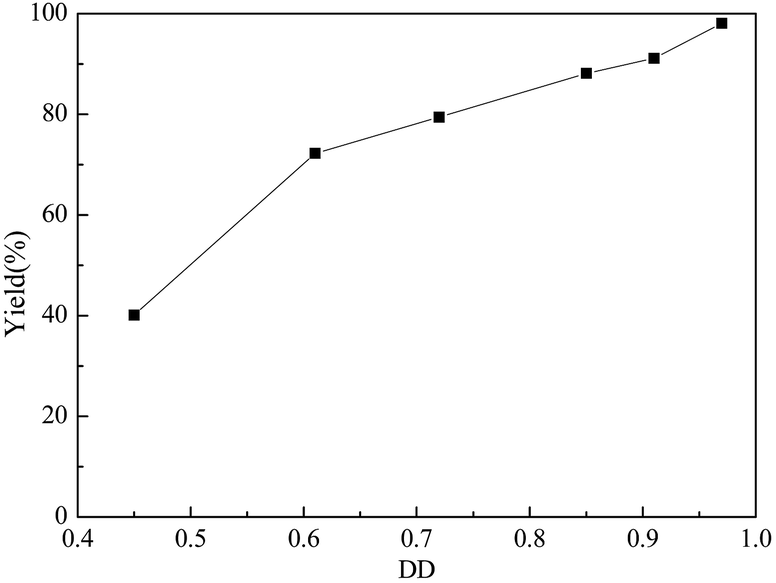 | ||
| Fig. 4 Effect of DD on the yield of D-glucosamine (chitosan: 1.0 g, solid acid: 0.3 g, reaction time: 6.0 h, reaction temperature: 110 °C, nHCl:nchitosan: 1.1). | ||
Besides, the D-glucosamine yield with DD of 0.45 was only ca. 40%, which was much less than the theoretical concentration of D-glucosamine. The extent of the crystallinity of chitosan was inversely associated with its DD.54 It was concluded that the chitosan chains with low DD could result in a more compact structure that might enhance the radical–radical combinations of the chains.55 In addition, acetyl groups in the chitosan chains with low DD can aggregate in solution, because of their hydrophobic interactions.56 This might have prevented the contact between the solid acid catalyst and chitosan molecules, which led to the unsatisfactory catalytic performance.
Recyclability of the GSA catalyst
Recyclability is considered as one of the important characteristics of a promising solid acid catalyst, to reduce the cost. Hence, the stability of the catalyst was investigated. As shown in Fig. 5, the yield of D-glucosamine maintained a constant value, even after the catalyst was repeatedly being used for 6 times. It can be concluded that the GSA catalyst could easily be reused without any remarkable deactivation.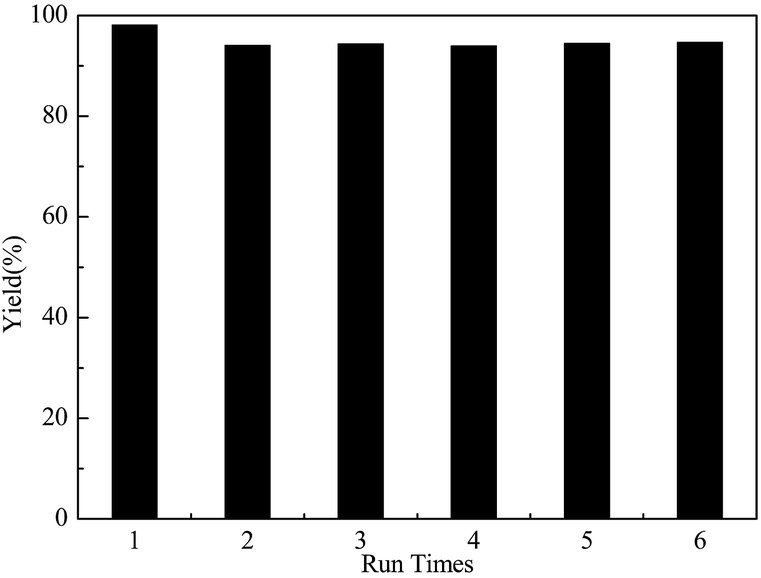 | ||
| Fig. 5 Recyclability of the GSA sample (chitosan: 1.0 g, mass of GSA: 0.3 g, reaction time: 6.0 h, reaction temperature: 110 °C, nHCl:chitosan: 1.1). | ||
Conclusions
Spherical, glucose-derived solid acid (GSA) catalyst bearing –SO3H, –OH and –COOH groups, was demonstrated to be an efficient catalyst system for the hydrolysis of chitosan to produce D-glucosamine. A maximum yield of D-glucosamine (98.1%) could be achieved. The GSA sample was easily synthesized, and no obvious deactivation was observed even after six consecutive runs. This might open a relatively green route for chitosan hydrolysis over heterogeneous catalysts, instead of conventional liquid acid catalysts.Conflicts of interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work, there is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled, “A method of hydrolyzing chitosan to produce D-glucosamine by carbon-based solid acid”.Acknowledgements
This work was supported by the Scientific Research Fund of Liaoning Provincial Education Department (grant number L2014223); and the Youth Foundation of Dalian Polytechnic University (grant number QNJJ201303).References
- X. Chen, H. Yang and N. Yan, Chem.–Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed.
- N. Yan and X. Chen, Nature, 2015, 13, 155–157 CrossRef PubMed.
- M. R. Kasaai, J. Agric. Food Chem., 2009, 57, 1667–1676 CrossRef CAS PubMed.
- R. L. Carmen and B. Roland, J. Controlled Release, 1997, 44, 215–225 CrossRef.
- J. Rhoades, G. Gibson, K. Formentin, M. Beer and R. Rastall, Carbohydr. Polym., 2006, 64, 57–59 CrossRef CAS.
- A. Sawaguchi, S. Ono, M. Oomura, K. Inami, Y. Kumeta, K. Honda, R. Sameshima-Saito, K. Sakamoto, A. Ando and A. Saito, Soil Sci. Plant Nutr., 2015, 61, 471–480 CrossRef CAS.
- S. Yu, P. Chen, X. Liu and L. Li, Catal. Lett., 2015, 145, 1845–1850 CrossRef CAS.
- B. Li, J. Zhang, F. Bu and W. Xia, Carbohydr. Res., 2013, 366, 50–54 CrossRef CAS PubMed.
- X. Yan and H. M. Evenocheck, Carbohydr. Polym., 2012, 87, 1774–1778 CrossRef CAS.
- J. Zhang and N. Yan, Green Chem., 2016, 18, 5050–5058 RSC.
- Z. Han, Y. Zeng, H. Lu and L. Zhang, Carbohydr. Res., 2015, 413, 75–84 CrossRef CAS PubMed.
- L. A. Nudga, V. A. Petrova, E. E. Kever and T. V. Makarova, Russ. J. Appl. Chem., 2002, 75, 1864–1867 CrossRef CAS.
- V. Georgieva, D. Zvezdova and L. Vlaev, Chem. Cent. J., 2012, 6, 81–90 CAS.
- Z. Ma, Y. Wu, Y. He and T. Wu, RSC Adv., 2013, 3, 12049–12051 RSC.
- Y. Sun, J. Zhang, S. Wu and S. Wang, Int. J. Biol. Macromol., 2013, 61, 160–163 CrossRef CAS PubMed.
- Q. Lu, Y. Shi, S. Wang, Y. Yang, B. Han, W. Liu, D. N. M. Jones and W. Liu, Biochim. Biophys. Acta, 2015, 18(50), 1953–1961 CrossRef PubMed.
- Q. Chen, W. Xiao, L. Zhou, T. Wu and Y. Wu, Polym. Degrad. Stab., 2012, 97, 49–53 CrossRef CAS.
- E. Savitri, S. R. Juliastuti, A. H. Sumarno and A. Roesyadi, Polym. Degrad. Stab., 2014, 110, 344–352 CrossRef CAS.
- M. Mahmud, M. I. Naziri, N. Yacob, N. Talip and Z. Abdullah, Am. Inst. Phys., 2014, 19, 136–140 Search PubMed.
- K. Li, R. Xing, S. Liu, Y. Qin, X. Meng and P. Li, Int. J. Biol. Macromol., 2012, 51, 767–773 CrossRef CAS PubMed.
- J. M. Wasikiewicz and S. G. Yeates, Polym. Degrad. Stab., 2013, 98, 863–867 CrossRef CAS.
- M. Yabushita, H. Kobayashi, K. Kuroki, S. Ito and A. Fukuoka, ChemSusChem, 2015, 8, 3760–3763 CrossRef CAS PubMed.
- X. Chen, H. Yang, Z. Zhong and N. Yan, Green Chem., 2017, 19, 2783–2792 RSC.
- J. Zhang and N. Yan, ChemCatChem, 2017, 9, 2790–2796 CrossRef CAS.
- Y. Pierson, X. Chen, F. D. Bobbink, J. Zhang and N. Yan, ACS Sustainable Chem. Eng., 2014, 2, 2081–2089 CrossRef CAS.
- S. G. Kirkham and K. Samarasinghe, J. Orthop. Surg., 2009, 17, 72–76 CrossRef CAS PubMed.
- T. Sun, D. Zhou, J. Xie and M. Fang, Eur. Food Res. Technol., 2007, 225, 451–456 CrossRef CAS.
- M. Yang, H. Shen, Y. Li, C. Shen and P. Zhang, RSC Adv., 2014, 4, 286–295 RSC.
- L. Jia, Y. Wang, Y. Qiao, Y. Qi and X. Hou, RSC Adv., 2014, 4(44), 253–260 Search PubMed.
- T. Nidheesh, P. Gaurav Kumar and P. V. Suresh, Int. Biodeterior. Biodegrad., 2015, 97, 97–106 CrossRef CAS.
- S. Mekasha, H. Toupalová, E. Linggadjaja, H. A. Tolani, L. Anděra, M. Ø. Arntzen, G. Vaaje-Kolstad, V. G. H. Eijsink and J. W. Agger, Carbohydr. Res., 2016, 433, 18–24 CrossRef CAS PubMed.
- G. A. M. Peyron, Carbohydr. Res., 1995, 277, 257–272 CrossRef.
- K. L. Chang, M. C. Tai and F. H. Cheng, J. Agric. Food Chem., 2001, 49, 4845–4851 CrossRef CAS PubMed.
- Z. Zhang, C. Li, Q. Wang and Z. K. Zhao, Carbohydr. Polym., 2009, 78, 685–689 CrossRef CAS.
- M. Mei, K. Takashi, I. Sosaku and M. Sukekuni, Food Sci. Technol. Res., 2006, 12, 85–90 Search PubMed.
- K. M. Anna, V. M. F. Lai, F. Fiedorowicz, K. Gohar and T. Piotr, Biotechnol. Prog., 2008, 24, 385–388 CrossRef PubMed.
- M. Ming, T. Kuroiwa, S. Ichikawa, S. Sato and S. Mukataka, Biochem. Eng. J., 2006, 28, 289–294 CrossRef CAS.
- A. Konieczna-Molenda, M. Fiedorowicz, W. Zhong and P. Tomasik, Carbohydr. Res., 2008, 343, 3117–3119 CrossRef CAS PubMed.
- K. M. V. Aslak Einbu, Biomacromolecules, 2008, 9, 1870–1875 CrossRef PubMed.
- A. Einbu, H. Grasdalen and K. M. Vårum, Carbohydr. Res., 2007, 342, 1055–1062 CrossRef CAS PubMed.
- K. M. Varum and O. Smidsrod, Carbohydr. Polym., 2001, 46, 89–98 CrossRef CAS.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1039 RSC.
- I. F. Nata, C. Irawan, P. Mardina and C.-K. Lee, J. Solid State Chem., 2015, 230, 163–168 CrossRef CAS.
- S. Satoshi, N. Kiyotaka, K. Masaaki, Y. Daizo, K. Hideki, H. Shigenobu and H. Michikazu, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef PubMed.
- D. Yamaguchi and M. Hara, Solid State Sci., 2010, 12, 1018–1023 CrossRef CAS.
- R. S. Thombal, A. R. Jadhav and V. H. Jadhav, RSC Adv., 2015, 5, 12981–12986 RSC.
- G. Chen and B. Fang, Bioresour. Technol., 2011, 102, 2635–2640 CrossRef CAS PubMed.
- R. A. Krishnan, P. Deshmukh, S. Agarwal, P. Purohit, D. Dhoble, P. Waske, D. Khandekar, R. Jain and P. Dandekar, Carbohydr. Polym., 2016, 151, 417–425 CrossRef CAS PubMed.
- J. Zou, D. Cao, W. Tao, S. Zhang, L. Cui, F. Zeng and J. Cai, RSC Adv., 2016, 6, 49528–49536 RSC.
- M. L. Ibrahim, R. Umer and H. T. Y. Yun, Arabian J. Chem., 2016, 9, 178–189 CrossRef.
- I. M. Lokman, M. Goto, U. Rashid and Y. H. Taufiq-Yap, Chem. Eng. J., 2016, 284, 872–878 CrossRef CAS.
- M. H. Zong, Z. Q. Duan, W. Y. Lou, T. J. Smith and H. Wu, Green Chem., 2007, 9, 434 RSC.
- H. Liu, J. Bao, Y. Du, X. Zhou and J. F. Kennedy, Carbohydr. Polym., 2006, 64, 553–559 CrossRef CAS.
- M. Dash, F. Chiellini, R. M. Ottenbrite and E. Chiellini, Prog. Polym. Sci., 2011, 36, 981–1014 CrossRef CAS.
- T. Pinar, C. Hazal and M. Sen, Radiat. Phys. Chem., 2014, 94, 236–239 CrossRef.
- Y. Wang, B. Ju and S. Zhang, Carbohydr. Polym., 2012, 90, 696–702 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |