
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural phase transition and bonding properties of high-pressure polymeric CaN3†
Pugeng Hou*a,
Lili Lianb,
Yongmao Caia,
Bao Liua,
Bo Wanga,
Shuli Weic and
Da Li *c
*c
aCollege of Science, Northeast Electric Power University, No. 169 Changchun Road, Jilin City, 132012, P. R. China. E-mail: houpugeng@126.com
bThe First Hospital of Jilin University, Changchun, 130021, P. R. China
cState Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun, 130012, P. R. China. E-mail: dali@jlu.edu.cn
First published on 23rd January 2018
Abstract
Alkaline-earth metal polynitrides can be used as a type of starting materials in the synthesis of polymeric nitrogen, which is used as a potential high-energy-density material. The structural evolutionary behaviors of nitrogen in CaN3 were studied at the pressure up to 100 GPa using a particle-swarm optimization structure search combining with density functional theory. Two new stable phases with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and C2/m space groups at the pressures of 26 and 60 GPa were identified for the first time. Throughout the stable pressure range, these two structures are semiconductors and consist of the N atoms in sp2-hybrid states. To the best of our knowledge, this is the first time an N6 chain is reported in case of alkaline-earth metal polynitrides. The stable polynitrogen compounds and polymeric nitrogen as high-energy-density materials have potential applications. The present results open a new possible avenue to synthesize high-energy-density polynitrogen.
and C2/m space groups at the pressures of 26 and 60 GPa were identified for the first time. Throughout the stable pressure range, these two structures are semiconductors and consist of the N atoms in sp2-hybrid states. To the best of our knowledge, this is the first time an N6 chain is reported in case of alkaline-earth metal polynitrides. The stable polynitrogen compounds and polymeric nitrogen as high-energy-density materials have potential applications. The present results open a new possible avenue to synthesize high-energy-density polynitrogen.
1 Introduction
To discover and synthesize environmentally harmless high energy-density materials (HEDMs) is becoming increasingly important at present. Among the myriad HEDMs, the polynitrogen compounds are of significant interest due to their particularly high energy and high enthalpy of formation among other features.1–9 Nitrogen exists in form of N2 molecules in nitrogen gas with N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds. It is significantly interesting if the abundant N2 molecules can be transformed into polynitrogen with N–N bonds and N
N bonds. It is significantly interesting if the abundant N2 molecules can be transformed into polynitrogen with N–N bonds and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds. The N–N and NN or NN bonds have large energy difference between them, which can be used in industrial applications. Although numerous theoretical studies predicting that some high-pressure nitrogen phases might be stable,7,10 it was not until 2004 that the cubic gauche phase of nitrogen (cg-N) was experimentally synthesized using high pressure and high temperature treatment (110 GPa, 2000 K).11 It has been predicted that the single-bond phase of solid nitrogen has more than three times higher energy storage capacity than that of the most powerful energetic materials.12
N bonds. The N–N and NN or NN bonds have large energy difference between them, which can be used in industrial applications. Although numerous theoretical studies predicting that some high-pressure nitrogen phases might be stable,7,10 it was not until 2004 that the cubic gauche phase of nitrogen (cg-N) was experimentally synthesized using high pressure and high temperature treatment (110 GPa, 2000 K).11 It has been predicted that the single-bond phase of solid nitrogen has more than three times higher energy storage capacity than that of the most powerful energetic materials.12
Recently, metal azides, as a type of precursors, have been proposed to be used in the formation of polymeric nitrogen. One feasible approach to obtain such polymeric nitrogen phases is to form nitrogen-rich alloys with other elements. Such “chemical precompression” may reduce the pressure of polymerization significantly. Theoretical and experimental studies are inspired by elusive polymeric nitrogen and considerable progress has been achieved in the exploration of stable nitrogen-rich compounds. Recently, metal azides can be used as the starting materials to synthesize numerous types of potential high-energy-density materials because the synthesis pressure of metal azides is potentially lower than that of pure nitrogen gas. Over the years, various metal azides have been proposed in numerous experimental and theoretical studies, for example, LiN3,13–16 LiN5,13 NaN3,17–21 KN3,20,22–27 CsN3,23,28 and AlN3.29 Moreover, N2H,30,31 C–N system,32,33 S–N system,34 and the P–N system35 are also studied as high-energy materials. These successes precipitate an increasing interest in searching for other forms of stable polynitrogen in metal polynitrogen compounds in the recent years.
The chemistry of calcium is relatively underdeveloped compared to that of its analogues (Be, Mg, Sr, and Ba). Recently, we theoretically explored the phase diagram of the Mg–N system at the pressures ranging from 0 to 100 GPa36 and discovered a series of new compounds in this family, including Mg2N3, MgN, MgN3, MgN4, and MgN5. The predicted compounds contain a rich variety of polynitrogen forms ranging from small molecules (N2, N3, N4, and N6) to the extended nitrogen chains. Alkaline-earth metal calcium and alkali metal potassium are adjacent in the periodic elements table. In addition, calcium can exhibits a similar chemical activity as compared to that of alkali metal element potassium. Thus, we can expect that mixing reactive calcium element with nitrogen will realize more diverse structures that obtain higher energy densities of the products. Therefore, a study of calcium polynitride would help to theoretically investigate more promising high-energy materials among alkaline-earth metal polynitrides. High pressure-treatment has emerged as a powerful tool to investigate the physical and chemical behaviours of materials and it can be a crucial thermodynamic parameter. This study has led to the synthesis of new and unexpected products. A new orthorhombic phase was predicted as the thermodynamic ground state of CaN2, which has considerably lower energy than that of the synthesized phase as well as the theoretical models considered earlier.37 Zhang et al. identified a tetragonal Ca2N structure under high pressures, giving an excellent agreement between the theoretical and experimental XRD data.38 Recently, Zhu et al. theoretically explored the phase diagram of the Ca–N system, including Ca2N3, CaN, CaN3, CaN4, and CaN5.39 The predicted CaNx compounds contained a rich variety of polynitrogen forms ranging from small molecules (N2, N4, N5, and N6) to the extended chains (N∞). The CaNx crystals with polynitrogens were expected to be highly exothermic, making them as the potential high-energy-density materials. The newly predicted CaN3 phase was calculated to become thermodynamically stable under low pressure (P = 8 GPa). At 8 GPa, CaN3 has an orthorhombic Pmma structure.
In the present study, the possible formation and the stability of the polymeric nitrogen compounds were investigated in the binary CaN3 under high pressure. The structure searches for the stable CaN3 compound were performed using an unbiased structure prediction method based on particle-swarm optimization algorithm in conjunction with density-functional calculations. Then, the electronic properties and chemical bonding of the identified Ca–N compounds were investigated systematically in order to study the properties and the potential applications of the binary CaN3 compound as high energy-density materials. The lattice dynamics, structural stability, electronic structure, and bonding nature were studied, providing more insights into the mechanism of the pressure-induced physical properties. The understanding of metal–nitrogen interactions and the bonding nature in these polynitrogens is of great importance to the research on nitrogen chemistry.
2 Computation details
The crystal structure prediction for the CaN3 system via global minimization of free energy surfaces was performed through CALYPSO code, which has demonstrated recent success in predicting the high-pressure structures of numerous substances from the elemental to binary and ternary compounds and validated with various known compounds.40–42 The lowest-energy structures for CaN3 were determined at 0, 20, 50, 80, and 100 GPa with the systems containing one to six formula units (f.u.) in the simulation cell. The underlying ab initio structural relaxations and electronic band structural calculations for CaN3 were performed in the framework of the density functional theory (DFT) within generalized gradient approximation Perdew–Burke–Ernzerhof (GGA-PBE)43 as implemented in the VASP (Vienna ab initio simulation package) code.44 The projector-augmented wave (PAW)45 pseudopotentials are used from the VASP potential library. The 3s23p64s2 for Ca and 2s22p3 for N were treated as the valence states. The computational parameters were tested such that the energy calculations were well converged to better than 1 meV per atom. A plane-wave energy cutoff of 620 eV and appropriate Monkhorst–Pack46 k-meshes (k-points grid of 0.03 Å−1) were used in the Brillouin zone. We calculated the net charge based on Bader analysis.47,48 The supercell method with PHONOPY code was used to calculate the phonon frequencies for all structures.49 In addition, we used the DFT + D2 approach,50–52 which is a simple and efficient way to approximately account for the long-range van der Waals interactions. In this method, the sum of the induced dipole–dipole interactions (Edisp) was added to the self-consistent total energy calculated within Kohn–Sham DFT (EKS-DFT), EDFT+D2 = EKS-DFT + Edisp. We choose a cutoff radius of 30 Åfor the van der Waals interactions and the global scaling parameter s6 was set to 0.75.3 Results and discussions
3.1 Phase stability
The most favorable Ca–N compounds obtained in the structure search at various pressures are presented in Fig. 1a.39 For the energetically most favorable CaN3 structures, the calculated formation enthalpies ΔHf in the pressure range from 0 to 100 GPa are shown in Fig. 1b. The ΔHf of each CaN3 structure was calculated by ΔHf (CaN3) = H(CaN3) − H(Ca) − 3H(N) at T = 0 K. The known solid calcium (α, β, and γ phase) and solid nitrogen (α-, Pbcn-, P2/c-, and cg-phases)53 were used as the reference structures. These structures are energetically most favorable in their corresponding stable pressure ranges. In order to determine the phase transition pressure for the predicted CaN3, we have plotted out the enthalpy curves relative to the ambient pressure P-CaN3. The enthalpies of the most energetically competitive structures are compared over the pressure range from 0 GPa to 100 GPa as shown in Fig. 1. The most stable structure has a monoclinic phase with P symmetry from 26 GPa up to 40 GPa, which is then replaced by a lower-enthalpy C2/c structure that has been proposed by Zhu et al.39 With further compression above 60 GPa, another monoclinic phase, C2/m-CaN3, becomes the most thermodynamically stable phase and remains the lowest-enthalpy phase up to 100 GPa. Since the two low-pressure P-CaN3 and high-pressure C2/m-CaN3 phases are novel for alkali-earth metal polynitrides and metal azides, the investigation about the properties of the two high-pressure phases in detail is essential. The ambient-pressure decomposition of P-CaN3 and C2/m-CaN3 to CaN2 and N2 are estimated to release 1.04 eV and 1.34 eV energy per formula unit (f.u.), which corresponds to an energy density of approximately 1.14 kJ g−1 and 1.47 kJ g−1, respectively. Such high energy content can establish that CaN3 is indeed a high energy density material. It can replace modern high explosives, such as TATB, RDX, and HMX, which typically have energy densities ranging from 1 to 3 kJ g−1.54 Thus, we may find their applications as a high energy materials. This study, therefore, represents an exciting prediction and can hopefully encourage the experimental efforts in their synthesis.
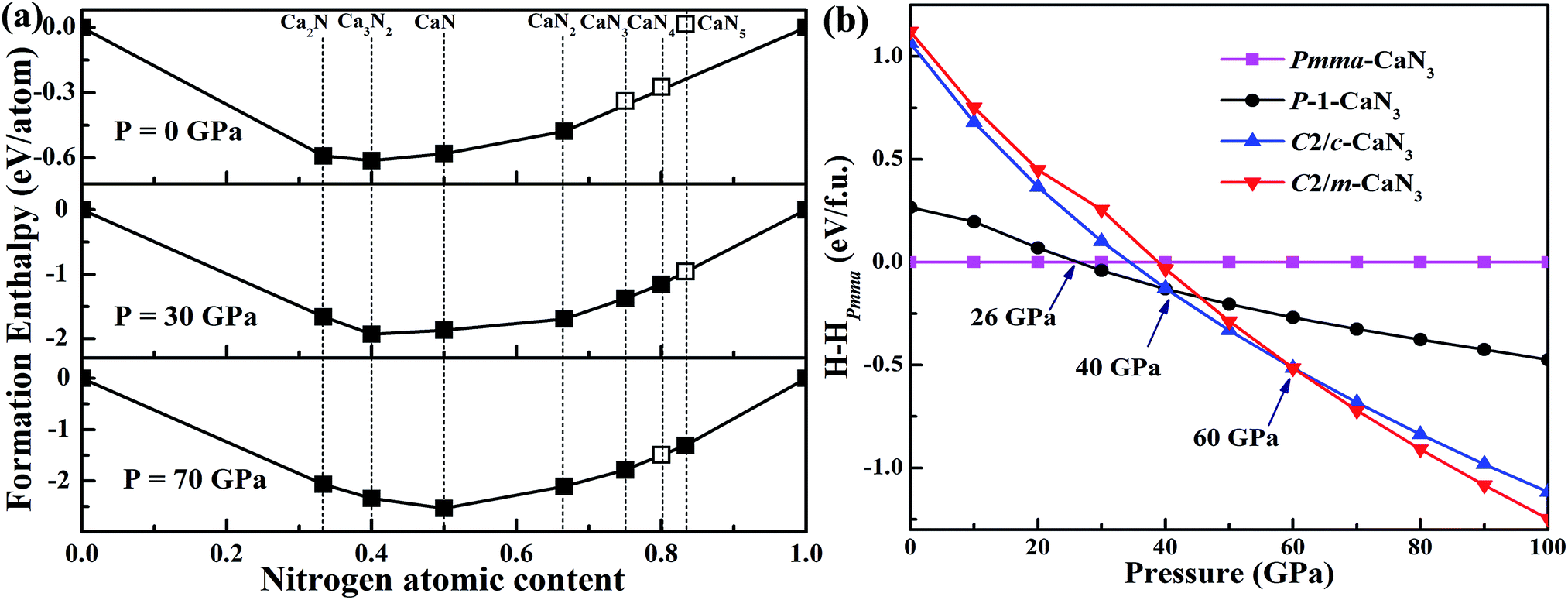 | ||
| Fig. 1 (a) Relative enthalpies of the formation of Ca–N phases with respect to solid calcium and nitrogen. Reference to the results of Zhu et al.37 (b) Relative enthalpies of the CaN3 relative to the Pmma-CaN3 calculated by the DFT + D2 approach as a function of pressure. | ||
The calculated relations of phonon dispersion of low-pressure P-CaN3 and high-pressure C2/m-CaN3 also confirmed their mechanical and dynamical stability as shown in Fig. 2. No imaginary phonon mode can be observed, which proves clearly their lattice dynamical stability. Under some kinetic regimes at ambient conditions, the calcium polynitride CaN3 also has the possibility of stabilizing. Advantageously, these two structures P-CaN3 and C2/m-CaN3 are mechanically stable at ambient conditions and they may possibly quench under an ambient pressure.
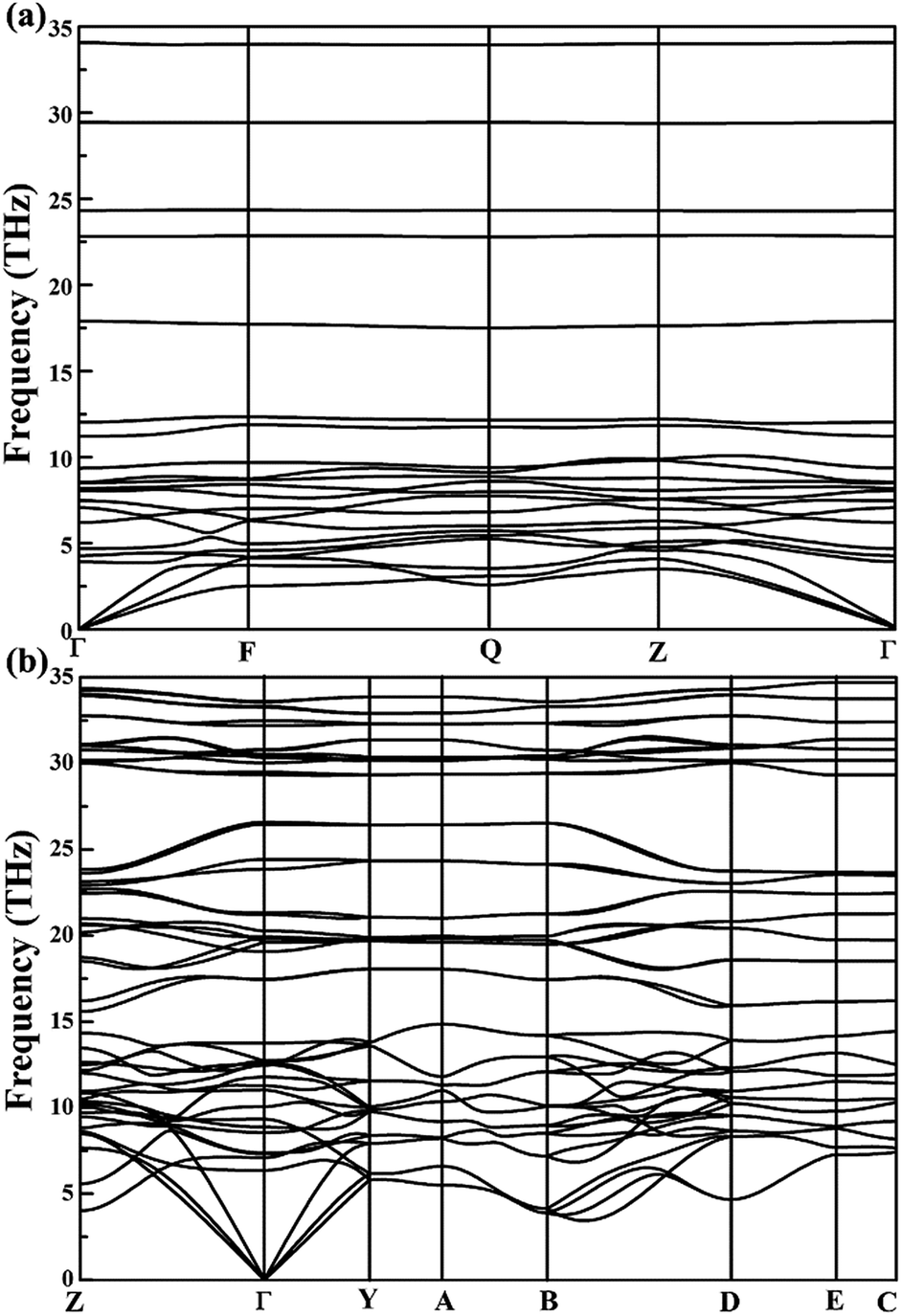 | ||
| Fig. 2 Phonon dispersion curves for (a) P-CaN3 at ambient pressure and (b) C2/m-CaN3 at 60 GPa. | ||
Another important indicator of the existence of these structures is their mechanical stability. According to the Born–Huang criterion,55 it was found that P-CaN3 and C2/m-CaN3 were mechanically stable at an ambient pressure and 60 GPa. In order to be used in potential technological and industrial applications, the understanding of the mechanical properties (elastic constant and anisotropy) of the predicted CaN3 structures is important. The elastic constants (Cij), bulk modulus (B), shear modulus (G), Young's modulus (Y), and Poisson's ratio (ν) of the P-CaN3 and C2/m-CaN3 structures are summarized in Table 1. The elastic constants for both P-CaN3 and C2/m-CaN3 were calculated using the strain–stress method. For the monoclinic crystals, if they are mechanically stable, the elastic constants need to satisfy the following mechanical criteria:
| C11 > 0, C22 > 0, C33 > 0, C44 > 0, C55 > 0, C66 > 0 |
| [C11 + C22 + C33 + 2(C12 + C13 + C23)] > 0 |
| (C33C55 − C352) > 0, (C44C66 − C462) > 0, (C22 + C33 − 2C23) > 0 |
| [C22(C33C55 − C352) + 2C23C25C35 − C232C55 − C232C33] > 0. |
-CaN3 and C2/m-CaN3 structures under P = 0 GPa and 60 GPa
| C11 | C22 | C33 | C44 | C55 | C66 | C12 | C13 | C23 | C35 | C46 | B0 | G | Y | ν | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P-CaN3 |
216 | 208 | 122 | 64 | 50 | 86 | 77 | 46 | 82 | −1.4 | −5.5 | 101 | 59 | 149 | 0.25 |
| C2/m-CaN3 | 546 | 546 | 596 | 246 | 193 | 190 | 235 | 194 | 233 | −0.3 | 18.0 | 333 | 191 | 481 | 0.26 |
As illustrated in Table 1, we can observe that the elastic constants for the monoclinic P-CaN3 and C2/m-CaN3 structures satisfy the above conditions, which implies that the two phases, P-CaN3 and C2/m-CaN3, are elastically stable at ambient pressure and 60 GPa, respectively. Hence, the calculations indicate that P-CaN3 and C2/m-CaN3 are elastically stable. It is valuable to be mentioned that the bulk modulus for the C2/m-CaN3 phase, which was calculated from the elastic constants, is 333 GPa (Table 1). Hence, the C2/m-CaN3 phase is also a high-incompressibility material.
3.2 Structural features
CaN3 is predicted to take the monoclinic P space group at 0 GPa as shown in Fig. 3a. The lattice constants of the predicted P-CaN3 are a = 4.744 Å, b = 5.307 Å, c = 3.182 Å, α = 76.70°, β = 97.94°, and γ = 80.80° at P = 30 GPa. Four inequivalent atoms occupy the crystallographic structure: Ca at 2i (0.696, 0.198, 0.077), N1 at 2i (0.307, 0.321, 0.399), N2 at 2i (0.140, 0.517, 0.507) and N3 at 2i (0.204, 0.136, 0.262) positions. The C2/m-CaN3 crystal is predicted to become energetically stable near 70 GPa. This phase has the optimized lattice parameters: a = 8.125 Å, b = 5.175 Å, c = 3.206 Å, α = 90.0°, β = 67.2°, and γ = 90.0°. The atomic positions are as follows: Ca atoms at 4i (0.673, 0.00, 0.185) and N1 atom at 8j (0.422, 0.275, 0.204), N2 atom at 4i (0.085, 0.0, 0.542).
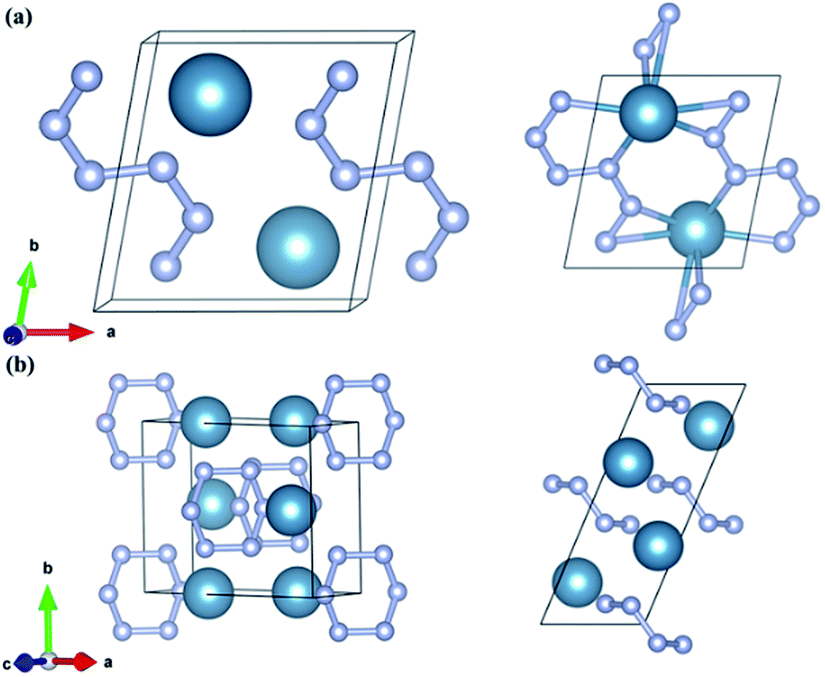 | ||
| Fig. 3 Crystal structures of the predicted stable (a) P-CaN3 and (b) C2/m-CaN3. The large and small spheres denote calcium and nitrogen atoms, respectively. | ||
The potential energy storage capabilities for the two predicted new P-CaN3 and C2/m-CaN3 have a close relationship with their structure motifs and the patterns of the N atoms. Fig. 3a shows the crystal structure of P-CaN3, in which the N atoms adopt the N6 chain in the (001) plane. The N atoms in this structure are sp2-hybridized containing a high content of N–N single bonds in P-CaN3. To the best of our knowledge, this is the first time the N6 chain is reported in case of alkaline-earth metal polynitrides. In addition, the bond lengths of N–N are from 1.323 Å to 1.352 Å, which are greater than that of an NN bond (1.20 Å) and shorter than that of an N–N single bond (1.45 Å). This intermediate distance has a connection with the electron delocalization, in which the π-electrons are distributed equally among six the N atoms. Each N atom of the N6 chain is coordinated with two nearest Ca atoms and each Ca atom is six-coordinated with nearby N atoms (dCa–N = 2.51 Å at 0 GPa). With an increase in the pressures to 60 GPa, a promising monoclinic C2/m phase including partial single N–N bonds was identified for CaN3. The high-pressure C2/m-CaN3, interestingly, has a puckered layer-like structure with six N atoms forming a nonplanar N6 ring as shown in Fig. 3b. The hexagonal N6 ring has been researched in numerous theoretical studies, but they are not satisfactory.56,57 By adding the coordinate-covalent bonds from oxygen, the isoelectronic with benzene N6 ring can be formed.58 Moreover, the planar N6 ring can be remarkably enhanced using the incorporation of a metal atom in these metal–N6 molecule compounds.59 We can obtain the N6 rings by theoretical simulation calculation in the bulk materials. This shows that additional metal atoms can play a very important role in stabilizing the N6 ring. More recently, a C2/m-CaN3 structure containing the N6 ring has been reported, which has been also predicted in LiN3,16,60 NaN3,21 MgN3,36 and CaN3.39 Moreover, the pressure effect on calcium azide might have a great impact in the rational design of potential polymeric nitrogen.
3.3 Electronic structures
To better understand the nature of the chemical bonding between two atoms and the formation mechanism of low-pressure P-CaN3 and high-pressure C2/m-CaN3, we have calculated their projected density of states (PDOS) at ambient pressure and high pressure P = 60 GPa as shown in Fig. 4. We can observe that P-CaN3 and C2/m-CaN3 are semiconductors characterized by an indirect band gap of ∼0.76 eV and ∼2.38 eV, respectively. The atom-resolved DOS of the low-pressure P-CaN3 and high-pressure C2/m-CaN3 structures reveal that the densities of states near Fermi levels are primarily originated from the N orbital electrons. In the “N6” chain of P-CaN3 (Fig. 3a), all N atoms are sp2-hybridized, forming five N–N σ bonds with neighboring N atoms and six lone pairs. The sp2-hybridized orbitals of the four central N atoms in the “N6” chain form two σ bonds with the sp2-hybridized orbital located at both N atoms at each side. The remaining sp2-hybridized orbitals at four central N atoms as well as the two sp2-hybridized orbitals at both terminal N atoms are filled and form lone pairs. Therefore, all of the bonding states and lone pair states are filled and all of the anti-bonding states are unoccupied in P-CaN3, leading to a semiconductive state. The strong covalent bondings between different inequivalent N atoms and the lone pairs on the side N atoms are also revealed by the ELF (Fig. 5a). This 6-membered ring with 10 π-electrons in high-pressure C2/m-CaN3 follows the aromatic 4n + 2 Hückel rule and it is similar to the well-known isoelectronic inorganic cyclic S3N3− and P64− species.61 In case of MgN3, Yu et al. reported that three bonding π-molecular orbitals (MO) as well as two degenerated antibonding π*-molecular orbitals in the N6 rings were fully occupied over the six π-levels, leading to only one π-bond delocalized over the 6 nitrogen atoms.62 Their electronic situation explains well the structural feature encountered in N64−, in which the N–N bonds are slightly stronger than the known N–N single bonds. Thus, in C2/m-CaN3, an energy gap exists in between 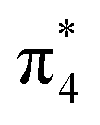 and
and  MO—top of the valence band and the empty
MO—top of the valence band and the empty 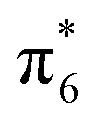 MO—bottom of the conduction band; hence, it behaves as a semiconductor.
MO—bottom of the conduction band; hence, it behaves as a semiconductor.
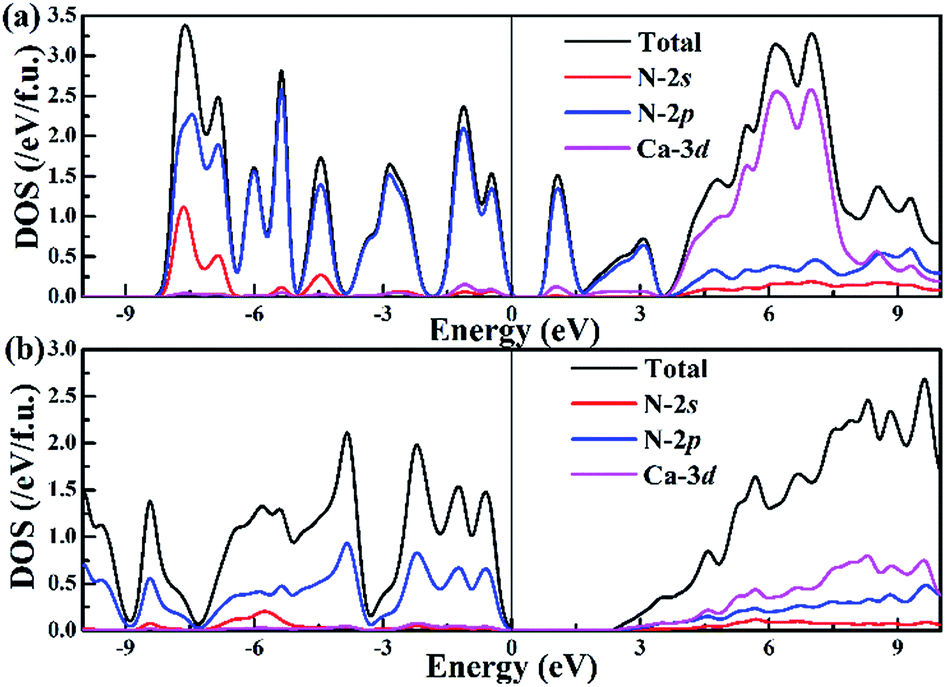 | ||
| Fig. 4 Band structure and projected density of states of (a) P-CaN3 at ambient pressure and (b) C2/m-CaN3 at 60 GPa. | ||
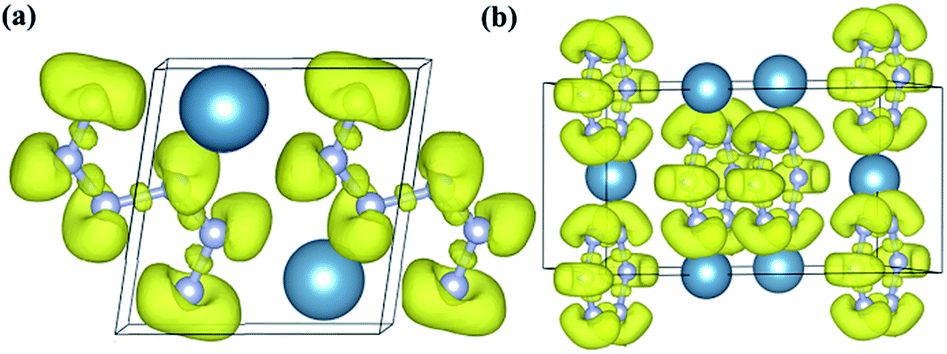 | ||
| Fig. 5 The ELF distributions of (a) P-CaN3 at ambient pressure and (b) C2/m-CaN3 at 60 GPa. The value of isosurface is 0.75. | ||
In addition, to study the impact of the partial occupation of N-2p orbital on the electronic properties, we calculated the charge of Ca and N atoms of P-CaN3 and C2/m-CaN3 based on Bader analysis. The Bader method is used to analyze the charge transfer by implementing an algorithm developed by Henkelman et al.47,48 The result reveals that the Ca atoms contribute almost 1.3e to the N atoms forming the charged “N6” chain, which suggests that P-CaN3 has ionic characteristics in the chemical bonds. In the predicted P-CaN3 and C2/m-CaN3 crystal, the Ca atoms behave as electron donors, whose concentration strongly influences the N–N bonding. This finding contributes to better understanding the chemical bonding behaviors.
3.4 Chemical bonding
Subsequently, in order to understand the bonding character of two CaN3 structures, the electronic localization function (ELF)63 was calculated as shown in Fig. 5. For two CaN3 structures, the strong covalent bonding between nitrogen atoms and the lone pair electrons is revealed clearly by the ELF (Fig. 5). The large areas with the large ELF values between N and N atoms are shown in Fig. 5, which is typical of a strong covalent bonding. In the “N6” chain of P-CaN3 shown in Fig. 1b, all N atoms are sp2-hybridized. For intermediate four N atoms, two sp2-hybridized orbitals form two N–N σ bonds with neighboring two atoms. The remaining sp2-hybridized orbital of this N atom is filled and forms one lone pair. For bicephalous two N atoms, one sp2-hybridized orbital forms one σ bond with the sp2-hybridized orbital of one neighboring N atom. The remaining two sp2-hybridized orbitals of the N atoms are filled and form lone pairs. The covalent bondings between different inequivalent N atoms and the lone pairs on the side N atoms are also revealed by the ELF (Fig. 5). Therefore, all of the bonding states and lone pair states are filled and all of the anti-bonding states are unoccupied in P-CaN3, leading to a semiconducting state.
To get further insight into the N–N bonding information in the P-CaN3 compound, we performed crystal orbital Hamilton population (COHP) analysis. COHP can partition the band-structure energy into the orbital–pair interactions and it is useful to indicate bonding, nonbonding, and antibonding contributions for the band-structure energy. Herein, the familiar COHP approach that stems from a plane-wave calculation was adopted and it was dubbed “projected COHP” (pCOHP).64,65 Then, we analyzed the COHP and integral COHP (ICOHP) to characterize the bonding nature of the N–N pairs in P-CaN3. Fig. 6 shows that the nitrogen atom pairs with different distances exist in the form of the covalent bonds. The COHP plot of the N1–N3 pairs and N1–N5 pairs are shown in Fig. 6a and b, respectively. It is clear to see that the bonding states in P-CaN3 are fully occupied and the antibonding states are unoccupied, which can lead to the covalent bonding between two adjacent nitrogen atoms. The bonding strength based on counting the energy-weighted population of wave functions between two atomic orbitals in P-CaN3 can be expressed by the theoretical values of ICOHP. The values of ICOHP corresponding to those shown in Fig. 6 are listed in the order −4.243 and −3.044. It is shown that the interaction between N1–N5 nitrogen atoms is stronger than that between N1–N3 atoms. For high-pressure C2/m-CaN3, all N atoms in the “N6” ring are sp2-hybridized, two sp2-hybrid orbitals form two N–N σ bonds with neighboring two atoms, and the remaining sp2-hybrid orbital of this N atom is filled and forms one lone pair. The single-bond feature can be inferred from the similar bond lengths and strengths of these N–N bonds. As a potential high-energy-density material, the dissociation of P-CaN3 and C2/m-CaN3 is highly exothermic because of the existence of the single-bond feature.
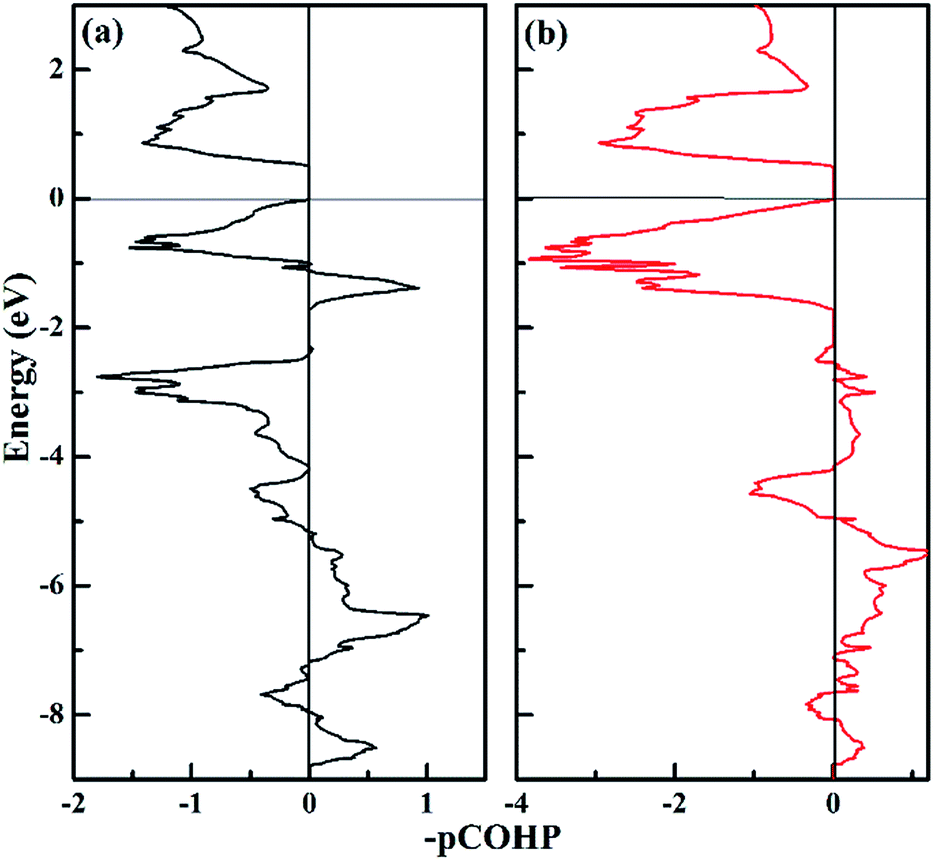 | ||
| Fig. 6 Plot of COHP for P-CaN3. The positive and negative COHP values denote bonding and antibonding interactions, respectively. (a) N1–N3 pairs separated by 1.34 Å. (b) N1–N5 pairs separated by 1.30 Å. | ||
4 Conclusions
In summary, the high-pressure polymeric nitrogen phase of CaN3 was explored using first-principles calculations up to a high-pressure of 100 GPa. One new monoclinic P-CaN3 was predicted to become energetically stable under low pressure. For the first time, we identified a novel phase featuring the charged “N6” chain in the P-CaN3 structure. On further compression above 60 GPa, another monoclinic phase C2/m-CaN3 becomes the most thermodynamically stable phase and remains the lowest-enthalpy phase up to 100 GPa. Moreover, this new predicted high-pressure structure can be mechanically stable at ambient conditions, which may make an ambient-pressure recovery possible. The nitrogen atoms are sp2-hybridized in the “N6” chain and the “N6” ring, which indicates that each N atom forms one or two σ bonds with its neighboring N atoms, suggesting an exceptionally high content of the single N–N bonds. The present study provides new insights into the understanding of polynitrogens and encourages the experimental exploration of these promising materials in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 11747003, 11404134), Jilin Provincial Science and Technology Development Project of China (20160520016JH). Project 2017005 supported by Graduate Innovation Fund of Jilin University.References
- Y. Li, J. Hao, H. Liu, S. Lu and J. S. Tse, Phys. Rev. Lett., 2015, 115, 105502 CrossRef PubMed.
- Z. Raza, C. J. Pickard, C. Pinilla and A. M. Saitta, Phys. Rev. Lett., 2013, 111, 235501 CrossRef PubMed.
- L. Andrews, M. Zhou, G. V. Chertihin, W. D. Bare and Y. Hannachi, J. Phys. Chem. A, 2000, 104, 1656–1661 CrossRef CAS.
- M. M. G. Alemany and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 024110 CrossRef.
- B. Hirshberg, R. B. Gerber and A. I. Krylov, Nat. Chem., 2014, 6, 52–56 CrossRef CAS PubMed.
- F. Zahariev, S. V. Dudiy, J. Hooper, F. Zhang and T. K. Woo, Phys. Rev. Lett., 2006, 97, 155503 CrossRef CAS PubMed.
- X. Wang, Y. Wang, M. Miao, X. Zhong, J. Lv, T. Cui, J. Li, L. Chen, C. J. Pickard and Y. Ma, Phys. Rev. Lett., 2012, 109, 175502 CrossRef PubMed.
- F. Zahariev, A. Hu, J. Hooper, F. Zhang and T. Woo, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 214108-6 CrossRef.
- C. Mailhiot, L. H. Yang and A. K. McMahan, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 14419–14435 CrossRef CAS.
- Y. Ma, A. R. Oganov, Z. Li, Y. Xie and J. Kotakoski, Phys. Rev. Lett., 2009, 102, 065501 CrossRef PubMed.
- M. I. Eremets, A. G. Gavriliuk, I. A. Trojan, D. A. Dzivenko and R. Boehler, Nat. Mater., 2004, 3, 558–563 CrossRef CAS PubMed.
- J. Uddin, V. Barone and G. E. Scuseria, Mol. Phys., 2006, 104, 745–749 CrossRef CAS.
- F. Peng, Y. Yao, H. Liu and Y. Ma, J. Phys. Chem. Lett., 2015, 6, 2363–2366 CrossRef CAS PubMed.
- X. Huang, D. Li, F. Li, X. Jin, S. Jiang, W. Li, X. Yang, Q. Zhou, B. Zou, Q. Cui, B. Liu and T. Cui, J. Phys. Chem. C, 2012, 116, 9744–9749 CAS.
- S. A. Medvedev, I. A. Trojan, M. I. Eremets, T. Palasyuk, T. M. Klapotke and J. Evers, J. Phys.: Condens. Matter, 2009, 21, 195404 CrossRef CAS PubMed.
- X. Wang, J. Li, J. Botana, M. Zhang, H. Zhu, L. Chen, H. Liu, T. Cui and M. Miao, J. Chem. Phys., 2013, 139, 164710 CrossRef PubMed.
- M. I. Eremets, M. Y. Popov, I. A. Trojan, V. N. Denisov, R. Boehler and R. J. Hemley, J. Chem. Phys., 2004, 120, 10618–10623 CrossRef CAS PubMed.
- K. Ramesh Babu and G. Vaitheeswaran, Chem. Phys. Lett., 2013, 586, 44–50 CrossRef CAS.
- B. A. Steele and I. I. Oleynik, Chem. Phys. Lett., 2016, 643, 21–26 CrossRef CAS.
- H. Zhu, F. Zhang, C. Ji, D. Hou, J. Wu, T. Hannon and Y. Ma, J. Appl. Phys., 2013, 113, 033511 CrossRef.
- M. Zhang, K. Yin, X. Zhang, H. Wang, Q. Li and Z. Wu, Solid State Commun., 2013, 161, 13–18 CrossRef CAS.
- J. Hooper and E. Zurek, J. Phys. Chem. C, 2012, 116, 13322–13328 CAS.
- D. Hou, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 064127 CrossRef.
- D. Hou, F. Zhang, C. Ji, T. Hannon, H. Zhu, J. Wu, V. I. Levitas and Y. Ma, J. Appl. Phys., 2011, 110, 023524 CrossRef.
- C. Ji, F. Zhang, D. Hou, H. Zhu, J. Wu, M.-C. Chyu, V. I. Levitas and Y. Ma, J. Phys. Chem. Solids, 2011, 72, 736–739 CrossRef CAS.
- J. Li, X. Wang, N. Xu, D. Li, D. Wang and L. Chen, Europhys. Lett., 2013, 104, 16005 CrossRef.
- J. Zhang, Z. Zeng, H.-Q. Lin and Y.-L. Li, Sci. Rep., 2014, 4, 4358 CrossRef PubMed.
- X. Wang, J. Li, H. Zhu, L. Chen and H. Lin, J. Chem. Phys., 2014, 141, 044717 CrossRef PubMed.
- Z. Liu, D. Li, S. Wei, W. Wang, F. Tian, K. Bao, D. Duan, H. Yu, B. Liu and T. Cui, Inorg. Chem., 2017, 56, 7494–7500 CrossRef CAS PubMed.
- A. F. Goncharov, N. Holtgrewe, G. R. Qian, C. H. Hu, A. R. Oganov, M. Somayazulu, E. Stavrou, C. J. Pickard, A. Berlie and Y. Fei, J. Chem. Phys., 2015, 142, 648–651 CrossRef PubMed.
- K. Yin, Y. Wang, H. Liu, F. Peng and L. Zhang, J. Mater. Chem. A, 2015, 3, 4188–4194 CAS.
- A. Hu and F. Zhang, J. Phys.: Condens. Matter, 2010, 22, 505402 CrossRef PubMed.
- J. Wang, L. Zhang, F. Long, W. Wang, Y. Gu, S. Mo, Z. Zou and Z. Fu, RSC Adv., 2016, 6, 23272–23278 RSC.
- D. Li, F. Tian, Y. Lv, S. Wei, D. Duan, B. Liu and T. Cui, J. Phys. Chem. C, 2017, 121, 1515–1520 CAS.
- Z. Raza, I. Errea, A. R. Oganov and A. M. Saitta, Sci. Rep., 2013, 4, 5889 CrossRef PubMed.
- S. Wei, D. Li, Z. Liu, X. Li, F. Tian, D. Duan, B. Liu and T. Cui, Phys. Chem. Chem. Phys., 2017, 19, 9246–9252 RSC.
- H. Wang, Y. Yao, Y. Si, Z. Wu and G. Vaitheeswaran, J. Phys. Chem. C, 2014, 118, 650–656 CAS.
- Y. Zhang, W. Wu, Y. Wang, S. A. Yang and Y. Ma, J. Am. Chem. Soc., 2017, 139, 13798–13803 CrossRef CAS PubMed.
- S. Zhu, F. Peng, H. Liu, A. Majumdar, T. Gao and Y. Yao, Inorg. Chem., 2016, 55, 7550–7555 CrossRef CAS PubMed.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, Phys. Rev. Lett., 2011, 106, 015503 CrossRef PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1998, 80, 891 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- T. Bŭcko, J. Hafner, S. Lebégue and J. G. Angyán, J. Phys. Chem. A, 2010, 114, 11814 CrossRef PubMed.
- J. P. Prates Ramalho, J. R. B. Gomes and F. Illas, RSC Adv., 2013, 3, 13085 RSC.
- C. J. Pickard and R. J. Needs, Phys. Rev. Lett., 2009, 102, 125702 CrossRef PubMed.
- W. J. Evans, J. Lipp, C.-S. Yoo, H. Cynn, J. L. Herberg, R. S. Maxwell and M. F. Nicol, Chem. Mater., 2006, 18, 2520 CrossRef CAS.
- Z.-J. Wu, E.-J. Zhao, H.-P. Xiang, X.-F. Hao, X.-J. Liu and J. Meng, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 054115 CrossRef.
- E. D. Raczyńska, Comput. Theor. Chem., 2011, 971, 38–41 CrossRef.
- M. N. Glukhovtsev and P. von Ragué Schleyer, Chem. Phys. Lett., 1992, 198, 547–554 CrossRef CAS.
- K. J. Wilson, S. A. Perera, R. J. Bartlett and J. D. Watts, J. Phys. Chem. A, 2001, 105, 7693–7699 CrossRef CAS.
- H.-X. Duan and Q.-S. Li, Chem. Phys. Lett., 2006, 432, 331–335 CrossRef CAS.
- M. Zhang, H. Yan, Q. Wei, H. Wang and Z. Wu, Europhys. Lett., 2013, 101, 26004 CrossRef.
- C. W. Allen, RSC Pub., 2009, 17031–17032 CAS.
- S. Yu, B. Huang, Q. Zeng, A. R. Oganov, L. Zhang and G. Frapper, J. Phys. Chem. C, 2017, 121, 11037–11046 CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2013, 34, 2557–2567 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11260b |
| This journal is © The Royal Society of Chemistry 2018 |