
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymeric platform for the growth of chemically anchored ZnO nanostructures by ALD†
Giovanna Pellegrino *a,
Sabrina Carola Carroccioab,
Francesco Ruffinoac,
Guglielmo G. Condorellid,
Giuseppe Nicotrae,
Vittorio Priviteraa and
Giuliana Impellizzeria
*a,
Sabrina Carola Carroccioab,
Francesco Ruffinoac,
Guglielmo G. Condorellid,
Giuseppe Nicotrae,
Vittorio Priviteraa and
Giuliana Impellizzeria
aCNR-IMM, Via S. Sofia 64, 95123 Catania, Italy. E-mail: giovanna.pellegrino@imm.cnr.it
bCNR-IPCB, Via P. Gaifami 18, 95126 Catania, Italy
cDipartimento di Fisica e Astronomia, Università di Catania, Via S. Sofia 64, 95123 Catania, Italy
dDipartimento di Scienze Chimiche, Università degli Studi di Catania and INSTM UdR Catania, V.le A. Doria 6, 95125 Catania, Italy
eCNR-IMM, Z.I. VIII Strada 5, 95121 Catania, Italy
First published on 2nd January 2018
Abstract
The synthesis of hybrid nano-composites in which an inorganic layer is grown on a polymeric surface via chemical bonds, is a challenging goal for many applications from photocatalysis, to sensing and optoelectronics. Herein, we describe on the growth of ZnO nanostructures by an Atomic Layer Deposition (ALD) technique on polyetherimide (poly(2,2′-bis(3,4-dicarboxyphenoxy)phenylpropane)-2-phenylendiimide ULTEM® 1000) substrates. A surface modification consisting of a thermal photo-oxidization process, applied for different exposure times, has been properly performed on the polymer to promote the production of ALD suitable functionalities as reactive surface-sites. The chemical anchoring of the inorganic species from the gas-phase is demonstrated by spectroscopy and mass analyses to exclusively occur on the pre-oxidized films, whilst the ZnO growth does not occur in the untreated ULTEM® films in the operative ALD conditions. Notably, we demonstrate that two different regimes of growth take place in the oxidized polymer as a function of the photo-exposure time. In particular, the formation of a nanostructured coating of ZnO on the polyetherimide surface is found in the case of short-time photo-exposed polyetherimide (ALD-like regime), whilst an intermixed organic/polymer layer is found on the long-time oxidized films (Vapor Phase Infiltration, VPI-like regime). The photocatalytic activity of the synthesized materials has been tested through the degradation of methylene blue dye in aqueous solution under UV light irradiation, to give a proof of concept of a possible application of the nano-composites.
Introduction
The development of nanotechnology has brought concrete benefits to our daily life, finding applications in several fields of common interest extending from the environment, energy, agriculture, medicine to computing and communication.1–14The huge technological progress registered in the last decade is mainly due to the capability of manipulating the materials at the nanoscale level and realizing functional nano-sized smart systems, having activities and properties deeply different from their bulk counterparts.
The advanced methods of manufacturing has allowed the development of nano-engineering, as robust and powerful tool for the fabrication of low-dimensional materials, previously inaccessible due to physical restrictions. Over the conventional methods, Atomic Layer Deposition (ALD) represents an ideal instrument, which permits the realization of materials with atomic scaling precision, thus guaranteeing a strict control of the structural and chemical properties of the final objects.15–20 The high quality of the ALD materials originate from the peculiar growth mechanism based on selective and self-limiting reactions between the ALD gaseous precursors and the reactive groups exposed at surface of the substrate.21–24 The growth of the material on the substrate is controlled at the atomic scale, uniformly and conformably on large areas.
In addition, compared with other deposition methods, (e.g. Chemical Vapor Deposition (CVD) and sputtering), ALD is performed under much milder conditions of pressure and temperature, and in absence of molecular-level bombardment, thus being compatible with the use of substrates sensitive both to the thermal and mechanical stress, as in the case of polymeric materials.17,25,26
In this respect, the realization of novel inorganic/polymeric hybrid materials by ALD is appealing, being beneficial in a variety of applications including photocatalysis, sensing, energy harvesting and storage, and optoelectronics. Indeed, hybrid systems can combine the structural properties and functionalities of the two organic and inorganic components, which are unattainable from the use of the single phases.27 The ability to control the reaction between the ALD precursors and polymers opens new opportunities in materials processing.28,29 For instance, the flexibility, transparency, lightness, and low costs typical of the plastic substrates can suitably encounter the photocatalytic, optical, and electronic functions of the metal oxides. Nevertheless, due to the severe chemical and physical differences between polymeric and inorganic components, the design of the ALD synthesis of a hybrid organic/inorganic material represents a significant challenge.17,30
It is well known, for instance, that ALD on many polymers can involve subsurface precursor diffusion and reaction, which are not encountered during ALD on inorganic surfaces.19 The mechanism of diffusion of the precursors into the film depends on the organic and structural nature (e.g. topology, composition, molar masses) of the macromolecular materials.31 Vapor phase infiltration (VPI) is an emerging gas phase processing for transforming polymers into organic–inorganic hybrid materials based on the metalorganic precursors diffusion and subsequently reactions with the organic polymers. Such processing technique originates from the atomic layer deposition (ALD) research community, and its processing conditions are similar to ALD, nevertheless, the fundamental phenomenological mechanisms of VPI and ALD are significantly different.32
For highly reactive polymers to the ALD precursors, the reaction rate between the precursors and the surface is fast so that the inorganic phase rapidly nucleates on the surface of the polymer with the consequent formation of a conformal inorganic coating on the surface of the polymer. On polymers such as PVA or cellulose where there is a large density of reactive groups, the ALD reaction can be limited to the top surface of the polymer.19,29
In the case of the unreactive polymers towards the ALD reagents, no growth of the inorganic layer is observed for short times of depositions (few ALD cycles). The formation of the inorganic phase occurs, instead, for higher times of deposition with the precursors that can diffuse into the polymer and react within the film, resulting in an inorganic phase embedded in the organic bulk.33–37 It has been shown that under VPI conditions, no covalent reactions can occur between the precursors and the polymer matrix.38,39 Thus, the weak interaction occurring at the interface between the two components in such organic/inorganic bi-phase, results unattractive for applications, especially in the field of heterogeneous photocatalysis for water purification, wherein the photoactive structures could be easily dispersed in solution.
In order to obtain an appealing system, flexible and functional, it is necessary immobilizing the inorganic photocatalysts by stable chemical bonds on the substrate, to guarantee structural stability and prevent their dispersion in the surrounding media. With this purpose, we properly designed the growth of ZnO nano-structures on a surface-modified polymeric substrate. As substrate was selected the poly(2,2′-bis(3,4-dicarboxyphenoxy)phenylpropane)-2-phenylendiimide ULTEM® 1000 (hereafter simply called ULTEM®), an engineering thermoplastic material with superior durability properties, high modulus, toughness, and chemical resistance. Furthermore, due to its amorphous nature, transparent ULTEM® resin balances both mechanical properties and processability.
The molecular structure of polyimide is the responsible of the high mechanical performance of the ULTEM® resin, but at the same time, it results unsuitable for the ALD grafting reactions, due to the lack of reactive functionalities. In some previous works, the thermal and photo-oxidation reaction pathways of this polymer, thanks to UV light irradiation, were clarified by using Matrix Assisted Laser Desorption Ionization-Time Of Flight (MALDI-TOF) mass spectrometry.40–43 A detailed map of the photo-cleavage mechanisms of polyetherimide ULTEM® was depicted for different time of exposure, permitting us to discriminate among the reaction products of interest. Crucial, in view of the ALD, is the formation of –OH and –COOH groups along the repeat units of the polymer. The presence of such reactive chemical groups could permit a direct growth of the ZnO nanostructures by exploiting chemical bonds at the organic–inorganic interface. In this paper, we originally exploit the formation of reactive functionalities in the polyetherimide to graft the inorganic precursors in the ALD regime, with the formation of the inorganic phase. We show that the assembly of the inorganic phase upon the polymer is addressed by the entity of the oxidative pre-treatment (duration). In fact, different ZnO/polymer hybrid habits are achieved depending on the number and availability of the functional groups in the polymer, these being strongly related to the chemical and structural modification of the macromolecular network during the photo-oxidation treatment.
Results and discussion
Characterization of oxidized ULTEM® films
Films of ULTEM® have been prepared according to the procedure described in the Experimental section. The thickness was measured by calipers, resulting of ∼100 μm ± 10 μm. The thermal photo-oxidative process was carried out on four samples having the shape of a disk with a diameter of 5 cm, for a duration, respectively of 3, 8, 24 and 48 hours. Afterward, in order to evaluate the effect of the photo-process on the properties of the polymer surface, measurements of contact angle and FTIR ATR were performed. Whereas MALDI TOF analysis were used to obtain information on the entire polymeric materials. Fig. 1a shows the values of the contact angle as found in the different samples as a function of the photo-exposure time.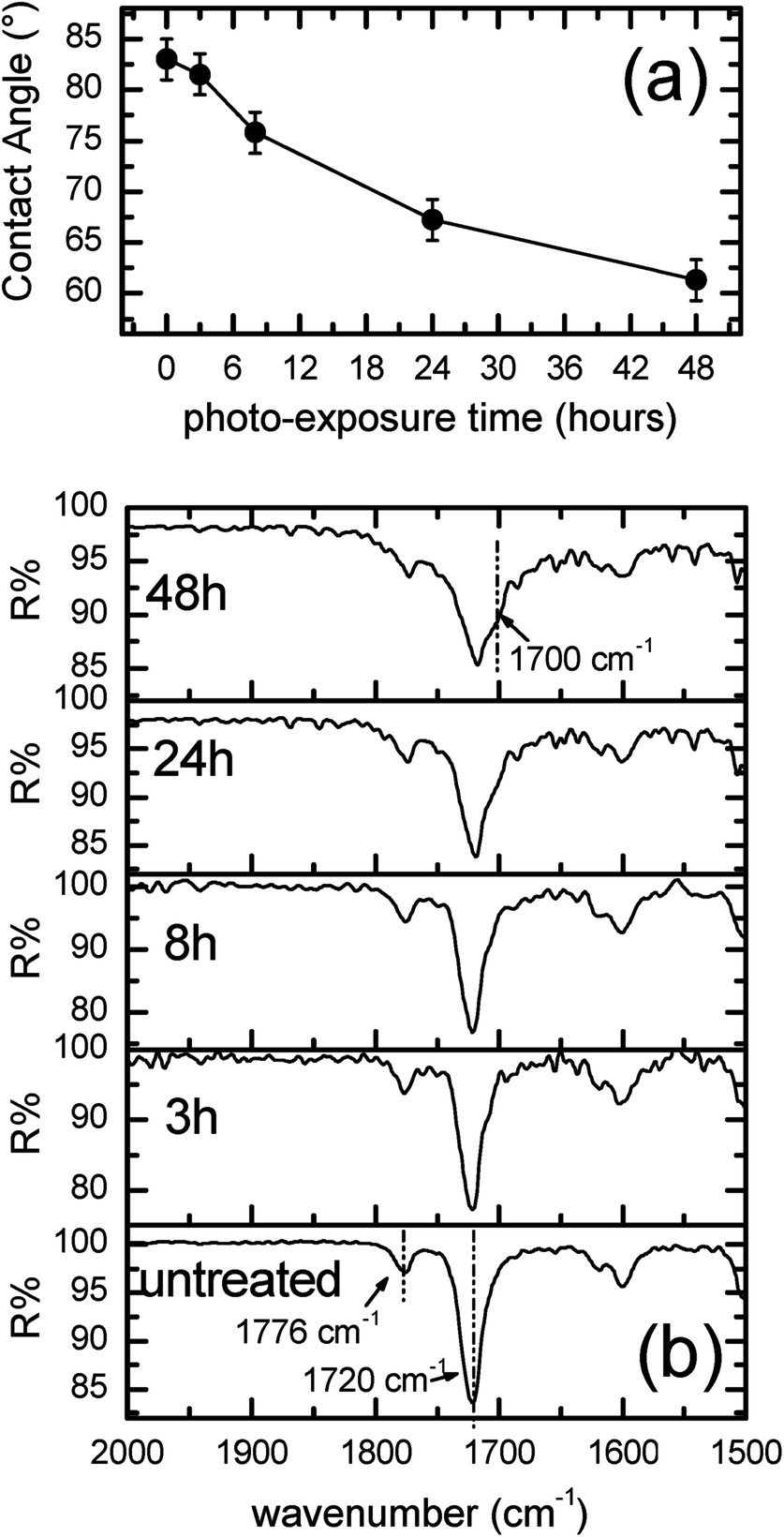 | ||
| Fig. 1 (a) Contact angle and (b) FTIR-ATR of the ∼100 μm-thick ULTEM® films as a function of the thermal photo-exposure duration. | ||
As can be observed from Fig. 1a, the value of contact angle measured in the case of the original ULTEM® film results 83° ± 2°, a value in line with the data reported in literature for poly-ether-imides.44 A gradual diminution of the contact angle is observed by increasing the photo-exposure time, thus indicating, consistently with the mass spectrometry results reported in the literature,41,42 the progressive formation of hydrophilic functionalities on the polymeric surface. MALDI analyses demonstrated, in fact, the formation of oxidized species such as alcoholic and acids moieties resulting from the oxidative procedure.40–42
Fig. 1b shows the evolution of the FTIR-ATR spectra in the diagnostic range between 2000 and 1500 cm−1 as a function of the duration of the photo-exposure. As can be seen from the figure, the FTIR-ATR spectrum of the original polymer shows the characteristic vibrational peaks due to the imides CO double bond. In particular, the contribution positioned at 1776 cm−1 is related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric stretch, whilst the second band lying at 1720 cm−1 is due to the CO symmetric stretch.45
O asymmetric stretch, whilst the second band lying at 1720 cm−1 is due to the CO symmetric stretch.45
As can be noticed, the employ of the photo-oxidation procedure on ULTEM®, produces an enlargement of the spectral IR contribution at ∼1700 cm−1. The variation is slightly visible at short time of photo-oxidation (e.g. 3 h and 8 h), becoming remarkable over 24 h. The broadening of the band at ∼1700 cm−1 has been ascribed to the formation of –COOH species, in strict correspondence with the mass spectrometry data.38–40
The presence of reactive chemical functionalities on the polymer surface is a fundamental requirement to graft metal species;46,47 we exploit such characteristic by growing ZnO by ALD on ULTEM®, focusing on the reactivity of the different photo-oxidized samples.
ALD deposition and characterization of ZnO/ULTEM® films
The ZnO deposition was carried out as described in the Experimental section by introducing in the ALD chamber the original polymer together with the photo-oxidized substrates.AFM analyses were performed on all the different films, before and after the ZnO deposition, with the aim of examining their morphology. Fig. 2a shows a 2 μm × 2 μm AFM image related to the original ULTEM® substrate before the ALD process. Before the ZnO deposition, the ULTEM® substrates, both the original and the photo-oxidized films reveal very low values of Root Mean Square (RMS), all included in the range between 0.3 and 0.7 nm and an average height in the range between 2 and 5 nm (see Fig. 2b, related to the 24 h-photo-oxidized ULTEM sample in comparison with the original ULTEM of Fig. 2a).
 | ||
| Fig. 2 AFM images of (a) original ULTEM and (b) 24 h-photo-oxidized ULTEM before the ALD process. (c) Original, (d) 3 h-, (e) 8 h-, (f) 24 h- and (g) 48 h-photo-oxidized ULTEM after the ZnO deposition. (e-1) shows the TEM cross section image related to 8 h-photo-oxidized sample after the ZnO deposition. | ||
After the ZnO deposition, the AFM image of the untreated sample shows the presence of few distinct features standing on the surface and having an average height of 18.3 nm (Fig. 2c). Such nanostructures are rarefied on the surface, showing a reciprocal distance of the order of ∼1–2 microns. The surface area surrounding such nanodomains presents a very low RMS value of ∼0.5 nm, comparable to that found in the case of the ULTEM® films before the ZnO deposition.
Differently, the AFM image related to the 3 h-photo-oxidized samples after the ZnO process (Fig. 2d), reveals the presence of nano-sized domains, which form a granular layer. The statistics on morphological parameters reveals a peak-to-peak (i.e. the highest probed height difference by the AFM tip) of ∼17.3 nm, comparable to that of Fig. 2c and an increased value of RMS to 1.7 nm. The AFM image related to the 8 h-oxidized film (Fig. 2e) shows values of peak-to-peak (∼17.7 nm) and RMS (1.8 nm) very close to those found in the AFM image of Fig. 2d. The formation of ZnO nano-sized structures is supported by Transmission Electron Microscopy (TEM) analyses (see Fig. 2e-1). The TEM image clearly shows the presence of ZnO nanostructures regularly distributed on the surface. On the basis of the AFM analyses, the surface density of the nanostructures appears increased with respect to the 3 h-sample, plausibly due to the increased number of ALD active functionalities on the surface, similarly to what found by Lee et al. by modulating the –OH/–CH3 ratio in self-assembled monolayer on silicon.48
The AFM images related to the 24- and 48-treated samples show the formation of nano-granular films with values of peak-to-peak and RMS significantly decreased if compared to the two previous cases. Surprisingly, in fact, the morphological parameters result of 5–7 nm and ∼0.5 nm (see Fig. 2f and g, respectively). The differences observed on the ZnO layers grown on long time-if compared to the short time-treated substrates, can arise from the alteration of the polymer structure induced by the prolonged photo-exposure time. Indeed, a modification of the mechanical and esthetic properties of ULTEM® when long photo-exposure is applied has been reported in literature, associated to the decrease of the average molar masses distribution within the polymer.40
In order to have a deeper comprehension of the phenomena occurring between the polymer and the inorganic precursors upon the ZnO deposition, we performed studies on the chemical composition of the ZnO/polymer interface by spectroscopic analyses.
After the ZnO deposition, FT-IR ATR measurements reveal diagnostic changes in the region associated to the CO functionalities (1600–1700 cm−1), with the decrease of the –COOH group in favor of the –COO− at 1600 cm−1 (see Fig. E1, ESI†), thus indicating the coordination of the metal species at the surface. As can be seen in Fig. E1,† an evident spectral variation is observed in the case of 48 h-treated sample, likely due to the higher amount of reactive terminations involved; on the other hand no significant differences are revealed in the spectrum of the original ULTEM® (Fig. E1a†), as expected. From the comparison of the spectra collected, however, no specific indications from FTIR findings can justify the morphological differences found by AFM in the short and long-time treated samples. With the aim of better understanding the data, XPS analyses were performed.
Table 1 reports the atomic percentages of the elements as detected from the spectra of: original, 8 h and 48 h, before and after the ALD process. In the case of the original ULTEM®, the percentages found by XPS are in line with the theoretical values expected for this polymer (C 82.2%, O 13.3%, and N 4.3%).49 The 8 h-photo-oxidized ULTEM® film does not show any appreciable variation in the atomic percentage values (in the range of the XPS uncertainty) whilst, in the case of the 48 h-treated sample, an increase of the oxygen relative atomic percentage is found, because of formation of oxidized species, a discussed in literature.38–40
| Original | 8 h | 48 h | ||||
|---|---|---|---|---|---|---|
| Before ALD | After ALD | Before ALD | After ALD | Before ALD | After ALD | |
| C (%) | 80.0 | 83.8 | 80.2 | 68.4 | 63.3 | 58.7 |
| O (%) | 14.9 | 11.4 | 14.9 | 24.7 | 30.6 | 32.8 |
| N (%) | 5.1 | 4.6 | 4.8 | <0.5 | 6.0 | 1.6 |
| Zn (%) | — | <0.2 | — | 6.5 | — | 6.8 |
With the aim of deeper analyzing the chemical structure of the polyetherimide macromolecules as a function of the photo-oxidation time, we collected high resolution spectra in the regions of interest: C 1s, O 1s, N 1s and Zn 2p, the latter after the ZnO deposition. The diagnostic C 1s and O 1s XPS regions are shown in the following, whilst N1 s and Zn 2p are reported in the ESI (Fig. E2†).
Fig. 3a shows the high resolution C 1s XPS region of the untreated ULTEM® film showing the characteristic components at 285.0 eV (due to the C–C, CC and C–H species), a shoulder at 286.4 eV (due to the ether groups), and a distinct band at 288.8 eV (imide groups). The presence of the shake up band at higher binding energy (BE) values (∼292 eV) is also visible in the spectrum, due to the π → π* transition.45
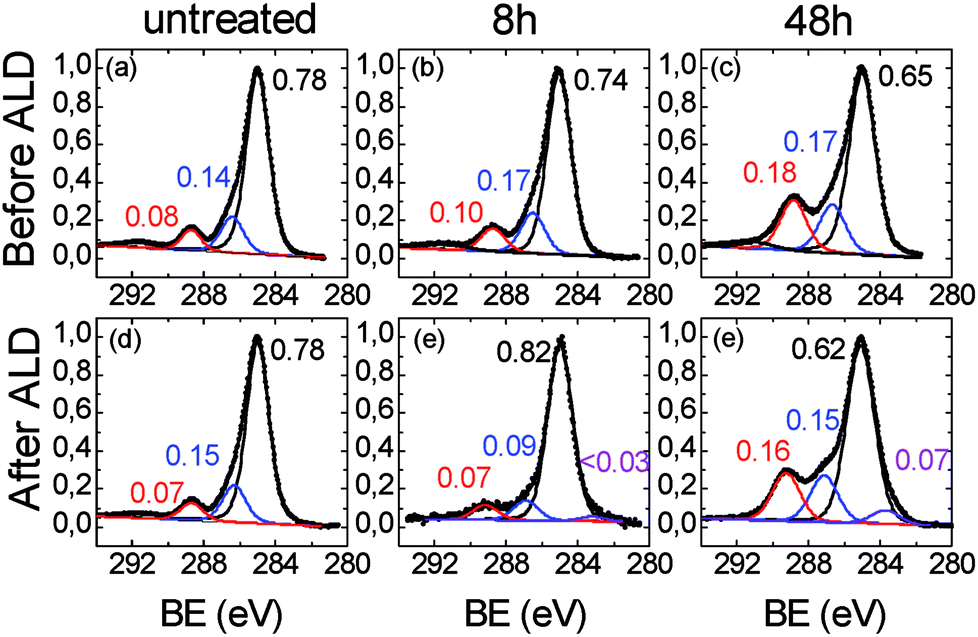 | ||
| Fig. 3 XPS spectra in the C 1s region related to (a), (d) untreated, (b), (e) 8 h- and (c), (f), 48 h-ULTEM® films, respectively before (top panel) and after (bottom panel) the ZnO ALD deposition. | ||
Fig. 3b relates to the 8 h-treated polymer. As can be seen, small differences emerge from the comparison with Fig. 3a, consisting in a slight enlargement of the bandwidths (1.5 vs. 1.6 eV) and in a slight increase of the intensity (∼10%) of both the components at ∼286.4 and ∼288.8 eV with respect to that at 285.0 eV. Such spectral variations are compatible with the formation of some C–OH/COOH species50,51 with the photo-exposure.
The C 1s region related to the 48 h-treated sample (Fig. 3c), shows a marked modification of the functional groups of the polymer if compared to the original film. The components associated to the oxidized species (i.e. C–OH at ∼287 eV and –COOH ∼289 eV), severely increased with respect to the main C–C/CC/CH contribution, consistently with the increase of the oxygen percentage in the overall spectrum (see Table 1) and with the reduction of the contact angles values due to the formation of hydrophilic functionalities, as visible in Fig. 1a. In addition, the C 1s signal can be fitted by further enlarging the FWHM of the components (1.75 eV), thus suggesting a certain chemical heterogeneity of the carbon species which constitute the organic chains, due to the formation of the C–OH and –COOH functionalities. The XPS spectra related to the O 1s region are reported in Fig. 4. In the case of the original ULTEM® film (Fig. 4a), according to the molecular structure of the polyetherimide, the O 1s XPS region shows a signal consisting of two components which are due, respectively, to the oxygen species of the imide (532.2 eV) and to the ether (533.7 eV) functionalities,45 both with a FWHM of 1.5 eV.
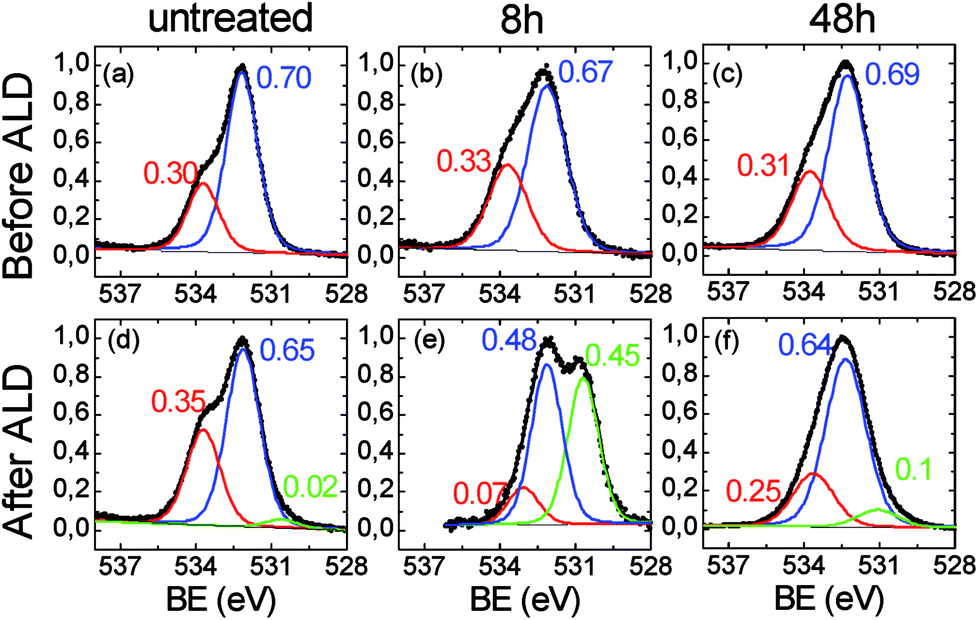 | ||
| Fig. 4 XPS spectra in the O 1s region related to (a), (d) untreated, (b), (e) 8 h-and (c), (f), 48 h-ULTEM® films, respectively before (top panel) and after (bottom panel) the ZnO ALD deposition. | ||
After the 8 hours of photo-exposure treatment, an enlargement of the bandwidths of both the components (1.7 vs. 1.5 eV) and an increase of the intensity of the band at ∼533.7 eV is observed in the spectrum (Fig. 4b) which can be associated to the presence of carboxylic functionalities.52 The O 1s region of the long-time treated polymer (48 h, Fig. 4c), shows similarities with that of the 8 h-treated sample, evidencing the same FWHM (1.7 eV) and similar ratio between the two components. Such spectroscopic findings suggest scarce differences between the typologies of oxygen present on the surface of the 8 h- and 48 h-treated samples.
The modification of the C 1s and O 1s spectral features, after the ZnO deposition by the ALD process, are shown in the bottom panels of Fig. 3 and 4, respectively. As expected, no differences emerge by comparing the C 1s spectra of the original ULTEM® film before and after the ZnO deposition (see Fig. 3a and d) since the negligible amount of ZnO present on the surface (see the Zn atomic percentage in Table 1 and the Zn 2p region in Fig. E2 in ESI†).
In the C 1s spectrum related to the 8 h-treated sample (Fig. 3e), the increase of the component at 285.0 eV with respect to the other two bands positioned slightly shifted at 287.0 eV and ∼289.1 eV, is observed, in comparison with the unprocessed sample (Fig. 3b). This finding can originate from the decrease of the carboxylic and alcoholic functionalities due to the formation of the C–O–Zn groups at the surface.53 In addition, the occurrence of a very small band at 283.6 eV is found in the spectrum after the deposition.
Differently, in the case of the 48 h-treated sample, after the ZnO deposition, no significant changes are observed in the intensity ratio between the component at 285.0 and the other two at bands, which result also in this case at 287.0 eV and ∼289.1 eV (Fig. 3f). In addition, the presence of the band at 283.6 eV, becomes more visible. The position of the band at such low value of binding energy suggests the presence of the C–Zn bond, as discussed by Gregorczyk and co-workers.53 The authors attributed such contribution to the presence of partially reacted DEZ species. The differences emerging from the comparison of the C 1s spectra, suggest different scenarios in the untreated, 8 h and 48 h-treated samples. Further information is gained from the analysis of the O 1s spectral features.
For what concerns the original sample, after the ALD deposition, the O 1s spectrum shows the presence of a very low intense band located at ∼530.6 eV, compatible with the presence of the oxygen bonded to metal (Fig. 4d).54 The low intensity of the band is compatible with the amount of zinc revealed by XPS which is just below the 0.2% (see Table 1, Fig. E2 in the ESI†). Indeed, the presence of a small number of ZnO nano-columns on the surface of the original ULTEM® is observed in the AFM image (see Fig. 2c). The formation of such nanostructures can be ascribed to the negligible amount of reactive functionalities on the ULTEM® surface, likely due to macromolecules having phthalic anhydrides as terminal group that can hydrolyze upon the effect of water during the ALD process.
For what concerns the 8 h-treated sample after the ALD deposition (Fig. 4e), the O 1s XPS spectrum reveals the presence of an intense contribution at 530.6 eV, as a clear indication of the presence of ZnO at the surface. In addition, as can be seen from the figure, the contribution at higher binding energy (–COOH/–OH) is reduced as a consequence of the surface reaction between ZnO and the carboxylic groups. In the spectrum of the 48 h-treated sample, the spectral contribution at 530.6 eV, associated to the Zn–O bond, is still present, but its relative intensity with respect to the other components is decreased in comparison with the case of 8 h-treated sample. Although, the O 1s XPS signals related to the 8 h and 48 h-treated unprocessed samples appear similar (see Fig. 4b and c), after the ZnO deposition, notable differences emerge, mostly related to the weight of the Zn–O component in the O 1s spectrum (see Fig. 4e and f).
The quantitative data in Table 1 further unveil dissimilarities between the 8 h and 48 h-samples before and after the ALD process. It is to be noticed, in fact, that, after the ZnO deposition, the oxygen percentage found in the spectrum of the 8 h-treated sample almost doubles, where just a small increment is seen in the 48 h-treated case. Correspondingly, the percentages of the elements characterizing the polymeric substrate (C 1s and N 1s) significantly decrease in the 8 h-treated sample likely due to the growth of an overlayer of ZnO on the polymer surface. Different is the case of the 48 h-treated sample where, the variation of the elemental percentage before and after the deposition process is less relevant, thus suggesting that the zinc is englobed into the organic bulk.31,33,34 This can affect the surface morphology of the hybrid samples, as revealed by AFM.
By summarizing, XPS analyses indicated that the chemically inert untreated polymer remains practically unaltered under the ALD operative conditions used, whilst the anchoring of the ALD precursor occurs selectively on the photo-oxidized samples thanks to the presence of reactive organic functionalities, such as –OH and COOH. Nevertheless, the present results clearly indicate the formation of the inorganic phase, not only due to the occurrence of the ALD surface reactions. Concurrently, other events, especially in the case of the 48 h treated sample, can take place. Phenomena of gas penetration and diffusion between the polymer chains of the degraded 48 h-treated sample occur, followed by subsurface reactions. This could explain the differences in the qualitative and quantitative XPS data shown in the work (see XPS spectra and table), between the slight surface-modified and the deeply modified ULTEM® films and, also, the differences observed in the surface morphological parameters (Fig. 2). In the first case (8 h) an inorganic nanostructured overlayer is formed on the surface, whilst in the second case (48 h), a hybrid intermixing of Zn–O and polymer is formed. In this respect, the band at 283.6 eV observed in the XPS C 1s region, suggests the presence of the C–Zn-bond, as an indication of partially reacted Zn-precursor present in the material. As can be seen, such contribution is negligible in the spectrum of the 8 h-treated sample (Fig. 3e) in comparison with that of the 48 h-treated sample (Fig. 3f). The altered structural properties of the polymer as a consequent of the long photo-oxidation process (density, flexibility, oligomers with short length), can facilitate the gas permeability and diffusion within the macromolecules, thus favoring the occurrence of uncompleted sub-reactions.
This idea is further confirmed by mass analyses. MALDI spectra of the ULTEM® film, as received and after photo-oxidation, have been previously reported.40–42 Here, for brevity of discussion, we present only the expanded portions of the MALDI spectra in the range of mass (m/z) 1336–1640, related to the 8 h-oxidized ULTEM® film before (Fig. 5a) and after the ZnO deposition (Fig. 5b). The sequel reports also the spectrum of the 48 h-sample after the ALD process (Fig. 5c).
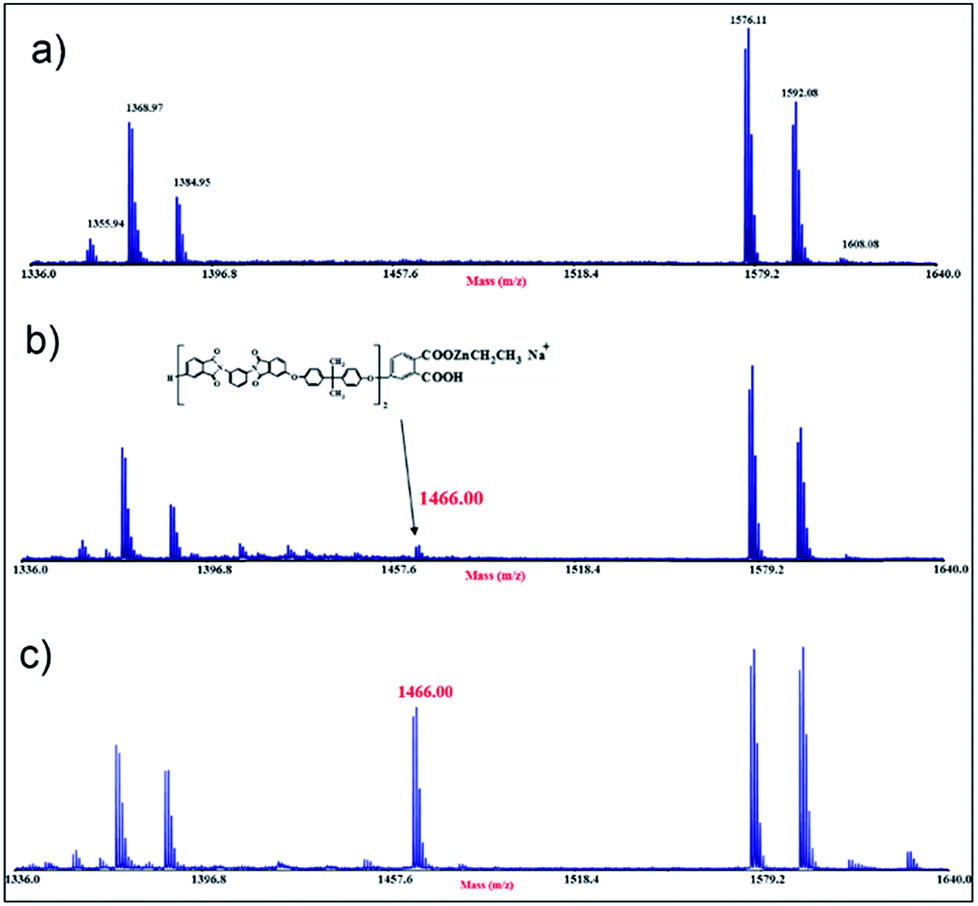 | ||
| Fig. 5 MALDI mass spectra in the m/z range 1336–1640 registered in reflectron and positive mode, of the commercial ULTEM® sample photo-oxidized for 8 h (a) before and (b) after the ZnO deposition; (c) 48 h-treated sample after the ZnO deposition. | ||
As visible in Fig. 5a, the MALDI spectrum of the 8 h oxidized – sample (∼100 μm-thick) shows the peaks due to both the oligomers from the bulk (e.g. m/z 1355.94, m/z 1368.97, m/z 1576.11) and from the oxidized topmost part of the polymer (m/z 1384.95, m/z 1592.08 and m/z 1608.08).40–42
From the comparison between this spectrum with those of the 8 h- and 48 h-treated samples after the ALD process (Fig. 5b and c), we notice the presence of an increased number of peaks in the mass scale, which indicates the formation of new species. In particular, the peak at m/z 1466.00, is reasonable assigned to the structure containing the ethyl zinc-moiety bonded to phthalic-acetate terminal group of the polymer (see the structure in Fig. 5). As can be seen, in line with the increment of the C–Zn band at 283.6 eV observed by XPS, the relative abundance of that peak in the mass spectrum of the 48 h-sample, is higher if compared to that of the 8 h-sample, plausibly due to the higher amount of suitable functionalities in the sample (FT-IR results). Based on the presented results, the idea is that in the case of 8 h-treated sample, the formation of the inorganic phase is predominantly due to surface reactions between the oxidized polymer at the topmost part of the film and the ALD precursors (ALD-like regime). Then, the underneath portion of the polymer acts as a barrier to the gas infiltration, according to the inert response of the untreated sample under the used ALD conditions. On the contrary, in the case of 48 h-treated sample, the polymer presented a significant decrement of molar masses40 that increased the free volume between the photo-degraded macromolecules.55–57 This would favor the infiltration and diffusion of the ALD reactive gas and the occurrence of sub-reactions with the accessible reactive functionalities. This mechanism has something similar to the growth of the inorganic structure embedded in the reactive polymer by infiltration (VPI-like regime).49 This could also explain the different morphological parameters found by AFM analyses in the polymeric platforms after the ZnO deposition (Fig. 2), with both the values of RMS and peak-to-peak higher in the cases of 3 h and 8 h-treated samples, with respect to the 24 h and 48 h-cases.
Photocatalysis tests
Tests of Methylene Blue (MB) photodegradation was performed with the aim of investigating the efficiency in terms of photocatalytic activity of the ZnO layers, deposited by ALD on the different typologies of polymeric substrates. The measurements have been carried out under UV light by including in the tests the unprocessed ULTEM® substrates, as blankets.According to the Langmuir–Hinshelwood model, we evaluated the photocatalytic reaction rate, k, given by the following equation: ln(C/C0) = −kt, where C is the concentration of the MB, C0 the starting concentration of the MB, and t the time.58
The values of the reaction rate k reported in the ordinate axis are normalized to the k value found in the absence of any catalyst materials (k/kMB). Fig. 6 reports the photocatalytic results after four hours of irradiation for all the investigated samples. As can be seen, the MB, the ULTEM® substrate and the untreated ULTEM® plus the ZnO deposition do not show any photo-catalytic activities, as expected.
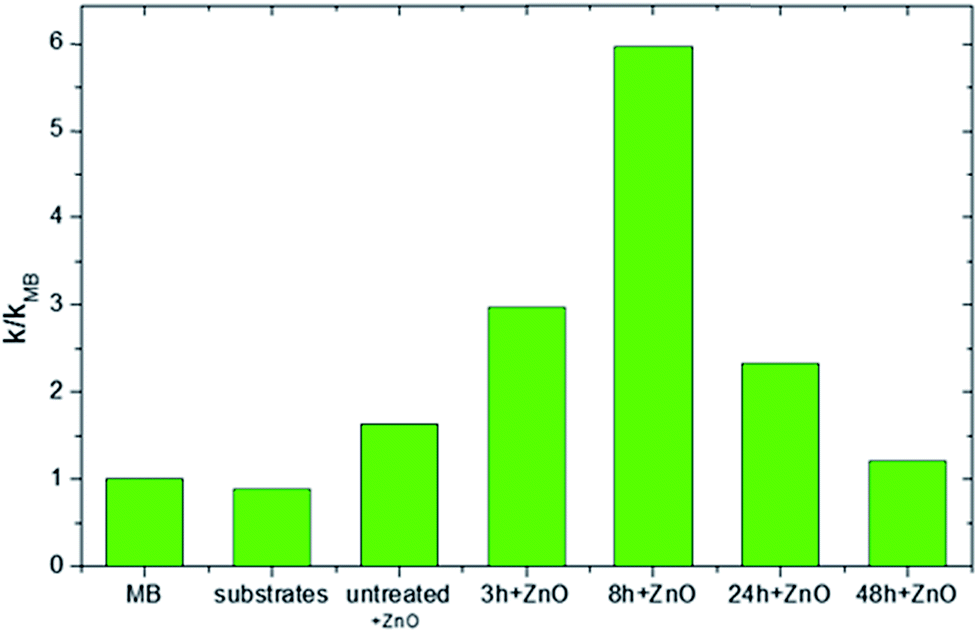 | ||
| Fig. 6 Photocatalytic rate of MB photodegradation, normalized to the value obtained in absence of photocatalysts (k/kMB), for the different typologies of ULTEM® samples (in the abscise axis) under UV irradiation. | ||
The best photo-activity response is displayed in the case of ZnO deposited on the 8 h-treated polymer. In this case, in fact, a nanostructured ZnO layer has been seen to grow over the surface of the polymer. On the other hand, a significant drop of the photocatalytic efficiency is registered in the cases of long-time treated samples. This is reasonably due to the presence of ineffective ZnO phase infiltrated into the polymer matrix or the possible release of low masses compounds from the degraded polymer, which might be competitive to MB during the photo-degradation processes.
In Fig. 7 is reported a sequel of diagnostic parameters as a function of the exposure time (a–d) and compared to the related photo-catalytic activity (e). As suitably displayed, photo-catalytic performance shows a non-linear trend with the contact angle measures and the COOH XPS amount (a, b), reaching the maximum in correspondence of 8 h, where the highest amount of ZnO nanostructures is accomplished (c, d).
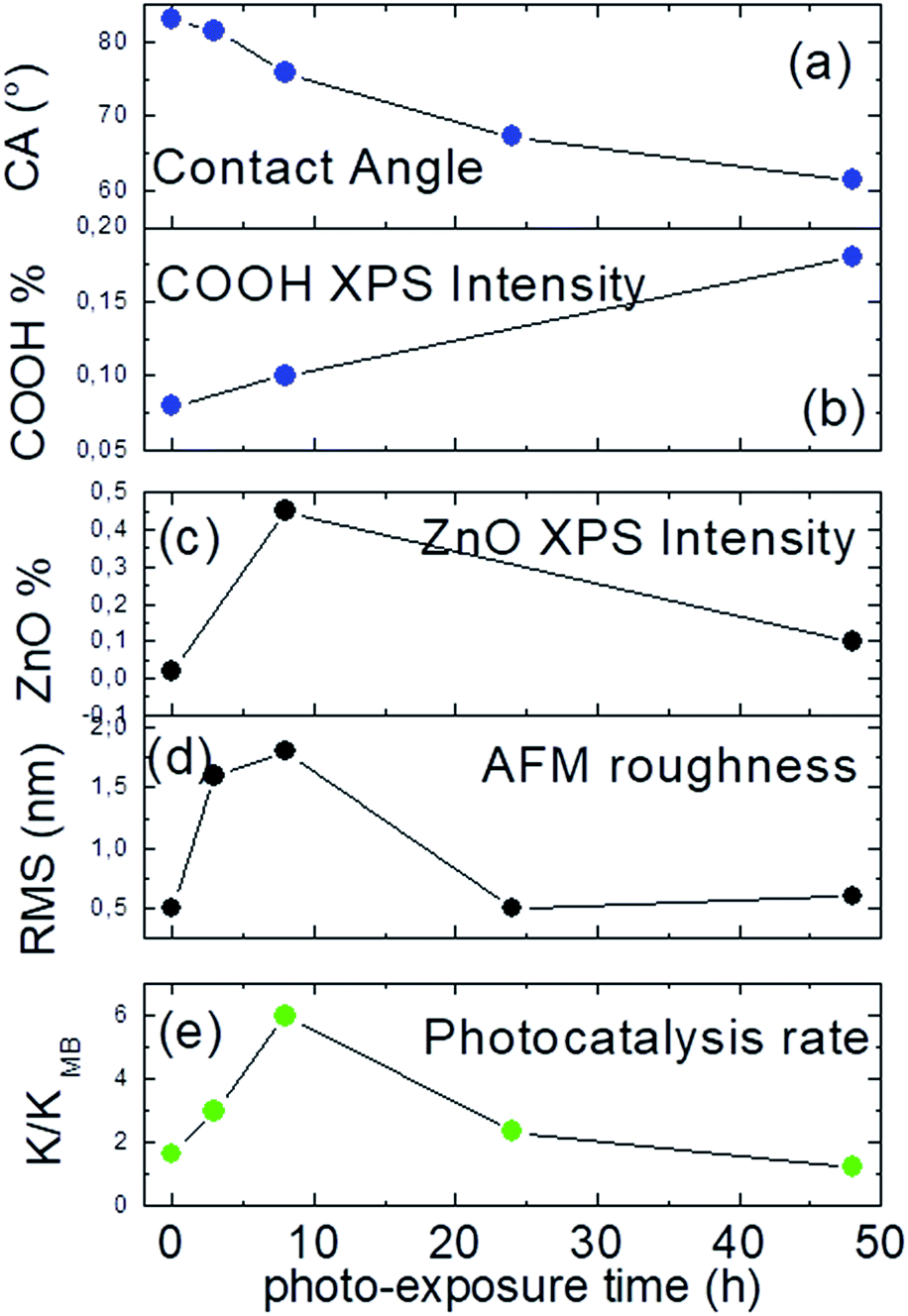 | ||
| Fig. 7 (a) Contact angle; (b) XPS COOH%; (c) O 1s at 530.6 XPS%; (d) RMS and (e) photocatalytic rate as a function of the photo-exposure time. | ||
Experimental
Preparation of the substrates
Poly(2,2′-bis(3,4-dicarboxyphenoxy)phenylpropane)-2-phenylendiimide ULTEM® 1000, herein simply called “ULTEM®”, was purchased from Sigma-Aldrich Chemical Co (Italy). ULTEM® films with a uniform thickness of ∼100 μm were obtained by casting 10 mL of a 2% solution of the polymer in CHCl3 in a Petri dish with 5.0 cm of diameter. After the evaporation of the solvent overnight at room temperature, the removal of the residual molecules of solvent was completed by treating the films at 60 °C for 4 hours in vacuum at 0.5 mBarr.Photo-oxidative degradation of ULTEM®
The photo-oxidation of the ULTEM® films was performed on the 100 μm-thick ULTEM® films in a QUV PANEL apparatus at 60 °C under continued UV exposure at 340 nm (UVA 340 lamps) for different times: 3, 8, 24 and 48 hours.ZnO deposition by ALD
ZnO films were deposited on the ULTEM® films by ALD technique by using a Picosun R-200 Advanced reactor. During the deposition, the temperature of the chamber was fixed at 80 °C, preceded by a one-hour long thermal stabilization. Diethyl zinc (DEZ, purity 100%) and de-ionized water were used as precursors (provided by air liquid), while N2 was used as carrier and purge gas (purity ≥ 99.999%). The pulse and purge times were kept constant at 0.1/3/0.1/5 s for DEZ/N2/H2O/N2, which corresponds to one ALD cycle. The entire ALD deposition described in the article consists of 304 cycles. The nominal thickness of the ZnO layers obtained on silicon slices was evaluated by ellipsometric analyses by using an M-2000 instrument provided by J.A. Woollam Co., Inc., by applying a Cauchy model in the 400–1700 nm range, giving a nominal thickness of ZnO of ∼15 nm. Due to the sensitivity limitations, the crystallinity of the deposited ZnO has been checked on a film obtained by doubling the ALD cycles, by means of X-ray diffraction (XRD) analyses, which reveals the peaks characteristic of the wurtzite crystalline structure, typical of polycrystalline ZnO.Contact angle
Wettability measurements have been carried out by using a DATAPHYSICS-OCA 15 PRO apparatus and water as liquid. Direct measurements of the contact angles were performed on four different areas of the same sample.FTIR-attenuated total reflectance (FTIR-ATR)
FTIR-ATR spectra were recorded on JASCO FTIR 430 equipped with a Harrick GATR germanium single reflection accessory, collecting the spectra with 100 scans at 4 cm−1 resolution. The original and photo-exposed films of ULTEM® were analyzed by pressing the polymer on the top of the Harrick GATR germanium crystal. Both the front and the back sides of the 100 μm-thick films were analyzed. Just the FTIR-ATR spectra obtained from the front side of the sample, subjected to the UV-irradiation, shows differences with respect to the untreated ULTEM® film.Atomic force microscopy (AFM)
AFM analyses were performed using a Bruker-Innova microscope operating in high-amplitude mode and ultra-sharpened Si tips were used MSNL-10 from Bruker Instruments, with anisotropic geometry, radius of curvature ∼2 nm, tip height ∼2.5 μm, front angle ∼15°, back angle ∼25°, side angle 22.5°. The Si tips were substituted as soon as a resolution loose was observed during the AFM images acquisition. The AFM images were analyzed by using the SPMLABANALYSES V7.00 software. In order to give representative parameters, statistics on the AFM images were carried out over different areas of the same sample.Transmission electron microscopy (TEM)
JEOL ARM200F aberration-corrected transmission electron microscope, operated at 200 keV in Conventional Bright Field TEM (BF-CTEM) mode, was used for the morphological characterization of the interface between the insulating substrate, made by ULTEM, and the deposited ZnO. To minimize the charging effect induced by the electron beam at the interface, such as, electron drift, and epilayer delamination, a very low beam dose of 30 e− per Å2 was applied to illuminate the sample. As for as the specimens preparation, a slice of ULTEM coated be the ZnO, was embedded in a liquid epoxy resin then sectioned by ultramicrotomy technique (with a Leica Ultracut UCT), collecting sections of 50–70 nm in thickness, than transferred on a copper mesh TEM grids.Matrix assisted laser desorption ionization-time of flight (MALDI-TOF)
Applied Biosystems 4800 MALDI TOF/TOFTM Analyzer (Framingham, MA, USA) mass spectrometer was used in this study to acquire MALDI spectra. The TOF/TOF instrument is equipped with a Nd:YAG laser (355 nm wavelength) of <500 ps pulse and 200 Hz repetition rate. MALDI-TOF/TOF-MS spectra were recorded in reflector positive ion mode. The resolution of MALDI spectra reported in the text is about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 FWHM. The laser irradiance was maintained slightly above threshold. 2-(4-hydroxyphenylazo)benzoic acid (0.1 M in THF/CHCl3 solution) was used as matrix. Appropriate volumes of polymer solution (5 mg mL−1 in CHCl3) and matrix solution were mixed to obtain 2:1, 1:1, and 1:2 ratios (sample/matrix v/v). Another set of analysis was performed by adding 1 mL of sodium trifluoroacetate (0.1 M in THF) to the analyte/HABA matrix mixtures. 1 μL of each sample/matrix mixture was spotted on the MALDI sample holder and slowly dried to allow matrix crystallization. At least three separate portions were analyzed at each photo-exposure time. The structural identification of MALDI peaks was mainly based on empirical formulas. However, isotopic resolution helps considerably in the peak-assignment process. Some plausible structures were also derived from and supported by previous studies40–43
000 FWHM. The laser irradiance was maintained slightly above threshold. 2-(4-hydroxyphenylazo)benzoic acid (0.1 M in THF/CHCl3 solution) was used as matrix. Appropriate volumes of polymer solution (5 mg mL−1 in CHCl3) and matrix solution were mixed to obtain 2:1, 1:1, and 1:2 ratios (sample/matrix v/v). Another set of analysis was performed by adding 1 mL of sodium trifluoroacetate (0.1 M in THF) to the analyte/HABA matrix mixtures. 1 μL of each sample/matrix mixture was spotted on the MALDI sample holder and slowly dried to allow matrix crystallization. At least three separate portions were analyzed at each photo-exposure time. The structural identification of MALDI peaks was mainly based on empirical formulas. However, isotopic resolution helps considerably in the peak-assignment process. Some plausible structures were also derived from and supported by previous studies40–43
X-ray photoelectron spectroscopy
XPS measurements were performed by a PHI ESCA/SAM 5600 Multi technique spectrometer with the use of a Mg standard X-ray source. The pressure in the chamber was ∼10−9 Torr. The analyses were carried out at 45° photoelectron takeoff angle relative to the sample surface with an acceptance angle of ±3°. The analyzer pass energy was set at 23.5 eV for the high-resolution of all the spectra in the regions of C 1s, N 1s, O 1s, and Zn 2p. The binding energy (BE) scale was calibrated by centering the C 1s signal of the aliphatic/aromatic component at 285.0 eV.Photo-degradation measurements
The photocatalytic activity of the ZnO/polymers composites was evaluated by the degradation of Methylen Blue (MB) in water under UV irradiation. Before the MB photo-degradation tests, the samples were irradiated by an UV lamp for 60 min in order to remove the hydrocarbons from the sample surface. The samples (1 cm × 1 cm in size), were thereafter immersed in a 2 mL of a MB 1.5 × 10−5 M solution. In order to achieve the physisorption equilibrium, the MB aqueous solution was left in dark for 120 min. After such preconditioning step, the samples were irradiated under UV (368 nm, power of ∼8 W, irradiance of ∼2 mW cm−2). The sample containers were covered with quartz slices with the aim of preventing the evaporation of the solvent during the experiments. Measurements of absorbance were performed by using a Lambda 45 spectrophotometer provided by Perkin-Elmer. The variation of the MB concentration as a function of the irradiation time was evaluated by applying the Lambert–Beer law to the absorbance value at λ = 664 nm. A reference, consisting of 2 mL MB solution without any photocatalyst materials immersed was measured. The tests were repeated three times on the set of samples.Conclusions
In this paper, we discuss on the synthesis of hybrid systems consisting of ZnO nanostructures directly grown on polyetherimide (ULTEM®) by ALD. The polymers have been properly surface-modified by photo-oxidation so to trigger the formation of ALD reactive sites for the covalent anchoring of the ALD species. Surface reactions between the vapor precursors and the reactive functionalities of the polymer are demonstrated, with the formation of chemical bonds at the organic–inorganic interface. By combining spectroscopy and mass analyses, we show that two different regimes of growth occur in the assembly of the inorganic phase during the deposition process, depending on time of the photo-oxidative process applied on the polymer. In particular, a nano-structured layer of ZnO is seated on the ULTEM surface when short-time pre-oxidative treatments are performed (ALD-like reactions). On the other hand, phenomena of gas diffusion result facilitated in the long-irradiated polyetherimide because of the decrease of molar masses of the polymer thus leading to sub-surface grafting of the metal-precursor and sub-surface reactions for the building of the ZnO phase englobed in the polymeric matrix (VPI-like regime). In conclusion, the extent of the polymer photo-exposure affects the macromolecule structure and tunes the density and accessibility of the functional groups, determining the arrangement of the organic/inorganic assembly in the final hybrid nano-composite. As a result, variability in the material properties is predictable, as in the case herein shown of photo-catalytic performance. We retain that our approach, based on tunable surface modification of the organic material, can be easily extended to other polymers and metal oxides, thus opening new vistas on the formulation of novel polymer/inorganic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Luciano Falqui for fruitful discussion, Maria Violetta Brundo for the TEM sample preparation by ultramicrotomy technique, and Giuseppe Panté for the technical assistance. This work has been supported by the FP7 European project WATER (Grant Agreement 316082).Notes and references
- X. Qu, P. J. J. Alvarez and Q. Li, Water Res., 2013, 47, 3931–3946 CrossRef CAS PubMed.
- M. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Mariñas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- A. S. Adeleye, J. R. Conway, K. Garner, Y. Huang, Y. Su and A. A. Keller, Chem. Eng. J., 2016, 286, 640–662 CrossRef CAS.
- Nanotechnology for Water Treatment and Purification, ed. A. Hu and A. Apblett, Springer, Switzerland, 2014 Search PubMed.
- H. N. Cheng, K. T. Klasson, T. Asakura and Q. Wu, ACS Symp. Ser., 2016, 1224, 233–242 CrossRef CAS.
- E. Serrano, G. Rus and J. Garcia-Martinez, Renewable Sustainable Energy Rev., 2009, 13, 2373–2384 CrossRef CAS.
- C. G. Thanos and D. F. Emerich, Expert Opin. Biol. Ther., 2003, 3(4), 655–663 CrossRef PubMed.
- G. P. Berman and G. D. Doolen, Superlattices Microstruct., 2000, 27(2–3), 89–104 CrossRef.
- V. Scuderi, G. Impellizzeri, M. Zimbone, R. Sanz, A. Di Mauro, M. A. Buccheri, M. Miritello, A. Terrasi, G. Rappazzo, G. Nicotra and V. Privitera, Appl. Catal., B, 2016, 183, 328–334 CrossRef CAS.
- V. Scuderi, G. Impellizzeri, L. Romano, M. Scuderi, G. Nicotra, K. Bergum, A. Irrera, B. G. Svensson and V. Privitera, Nanoscale Res. Lett., 2014, 9, 458 CrossRef PubMed.
- V. Scuderi, G. Impellizzeri, L. Romano, M. Scuderi, M. V. Brundo, K. Bergum, M. Zimbone, R. Sanz, M. a. Buccheri, F. Simone, G. Nicotra, B. G. Svensson, M. G. Grimaldi and V. Privitera, Nanoscale, 2014, 6, 11189–11195 RSC.
- F. Kayacia, S. Vempatia, C. Ozgit-Akguna, I. Donmeza, N. Biyikli and T. Uyara, Appl. Catal., B, 2015, 176–177, 646–653 CrossRef.
- F. Kayaci, S. Vempati, I. Donmez, N. Biyikliab and T. Uyar, Nanoscale, 2014, 6, 10224 RSC.
- A. Di Mauro, M. Zimbone, M. Scuderi, G. Nicotra, M. E. Fragalà and G. Impellizzeri, Nanoscale Res. Lett., 2015, 10, 484 CrossRef PubMed.
- Y. Li, R. Yao, H. Wang, X. Wu, J. Wu, X. Wu and W. Qin, ACS Appl. Mater. Interfaces, 2017, 9, 11711–11720 CAS.
- Y. Li, J. Wang, Y. Kong, J. Zhou, J. Wu, G. Wang, H. Bi, X. Wu, W. Qin and Q. Li, Sci. Rep., 2016, 6, 19187 CrossRef CAS PubMed.
- J. Gun Oh, Y. Shin, W. Cheol Shin, O. Sul and B. Jin Cho, Appl. Phys. Lett., 2011, 99(19), 193503 CrossRef.
- A. Di Mauro, M. Cantarella, G. Nicotra, V. Privitera and G. Impellizzeri, Appl. Catal., B, 2016, 196, 68–76 CrossRef CAS.
- N. Pinna and M. Knez, Atomic Layer Deposition of Nanostructured Materials, Wiley-VCH, Weinheim, Germany, 2012, pp. 201–203 Search PubMed.
- H. Kim, H. B. R. Lee and W. J. Maeng, Thin Solid Films, 2009, 517, 2563–2580 CrossRef CAS.
- G. N. Parsons, S. E. Atanasov, E. C. Dandley, C. K. Devine, B. Gong, J. S. Jur, K. Lee, C. J. Oldham, Q. Peng, J. C. Spagnola and P. S. Williams, Coord. Chem. Rev., 2013, 257, 3323–3331 CrossRef CAS.
- T. Suntola and J. Hyvarinen, Annu. Rev. Mater. Sci., 1985, 15, 177–195 CrossRef CAS.
- S. M. George and S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- J. Lu, J. Elam and P. Stair, Acc. Chem. Res., 2013, 46, 1806–1815 CrossRef CAS PubMed.
- A. Di Mauro, M. Cantarella, G. Nicotra, G. Pellegrino, A. Gulino, M. V. Brundo, V. Privitera and G. Impellizzeri, Sci. Rep., 2017, 7, 40895 CrossRef CAS PubMed.
- K. Gregorczyk and M. Knez, Prog. Mater. Sci., 2016, 75, 1–37 CrossRef CAS.
- N.-B. Singh and S. Agarwal, Emerging Mater. Res., 2016, 5(1), 5–43 CrossRef.
- L. Yao, L. E. Oquendo, M. W. Schulze, R. M. Lewis, W. L. Gladfelter and M. A. Hillmyer, ACS Appl. Mater. Interfaces, 2016, 8, 7431–7439 CAS.
- S.-M. Lee, E. Pippel, O. Moutanabbir, J.-H. Kim, H.-J. Lee and M. Knez, ACS Appl. Mater. Interfaces, 2014, 6, 16827–16834 CAS.
- K. J. Hughes and J. R. Engstrom, J. Vac. Sci. Technol., A, 2010, 28, 1033 CAS.
- M. Moshonov and G. L. Frey, Langmuir, 2015, 31, 12762–12769 CrossRef CAS PubMed.
- C. Z. Leng and M. D. Losego, Mater. Horiz., 2017, 4, 747–771 RSC.
- R. Saberi Moghaddam, S. Huettner, Y. Vaynzof, C. Ducati, G. Divitini, R. H. Lohwasser, K. P. Musselman, A. Sepe, M. R. J. Scherer, M. Thelakkat, U. Steiner and R. H. Friend, Nano Lett., 2013, 13, 4499–4504 CrossRef CAS PubMed.
- J. Frascaroli, E. Cianci, S. Spiga, G. Seguini and M. Perego, ACS Appl. Mater. Interfaces, 2016, 8, 33933–33942 CAS.
- B. Gong and G. N. Parsons, J. Mater. Chem., 2012, 22, 15672–15682 RSC.
- J. S. Jur, J. C. Spagnola, K. Lee, B. Gong, Q. Peng and G. N. Parsons, Langmuir, 2010, 26, 8239–8244 CrossRef CAS PubMed.
- R. P. Padbury and J. S. Jur, J. Vac. Sci. Technol., A, 2015, 33, 01A112 Search PubMed.
- M. Biswas, J. A. Libera, S. B. Darling and J. W. Elam, J. Phys. Chem. C, 2015, 119, 14585–14592 CAS.
- M. Biswas, J. A. Libera, S. B. Darling and J.W. Elam, Chem. Mater., 2014, 26, 6135–6141 CrossRef CAS.
- S. Carroccio, C. Puglisi and G. Montaudo, Macromolecules, 2005, 38, 6849–6862 CrossRef CAS.
- S. Carroccio, C. Puglisi and G. Montaudo, Macromolecules, 2005, 38, 6863–6870 CrossRef CAS.
- S. Carroccio, C. Puglisi and G. Montaudo, Polym. Degrad. Stab., 2003, 80, 459–476 CrossRef CAS.
- G. Montaudo, S. Carroccio, F. Samperi and C. Puglisi, Recent Advances in Maldi Mass Spectrometry of Polymers, Macromol. Symp., 2004, 218, 101 Search PubMed.
- S. Edge, W. J. Feast, W. F. Pacynko, L. Peston and S. Walker, Polym. Bull., 1992, 27, 441–445 CrossRef CAS.
- F. S. Li, W. Qiu, R. P. Lively, J. S. Lee, A. A. Rownaghi and W. J. Koros, ChemSusChem, 2013, 6, 1216–1223 CrossRef CAS PubMed.
- G. Pellegrino, A. Motta, A. Cornia, I. Spitaleri, I. L. Fragalà and G. G. Condorelli, Polyhedron, 2009, 28, 1758–1763 CrossRef CAS.
- G. G. Condorelli, A. Motta, C. Bedoya, A. Di Mauro, G. Pellegrino and E. Smecca, Inorg. Chim. Acta, 2007, 360, 170–178 CrossRef CAS.
- J. P. Lee, Y. J. Jang and M. M. Sung, Adv. Funct. Mater., 2003, 13, 873–876 CrossRef CAS.
- M. C. Burrell and J. J. Chera, Surf. Sci. Spectra, 1999, 6, 17–22 Search PubMed.
- G. G. Condorelli, A. Motta, G. Pellegrino, A. Cornia, L. Gorini, I. L. Fragalà, C. Sangregorio and L. Sorace, Chem. Mater., 2008, 20, 2405–2411 CrossRef CAS.
- G. Pellegrino, G. G. Condorelli, V. Privitera, B. Cafra, S. Di Marco and A. Alberti, J. Phys. Chem. C, 2011, 115, 7760–7767 CAS.
- P. V. Krasovskii, O. S. Malinovskaya, A. V. Samokhin, Y. V. Blagoveshchenskiy, V. A. Kazakov and A. A. Ashmarin, Appl. Surf. Sci., 2015, 339, 46–54 CrossRef CAS.
- K. E. Gregorczyk, D. F. Pickup, M. G. Sanz, I. A. Irakulis, C. Rogero and M. Knez, Chem. Mater., 2017, 27, 181–188 CrossRef.
- G. Pellegrino, G. G. Condorelli, F. De Rossi, T. M. Brown, F. Giovenale, C. Bongiorno and A. Alberti, Appl. Surf. Sci., 2014, 296, 69–78 CrossRef CAS.
- S. C. George and S. Thomas, Prog. Polym. Sci., 2001, 26, 985–1017 CrossRef CAS.
- T. Lodge, Phys. Rev. Lett., 1999, 83, 3218–3221 CrossRef CAS.
- S. Matteucci, Y. Yampolskii, B. D. Freeman and I. Pinnau, Mater. Sci. Membr. Gas Vap. Sep., 2006, 1–47 CAS.
- G. Impellizzeri, V. Scuderi, L. Romano, E. Napolitani, R. Sanz, R. Carles and V. Privitera, J. Appl. Phys., 2015, 117, 1053081–1053086 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. E1 – FTIR-ATR spectra related to the untreated, 8 h- and 48 h-treated ULTEM® films before and after ZnO ALD deposition. Fig. E2 – XPS spectra related to the N 1s and Zn 2p regions of untreated, 8 h- and 48 h-treated ULTEM® films after ZnO ALD deposition. See DOI: 10.1039/c7ra11168a |
| This journal is © The Royal Society of Chemistry 2018 |