
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize effect of oxygen reduction reaction on nitrogen-doped graphene quantum dots†
Peng Zhang a,
Qiang Hua,
Xuejing Yanga,
Xiuli Hou*a,
Jianli Mia,
Lei Liua and
Mingdong Dong*b
a,
Qiang Hua,
Xuejing Yanga,
Xiuli Hou*a,
Jianli Mia,
Lei Liua and
Mingdong Dong*b
aInstitute for Advanced Materials, School of Materials Science and Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: houxiuli@ujs.edu.cn
bCenter for DNA Nanotechnology (CDNA), Interdisciplinary Nanoscience Center (iNANO), Aarhus University, Aarhus DK-8000, Denmark. E-mail: dong@inano.au.dk
First published on 2nd January 2018
Abstract
N-doped graphene quantum dots (GQDs) are attracting great interest as promising Pt-free oxygen reduction reaction (ORR) catalysts. In this work, the mechanism of ORR on N-doped GQDs with three different sizes has been investigated based on density functional theory. Our results show that the size of the N-doped GQDs affects the ORR. The adsorption strength of ORR intermediates, the reaction free energy of the rate-determining step, and the overpotential increase with the increase of the size of N-doped GQDs. The N-doped GQDs with the smallest size possess the smallest overpotential, indicating the highest ORR catalytic activity. Our results will help to understand the size effect of N-doped GQDs on the catalytic activity of ORR.
Introduction
Fuel cells have attracted much attention as a clean and sustainable power source. They can directly convert the chemical energy contained in fuels into electric energy with high conversion efficiency, high power density, quiet operation, and no pollution.1,2 The current bottleneck of fuel cells lies in the sluggish oxygen reduction reaction (ORR) on the cathode side. To date, the preferred electrocatalysts are Pt and platinum group metals (PGM), which pose a significant cost barrier for fuel cell commercialization due to their scarcity and high price. Great efforts have been devoted to the development of low-cost alternative catalysts with high activity and durability in the ORR, including metal-free heteroatom doped carbon materials,3–5 transition metal–nitrogen–carbon materials,6–9 and transition metal oxides or sulfides.10–13Among various alternative ORR catalysts, heteroatom (N, B, P, F, and S)-doped carbon materials, including fullerenes, carbon nanotubes, graphene, and mesoporous carbon, are expected to be one of the most promising alternatives to Pt-based catalysts due to their low-cost, high electrocatalytic activity, selectivity and stability.14–20 Recent studies have confirmed that N-doped carbon nanotubes and graphene can catalyze a four-electron ORR process free from CO poisoning with a 3-time higher electrocatalytic activity, smaller crossover effect, and better long-term operation stability than that of Pt-based electrodes in alkaline electrolytes.21,22 The high catalytic activity of the doped carbon materials may be attributed to the polarized distribution of spin and charge density which are caused by the introduced heteroatoms.23–32
Among various carbon materials, zero-dimensional graphene quantum dots (GQDs) with abundant edges have attracted great interest.5,14,33–36 The edge-abundant features of GQDs are particularly advantageous for electrocatalysts as ORR is more readily electrochemically catalyzed at the edge than the basal plane.14,37–39 Recently, Qu et al. electrochemically prepared N-doped GQDs with N/C atomic ratio of ca. 4.3%, which possessed an electrocatalytic activity comparable to that of a commercial Pt/C catalyst toward ORR in alkaline media.40 Furthermore, B- and N-doped graphene quantum dots/graphene hybrid nanoplatelets exhibited excellent oxygen reduction performance with activity higher than that of commercial Pt/C in alkaline media.41 The high activity of this hybrid materials can be attributed to the abundant edges and doping sites, high electrical conductivity, and high surface area.41
It is well known that the electronic structure of GQDs is strongly dependent on the geometrical size,42 which will change the catalytic activity of GQDs. However, a complete understanding of size effect of ORR on GQDs has not been obtained so far. In this work, the size effect of ORR on N-doped GQDs was studied based on density functional theory (DFT). Our results show that the size of N-doped GQDs can alter the adsorption strength of ORR intermediates, and also the overpotential significantly. All the adsorption strength of ORR intermediates, the reaction free energy of rate-determining step, and the overpotential increase with the increasing size of GQDs.
Computational method
The spin-unrestricted DFT calculations are performed using DMol3 code, which uses numerical functions on an atom-centered grid as its atomic basis.43,44 The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) method is utilized as the exchange–correlation functional.45 The All Electron Relativistic (AER), which includes all core electrons explicitly and introduces some relativistic effects into the core, is adopted for the core treatment.46 In addition, double numerical plus polarization (DNP) is used as the basis set.43 In order to describe the van der Waals (vdW) interaction, the DFT + D2 method within Grimme scheme is adopted.47 The convergence tolerance of energy is 1.0 × 10−5 Ha, maximum force is 0.002 Ha Å−1, and maximum displacement is 0.005 Å in geometry optimization.Graphene is a crystalline allotrope of carbon consisting of a single layer of carbon atoms arranged in a hexagonal lattice. The packed carbon atoms bond with three neighbors through sp2 orbital hybridization. Recent investigations have shown that the high catalytic activity of N-doped carbon materials mostly contributes to the graphitic-type N-doped zigzag-shaped edge.48–52 Thus, only graphitic-type N-doped GQDs are considered here. Three hexagonal GQDs with different sizes are constructed. The location of N dopants is important to the ORR catalytic activity. The energy variation of different N-doped site is described in ESI (Fig. S1 and Table S1†). N atoms prefer to substitute the C atoms near the edge. This is consistent with recent published papers.48,51,52 The most stable atomic structures of N-doped GQDs (named C23H12N, C53H18N and C95H24N) are shown in Fig. 1. The orthorhombic supercells with dimension of 45 × 45 × 45 Å3 are constructed for N-doped GQDs. Such large cells are required to eliminate the interactions between GQDs in adjacent cells. The adsorption energy (Ead) values of ORR intermediates on N-doped GQDs are calculated as Ead = Emol + EN-GQDs − Emol-N-GQDs, where Emol, EN-GQDs, and Emol-N-GQDs are the DFT energies of an isolated adsorbate molecule, N-doped GQDs and the adsorption systems, respectively. Based on these definitions, a positive Ead value denotes an exothermic adsorption process.
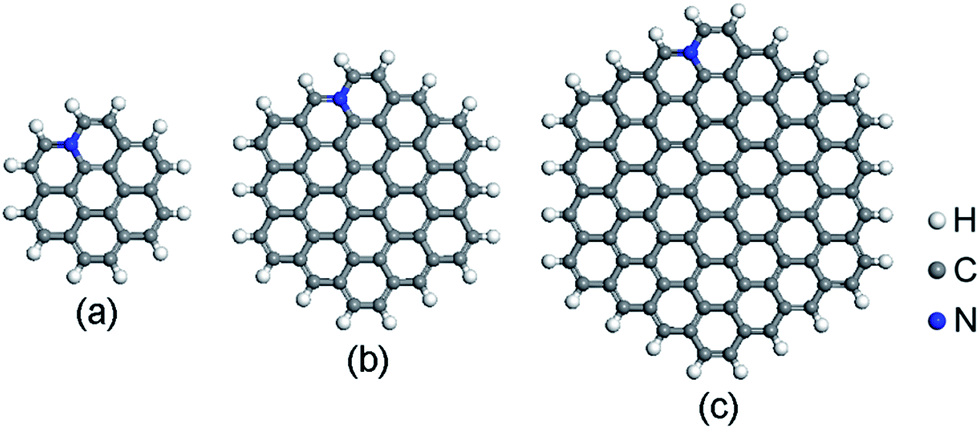 | ||
| Fig. 1 Optimized structures of N-doped graphene quantum dots: (a) C23H12N, (b) C53H18N, and (c) C95H24N. | ||
The reaction free energies (ΔG) of elemental steps involved in ORR are calculated based on the computational hydrogen electrode (CHE) model, developed by Nørskov et al.53–55 The ΔG for a elemental step is calculated as follows: ΔG = ΔE + ΔZPE − TΔS + ΔGU + ΔGpH, where ΔE is the reaction energy obtained from DFT calculations, ΔZPE is the difference in zero point energies, T is the temperature (T = 298.15 K in this paper), and ΔS is the change in entropy. The calculated electronic energies, zero point energies, and entropy values of N-doped GQDs with adsorbed ORR intermediates are summarized in Table S2 of ESI.† The effect of the electrode potential on ΔG is quantified as ΔGU = neU, where U is the electrode potential relative to the standard hydrogen electrode and n is the number of electrons transferred in the elemental steps. ΔGpH is related to the free energy contributed from pH value, and ΔGpH = kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln10 × pH, where kB is the Boltzmann's constant. pH is 0 in this work. A conductor-like screening model (COSMO) is adopted to describe the water solvent environment, and the dielectric constant is set as 78.54 for water solvent.56
ln10 × pH, where kB is the Boltzmann's constant. pH is 0 in this work. A conductor-like screening model (COSMO) is adopted to describe the water solvent environment, and the dielectric constant is set as 78.54 for water solvent.56
Results and discussion
Before the investigation of the ORR mechanisms, the adsorption properties of ORR intermediates on N-doped GQDs are studied. Fig. 2 displays the energy favorable adsorption structures of ORR intermediates (O2, OOH, O, OH and H2O) on N-doped GQDs. The Ead values of ORR intermediates are summarized in Table 1. The bond lengths of O–C bond for adsorbed ORR intermediates on N-doped GQDs are displayed in Table S3 of ESI.† The adsorption of O2 molecule is a prerequisite for the ORR proceeded on the surface of electrocatalysts. It is the first step in ORR progress, which always determines the ORR catalytic active sites of N-doped carbon materials. As shown in Fig. 2, the O2 molecule prefers to adsorb on C atom near doped N atom at the edge of N-doped GQDs. It is interesting to note that the Ead value of O2 molecule increases from 0.24 eV on C23H12N to 0.48 eV on C53H18N and 0.49 eV on C95H24N. This is agreement with the Mulliken charge population distribution for O2 adsorbed on N-doped GQDs. The quantities of charge transferred from the C atom to O2 molecule are quantified as 0.669, 0.686, and 0.693e for C23H12N, C53H18N, and C95H24N, respectively. Furthermore, the adsorption strength of O2 on N-doped GQDs is consistent with the bond length of O–C bond in Table S2.† It is clear that the stronger the adsorption strength of O2 is, the shorter the bond length becomes.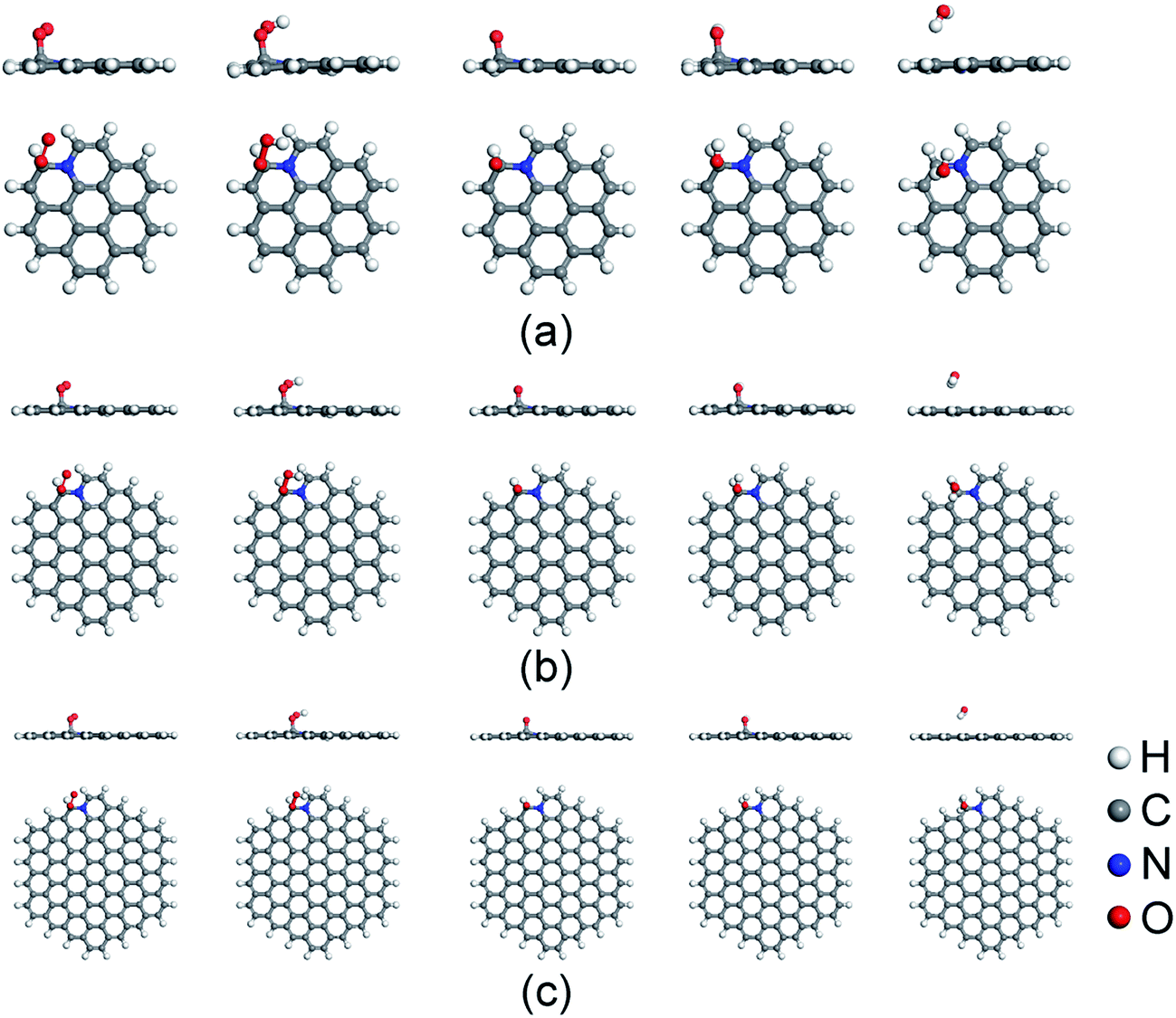 | ||
| Fig. 2 Favorable adsorption structures of ORR intermediates on C23H12N (a), C53H18N (b), and C95H24N (c). | ||
| Ead | O2 | OOH | O | OH | H2O |
|---|---|---|---|---|---|
| C23H12N | 0.24 | 1.45 | 4.06 | 2.69 | 0.18 |
| C53H18N | 0.48 | 1.65 | 4.26 | 2.97 | 0.12 |
| C95H24N | 0.49 | 1.65 | 4.27 | 3.00 | 0.13 |
| C94H24N2 | 0.79 | 1.68 | 4.63 | 3.01 | 0.14 |
| C92H24N4 | 0.84 | 1.56 | 4.75 | 2.88 | 0.16 |
Similar with O2, the favorable adsorption site of OOH, O and OH also locates at the C atom near doped N atom. This is consistent with previous reports about N-doped carbon nanotubes and graphene.48,51,52,55 The adsorption strength of OOH, O and OH also increases with the increase of the size of the N-doped GQDs. Furthermore, the H2O molecule is only physisorbed on the N-doped GQDs and can easily desorb from the reaction site once it forms, which will be helpful to renew the ORR catalysts.
Above results show that the size of N-doped GQDs has strong effect on the adsorption of ORR intermediates. As demonstrated by previous work on heterogeneous catalysts, the adsorption strength of ORR intermediates plays a critical role in the activity of ORR catalysts.57–60 The different adsorption strength of ORR intermediates on N-doped GQDs may correspond to different ORR kinetic properties.
Following the adsorption of O2, O2 can be reduced to H2O through four net coupled proton-electron transfer (CPET) steps. The whole ORR process can be expressed as follows:5,29,61,62
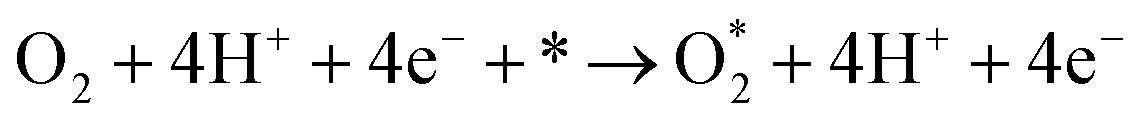 | (R1) |
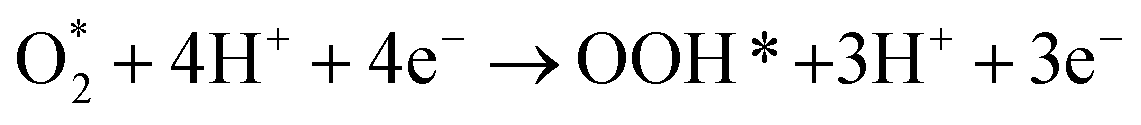 | (R2) |
| OOH* + 3H+ + 3e− → O* + 2H+ + 2e− + H2O | (R3) |
| O* + 2H+ + 2e− + H2O → OH* + H+ + e− + H2O | (R4) |
| OH* + H+ + e− + H2O → 2H2O + * | (R5) |
This pathway has been proved to be energy favorable on heteroatom-doped carbon materials with four CPET steps involved in the ORR process. The ΔG values of these steps are dependent on the electrode potential. The efficiency of the ORR process can be determined by the ΔG of the rate-determining step, which is the elemental step with the smallest ΔG in the ORR pathway. Here, the overpotential η is used to describe the catalytic activity of N-doped GQDs, which is defined as the difference between the equilibrium potential of the ORR process per step (1.23 V) and the lowest potential where the rate-determining step is downhill.5 The smaller the overpotential, the higher the activity of corresponding ORR catalyst.
Fig. 3 shows the free energy diagrams of ORR on N-doped GQDs with different size. When electrode potential U equals to 0 V, the ΔG values of the five elemental steps involved in ORR on C23H12N are calculated to be −0.35 eV for the adsorption of O2 molecule, −1.20 eV for the reduction of adsorbed O2 molecule to OOH, −1.87 eV for the reduction of OOH to form an adsorbed O atom and the first H2O molecule, −1.06 eV for the hydrogenation of O atom to OH, and −0.44 eV for the formation of the second H2O molecule, respectively. When the ideal electrode potential U increases to 1.23 V, the energy level of each net CPET step is shifted up by 1.23 eV. At U = 1.23 V, the ΔG values of the five elemental steps are −0.35, 0.03, −0.64, 0.17, and 0.79 eV, respectively. It can be seen that the formation of the second H2O molecule is the rate-determining step which represents the highest resistance for the whole ORR at high potential. For the ORR on C53H18N, the ΔG values of the five elemental steps are calculated to be −0.28, −1.24, −1.89, −1.15 and −0.35 eV, respectively, at U = 0 V, and −0.28, −0.01, −0.66, 0.08 and 0.88 eV, respectively, at U = 1.23 V. For ORR on C95H24N, the ΔG values of the five elemental steps are −0.42, −1.26, −1.87, −1.19 and −0.18 eV, respectively, at U = 0 V, and −0.42, −0.03, −0.64, 0.04 and 1.05 eV, respectively, at U = 1.23 V. Similar to the ORR on C23H12N, the rate-determining steps of the ORR on C53H18N and C95H24N are the formation of the second H2O molecule. This is consistent with our recent work about the ORR on N-doped zigzag graphene nanoribbons, where the rate-determining step also locates the step of the formation of the second H2O molecule.50
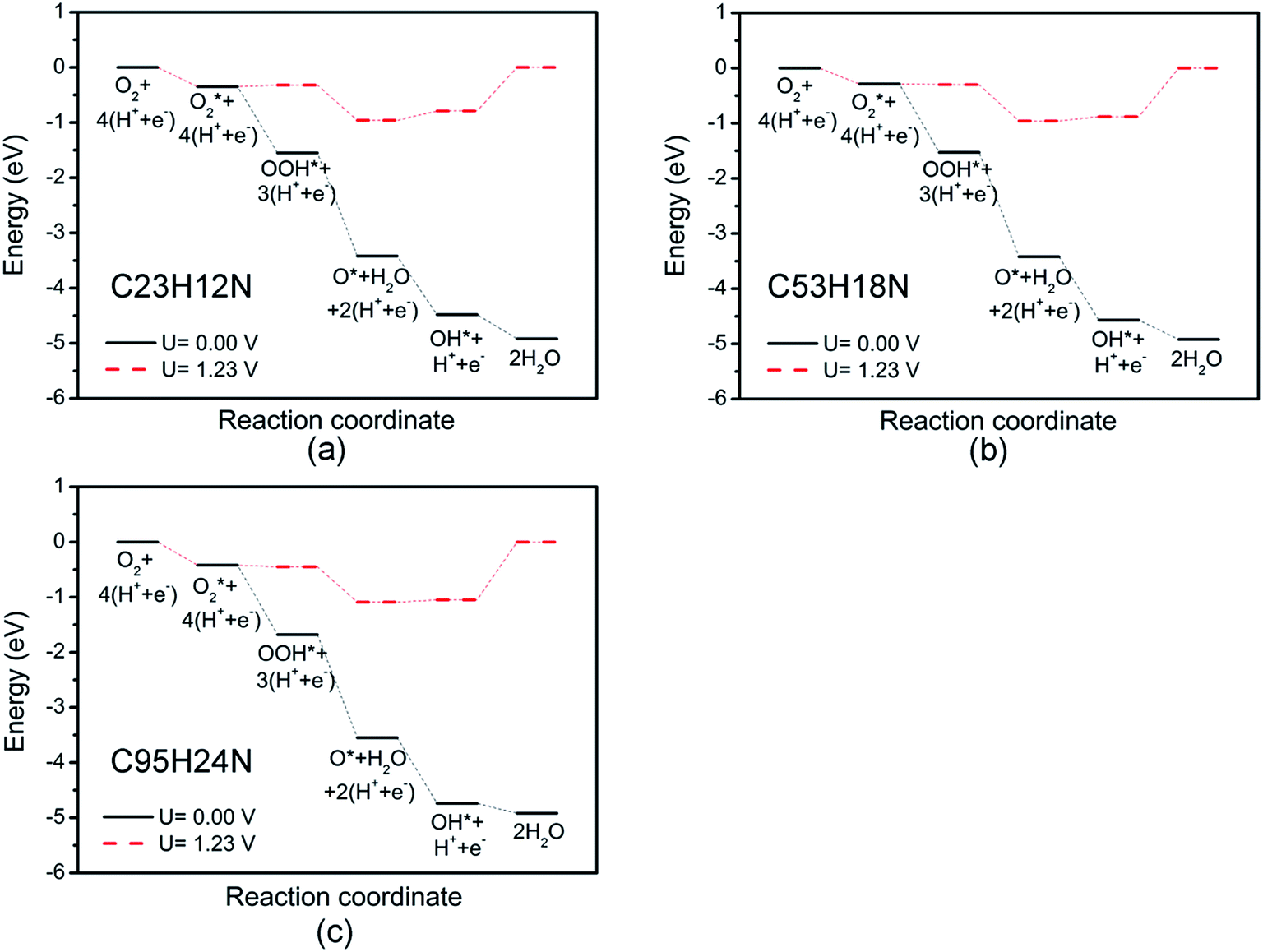 | ||
| Fig. 3 Schematic Gibbs free energy diagrams of ORR on C23H12N (a), C53H18N (b), and C95H24N (c). | ||
The overpotentials of N-doped GQDs are calculated to be 0.79, 0.88 and 1.05 V for C23H12N, C53H18N and C95H24N, respectively, suggesting that C23H12N exhibits the best ORR catalytic activity among the three N-doped GQDs from the theoretical viewpoint. This is consistent with the adsorption strength of ORR intermediates on N-doped GQDs. The ΔG value of rate-determining step increases with the adsorption strength on N-doped GQDs. The strong adsorption of ORR intermediates will make it hard to break the C–OH bond and increase the ΔG value of OH reduction to H2O.
We must keep in mind is that GQDs can be described as macromolecules and are typically semiconductive with a clear separation between the HOMO and LUMO orbits. We note that the energy gap (Eg) between the valence band maximum (VBM) and the conduction band minimum (CBM) for N-doped GQDs decreases from 2.502 eV of C23H12N to 1.171 eV of C95H24N, as shown in Fig. 4(a). The larger Eg of C23H12N may have negative effect on ORR due to their low electron conductivity.
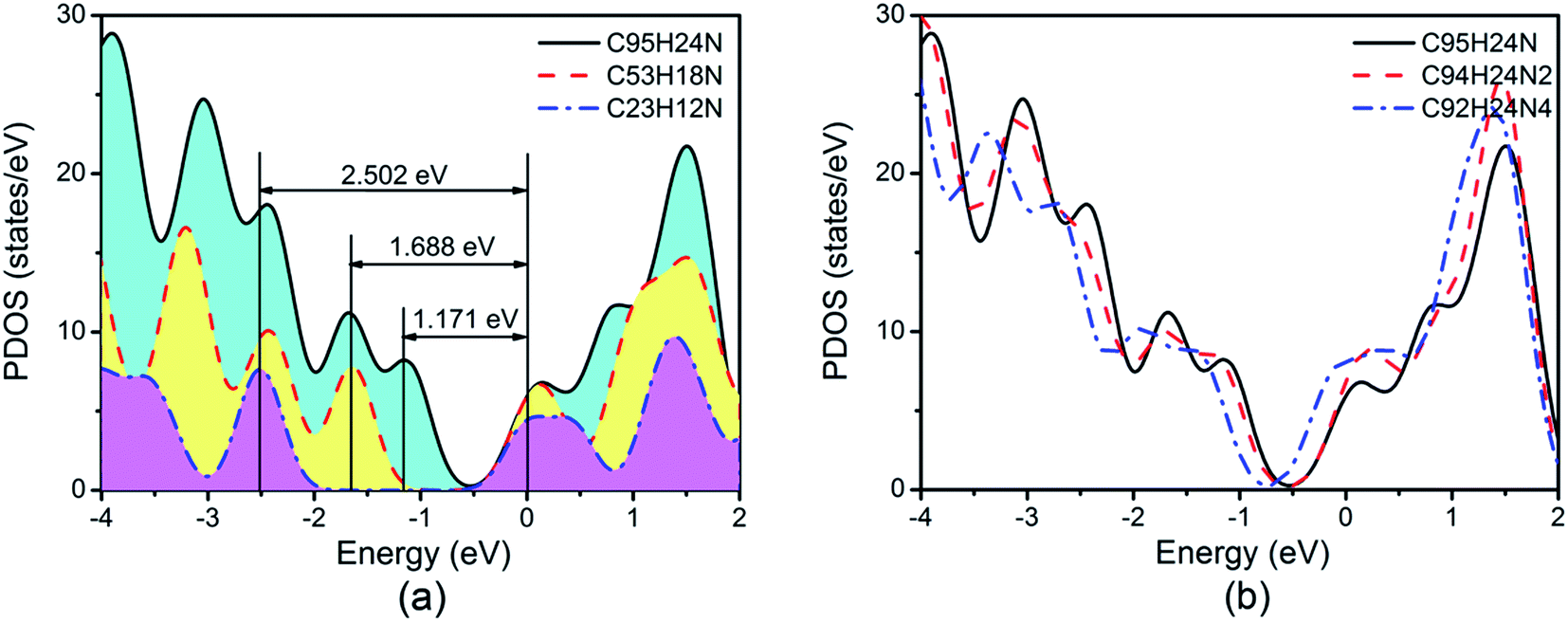 | ||
| Fig. 4 Partial density of states (PDOS) of N-doped graphene quantum dots. | ||
Based on recent experimental results, it is well known that the N concentration plays an important role in the catalytic activity of carbon materials toward ORR.63,64 In order to study the effect of the concentration of the graphitic-type N atoms on the catalytic activity of ORR, we investigated the catalytic activity of two graphitic-type N-doped GQDs with two and four N atoms named C94H24N2 and C92H24N4 (shown in Fig. 5). The corresponding schematic Gibbs free energy diagrams are shown in Fig. 6. The favorable adsorption structures of ORR intermediates on C94H24N2 and C92H24N4 are similar with those on C95H24N and are not shown again. Similar with ORR on C95H24N, the rate-determining step of ORR on C94H24N2 and C92H24N4 at high potential also locates at the step of the formation of the second H2O molecule. The ΔG values of the formation of the second H2O molecule at U = 1.23 V are calculated to be 1.12 and 1.06 eV on C94H24N2 and C92H24N4, respectively, which are consistent with that on C95H24N of 1.05 eV, suggesting that the concentration of graphitic-type N atom near the edge does not influence the overpotential of graphitic-type N-doped GQDs. This is agreement with comparable adsorption strength of OH on C95H24N, C94H24N2 and C92H24N4 (displayed in Table 1). Furthermore, the electronic structures of N-doped GQDs with different N atoms are checked and shown in Fig. 4(b). It is clear that the variation of the electronic structures of N-doped GQDs is very small, suggesting similar electrical conductivity. Although the concentration of graphitic-type N atom does not affect the overpotential and conductive property of GQDs clearly, enlarging the concentration of graphitic-type N atom can increase the number of active sites undoubtedly.
 | ||
| Fig. 5 Optimized structures of N-doped C96H24 graphene quantum dots: (a) C94H24N2, and (b) C92H24N4. | ||
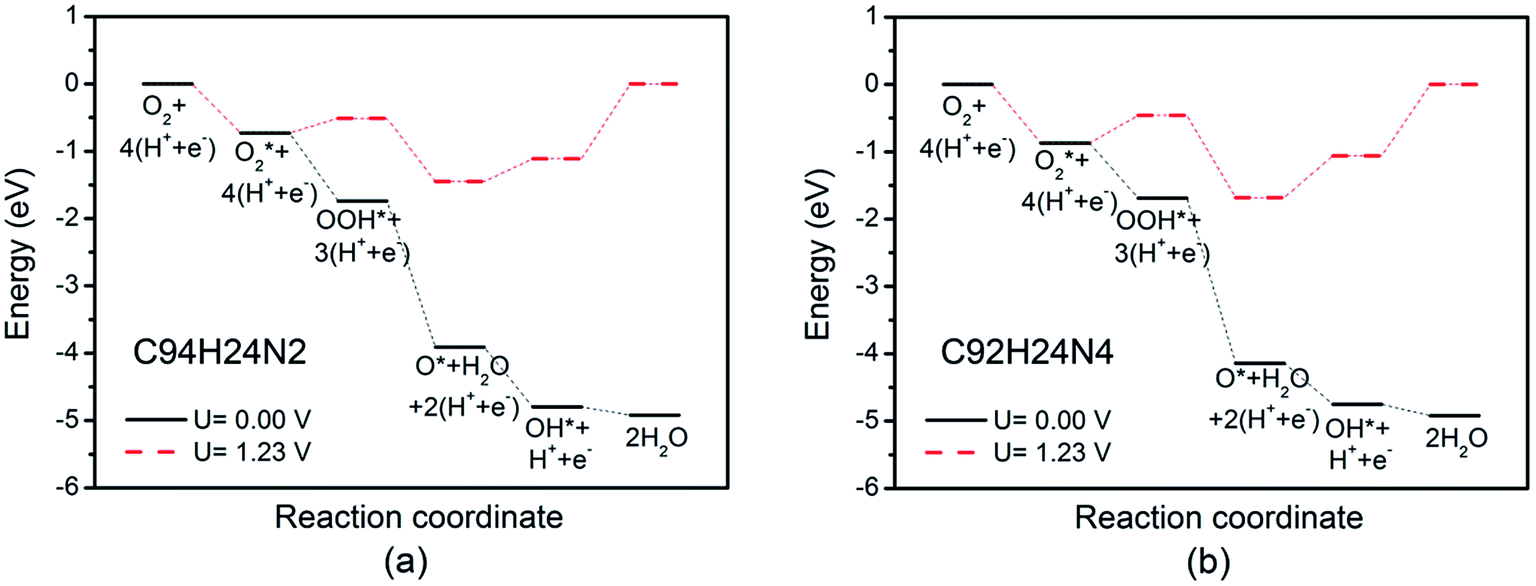 | ||
| Fig. 6 Schematic Gibbs free energy diagrams of ORR on C94H24N2 (a), and C92H24N4 (b). | ||
Conclusion
In summary, based on DFT calculation, the ORR on N-doped GQDs has been investigated systematically. It was found that N-doped GQDs with different size exhibits different catalytic activity. As the size of N-doped GQDs increases, the adsorption strength of ORR intermediates and the reaction free energy of rate-determining step involved in the ORR increase. These results could provide useful information to further develop novel N-doped GQDs with high ORR catalytic activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support from the National Natural Science Foundation of China (21403092 and 51401089), the Natural Science Foundation of Jiangsu (BK20140552), the Special Financial Grant from the China Postdoctoral Science Foundation (2015T80506), and the Senior Intellectuals Fund of Jiangsu University (12JDG094, 13JDG032 and 14JDG013). The authors would also like to thank Prof. Qing Jiang in Jilin University for the computer resources provided and helpful discussions.References
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060 RSC.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168 RSC.
- X. K. Kong, C. L. Chen and Q. W. Chen, Chem. Soc. Rev., 2014, 43, 2841 RSC.
- K. Chizari, A. Deneuve, O. Ersen, I. Florea, Y. Liu, D. Edouard, I. Janowska, D. Begin and C. Pham-Huu, ChemSusChem, 2012, 5, 102 CrossRef CAS PubMed.
- W. A. Saidi, J. Phys. Chem. Lett., 2013, 4, 4160 CrossRef CAS.
- A. Serov, K. Artyushkova and P. Atanassov, Adv. Energy Mater., 2014, 4, 1301735 CrossRef.
- H. W. Liang, W. Wei, Z. S. Wu, X. Feng and K. Mullen, J. Am. Chem. Soc., 2013, 135, 16002 CrossRef CAS PubMed.
- D. H. Lee, W. J. Lee, W. J. Lee, S. O. Kim and Y. H. Kim, Phys. Rev. Lett., 2011, 106, 175502 CrossRef PubMed.
- S. Kattel and G. Wang, J. Phys. Chem. Lett., 2014, 5, 452 CrossRef CAS PubMed.
- Y. Gorlin, C. J. Chung, D. Nordlund, B. M. Clemens and T. F. Jaramillo, ACS Catal., 2012, 2, 2687 CrossRef CAS.
- W. Yuan, J. Li, L. Wang, P. Chen, A. Xie and Y. Shen, ACS Appl. Mater. Interfaces, 2014, 6, 21978 CAS.
- P. Ganesan, M. Prabu, J. Sanetuntikul and S. Shanmugam, ACS Catal., 2015, 5, 3625 CrossRef CAS.
- X. Wang, Y. Ke, H. Pan, K. Ma, Q. Xiao, D. Yin, G. Wu and M. T. Swihart, ACS Catal., 2015, 5, 2534 CrossRef CAS.
- M. Favaro, L. Ferrighi, G. Fazio, L. Colazzo, C. Di Valentin, C. Durante, F. Sedona, A. Gennaro, S. Agnoli and G. Granozzi, ACS Catal., 2015, 5, 129 CrossRef CAS.
- X. Sun, Y. Zhang, P. Song, J. Pan, L. Zhuang, W. Xu and W. Xing, ACS Catal., 2013, 3, 1726 CrossRef CAS.
- C. Zhu and S. Dong, Nanoscale, 2013, 5, 1753 RSC.
- Z. Liu, H. Nie, Z. Yang, J. Zhang, Z. Jin, Y. Lu, Z. Xiao and S. Huang, Nanoscale, 2013, 5, 3283 RSC.
- Z. W. Liu, F. Peng, H. J. Wang, H. Yu, W. X. Zheng and J. Yang, Angew. Chem., Int. Ed., 2011, 50, 3257 CrossRef PubMed.
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, ACS Nano, 2011, 6, 205 CrossRef CAS PubMed.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J. B. Baek and L. Dai, ACS Nano, 2010, 4, 1321 CrossRef CAS PubMed.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084 CrossRef CAS PubMed.
- S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J. B. Baek and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 4285 CrossRef.
- S. Wang, E. Iyyamperumal, A. Roy, Y. Xue, D. Yu and L. Dai, Angew. Chem., Int. Ed., 2011, 50, 11756 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110 CrossRef CAS PubMed.
- D. Yu, Y. Xue and L. Dai, J. Phys. Chem. Lett., 2012, 3, 2863 CrossRef CAS PubMed.
- X. Hu, Y. Wu, H. Li and Z. Zhang, J. Phys. Chem. C, 2010, 114, 9603 CAS.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170 CAS.
- P. Lazar, J. Granatier, J. Klimes, P. Hobza and M. Otyepka, Phys. Chem. Chem. Phys., 2014, 16, 20818 RSC.
- S. Sarkar, M. Sudolská, M. Dubecký, C. J. Reckmeier, A. L. Rogach, R. Zbořil and M. Otyepka, J. Phys. Chem. C, 2016, 120, 1303 CAS.
- J. Bhattacharjee, J. Phys. Chem. Lett., 2015, 6, 1653 CrossRef CAS PubMed.
- Y. Liu and P. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 3362 CAS.
- H. Jin, H. Huang, Y. He, X. Feng, S. Wang, L. Dai and J. Wang, J. Am. Chem. Soc., 2015, 137, 7588 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2012, 134, 15 CrossRef CAS PubMed.
- J. Y. Cheon, J. H. Kim, J. H. Kim, K. C. Goddeti, J. Y. Park and S. H. Joo, J. Am. Chem. Soc., 2014, 136, 8875 CrossRef CAS PubMed.
- Z. Chen, D. Higgins, H. Tao, R. S. Hsu and Z. Chen, J. Phys. Chem. C, 2009, 113, 21008 CAS.
- D. Yu, E. Nagelli, F. Du and L. Dai, J. Phys. Chem. Lett., 2010, 1, 2165 CrossRef CAS.
- T. Xing, Y. Zheng, L. H. Li, B. C. C. Cowie, D. Gunzelmann, S. Z. Qiao, S. Huang and Y. Chen, ACS Nano, 2014, 8, 6856 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2012, 134, 15 CrossRef CAS PubMed.
- H. Fei, R. Ye, G. Ye, Y. Gong, Z. Peng, X. Fan, E. L. G. Samuel, P. M. Ajayan and J. M. Tour, ACS Nano, 2014, 8, 10837 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- D. D. Koelling and B. N. Harmon, J. Phys. C: Solid State Phys., 1977, 10, 3107 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- T. Ikeda, M. Boero, S. F. Huang, K. Terakura, M. Oshima and J. I. Ozaki, J. Phys. Chem. C, 2008, 112, 14706 CAS.
- T. Ikeda, M. Boero, S. F. Huang, K. Terakura, M. Oshima, J. I. Ozaki and S. Miyata, J. Phys. Chem. C, 2010, 114, 8933 CAS.
- X. Hou, Q. Hu, P. Zhang and J. Mi, Chem. Phys. Lett., 2016, 663, 123 CrossRef CAS.
- M. Li, L. Zhang, Q. Xu, J. Niu and Z. Xia, J. Catal., 2014, 314, 66 CrossRef CAS.
- H. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505 RSC.
- J. Rossmeisl, J. K. Nørskov, C. D. Taylor, M. J. Janik and M. Neurock, J. Phys. Chem. B, 2006, 110, 21833 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886 CrossRef.
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, J. Catal., 2011, 282, 183 CrossRef CAS.
- B. Delley, Mol. Simul., 2006, 32, 117 CrossRef CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed.
- P. Zhang, X. Hou, L. Liu, J. Mi and M. Dong, J. Phys. Chem. C, 2015, 119, 28028 CAS.
- X. Y. Lang, G. F. Han, B. B. Xiao, L. Gu, Z. Z. Yang, Z. Wen, Y. F. Zhu, M. Zhao, J. C. Li and Q. Jiang, Adv. Funct. Mater., 2015, 25, 230 CrossRef CAS.
- B. B. Xiao, Y. F. Zhu, X. Y. Lang, Z. Wen and Q. Jiang, Sci. Rep., 2014, 4, 5205 CrossRef CAS PubMed.
- P. Zhang, X. Hou, J. Mi, Y. He, L. Lin, Q. Jiang and M. Dong, Phys. Chem. Chem. Phys., 2014, 16, 17479 RSC.
- G. L. Chai, Z. Hou, D. J. Shu, T. Ikeda and K. Terakura, J. Am. Chem. Soc., 2014, 136, 13629 CrossRef CAS PubMed.
- Z. Chen, D. Higgins, H. Tao, R. S. Hsu and Z. Chen, J. Phys. Chem. C, 2009, 113, 21008 CAS.
- S. Kundu, T. C. Nagaiah, W. Xia, Y. Wang, S. V. Dommele, J. H. Itter, M. Santa, G. Grundmeier, M. Bron, W. Schuhmann and M. Muhler, J. Phys. Chem. C, 2009, 113, 14302 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10104j |
| This journal is © The Royal Society of Chemistry 2018 |