
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Paclitaxel prodrug based mixed micelles for tumor-targeted chemotherapy†
Dongyang Tang‡
a,
Xin Zhao‡b,
Tie Yangc and
Cheng Wang *d
*d
aDepartment of Experimental Center, Henan Institute of Science and Technology, Xinxiang, Henan 453003, P. R. China
bDepartment of Pharmacy, Xinxiang Central Hospital, Xinxiang, Henan 453000, P. R. China
cNanjing Research Center, Jiangsu Chiatai Tianqing Pharmaceutical Co. Ltd, Nanjing, Jiangsu 210042, P. R. China
dCollege of Pharmaceutical Sciences, Zhejiang University, 866 Yuhangtang Road, Hangzhou, Zhejiang 310058, P. R. China. E-mail: 11519016@zju.edu.cn
First published on 2nd January 2018
Abstract
An effective chemotherapy is usually subject to an insufficient loading of hydrophobic drugs as well as severe side effects. In order to address these dilemmas in one formulation, we herein construct paclitaxel prodrug based mixed micelles (MMs) for tumor-targeted chemotherapy. The paclitaxel prodrug containing a hydrophobic PTX and a hydrophilic PEG chain can self-assemble into uniform MMs with distearoyl phosphoethanolamine–polyethylene glycol–folate (DSPE–PEG–FA). The resultant MMs with preferable stability and hemolysis compatibility could improve the cellular uptake of nanoparticles via FA receptor-mediated endocytosis as compared to the single micelles (SMs). This tumor targetability was also confirmed in vivo by fluorescent imaging. MMs with a stable drug loading as well as tumor targetability displayed elevated in vitro cytotoxicity and in vivo antitumor efficacy compared with Taxol, which could be a potential formulation for cancer therapy.
1. Introduction
Until now, drug delivery in oncology remains of particular interest owing to the relatively narrow therapeutic window of many anti-neoplastic agents. The unfavorable effects, particularly the untargeted side effects as well as poor water solubility, significantly limit their further application in clinical practice. To address these dilemmas, micelles as a preferable drug delivery system (DDS) based on various types of polymeric materials have been proposed.1,2 Micelles have raised a special interest as nano-sized DDSs not only because they provide an increased solubility and stability of hydrophobic drugs,3,4 but also due to their superior advantages versus the free drug both in vitro and in vivo.5 Micelles with a size between 20 and 200 nm are large enough to prevent the premature elimination via glomerular filtration and sufficiently small to pass through certain blood vessels.6 Moreover, they are capable of improving the cellular uptake of encapsulated drugs as well as granting an alternative route of internalization (endosomes). The latter application is of vital importance in overcoming the multi-drug resistance (MDR) that is responsible for the poor performance of many anti-neoplastic agents.7However, most of the currently available micelles have usually succumbed to their poor drug loading capacity as well as uncontrollable drug leakage during the delivery process. One of the most studied approaches to compensate such flaws is to employ the drug molecules as the hydrophobic block of the micelle and as the therapeutic moiety,8 which is also referred as prodrug micelles (PMs). The prodrugs, by definition, are the derivatives of drugs that can be metabolized or activated in the body or cell to release or regenerate the active drugs.9,10 In the past decades, numerous research efforts have concentrated on conjugating antitumor drugs with a wide spectrum of low- and high-molecular-weight carriers including natural product and synthetic polymers.11 By selectively choosing the hydrophilic and hydrophobic parts of the prodrug, they can automatically flock together in an aqueous solution above the critical micelle concentration (CMC), resulting in the formation of PMs.12 The hydrophobic part of the drug can thereby gather to form the core of the micelle, in which the drug molecules can be entrapped by an outer hydrophilic shell, which isolates the encapsulated drug from the external medium that allows for the effective protection from the hostile in vivo environments.13
The incorporation of various functionalities such as a targeting ligand to seek out the overexpressed surface receptors on the diseased cells provides a more efficient and controlled therapy approach.14 However, the concomitant incorporation of multiple functionalities in a single copolymer micellar system is technically challenging to achieve. An interesting approach for optimizing the properties and overcoming some of the disadvantages of the single micelles is the combination of two or more distinct amphiphilic polymers in order to assemble the mixed micelles (MMs).15 In comparison to the single micelles (SMs), the mixed micellar systems exhibit multiple advantages, including the improvements in the colloidal stabilities,16 more accurate size control,17 and the easier ways to incorporate different modifications.18
Paclitaxel (PTX) conjugated with polyethylene glycol (PEG) to form a prodrug conjugate (PTX–PEG) has been well-established by some previous reports,10,16,19,20 in which PTX–PEG can self-assemble into the nanoscaled particles in an aqueous solution. However, the signal PTX–PEG micelles could not fully afford the sophisticated requirements for cancer therapy, such as an increased stability and tumor targeting.21,22 In order to construct a promising system that is suitable for cancer therapy, DSPE–PEG–FA was introduced to form the MMs with PTX–PEG. The as-prepared MMs showed a decreased critical micelle concentration as well as specific tumor targeting. The following advantages were expected to be translated in altered pharmacokinetics: longer mean residence time (MRT) of the drug in the bloodstream, increased bioavailability, reduced the administered dose, and possible diminished non-specific organ toxicity as a result of more precise drug delivery to the target tissues. The ability to engineer the mixed micelle based on PTX–PEG could open a new window to reveal the new potential of PTX–PEG and significantly boost the utility of this already promising polymer for a variety of applications.
2. Materials and methods
2.1. Materials
Polyethylene glycol monomethyl ether 2000 (PEG), paclitaxel (PTX), 4-dimethylaminopyridine (DMAP), N-hydroxysuccinimide (NHS), N,N′-dicyclohexylcarbodiimide (DCC), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), succinic anhydride, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), DiR, and coumarin-6 (C6) were purchased from Aladdin (Shanghai, China). Distearoyl phosphoethanolamine–polyethylene glycol–folate (DSPE–PEG–FA, 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) was obtained from Ponsure Biotechnology (Shanghai, China). All other chemical reagents used in our study of analytical grade were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China) without further purification.
000 Da) was obtained from Ponsure Biotechnology (Shanghai, China). All other chemical reagents used in our study of analytical grade were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China) without further purification.
2.2. Cell culture
Human cervical carcinoma cell line (Hela) was cultured in a folate (FA) free DMEM medium containing 10% fetal bovine serum (FBS, Hyclone, USA), 100 U mL−1 of penicillin, and 100 μg mL−1 of streptomycin at 37 °C using a humidified 5% CO2 incubator (311, Thermo Scientific, USA).2.3. Animal model
BALB/c mice and New Zealand rabbits were purchased from Shanghai Lab Animal Research Center. All animal experiments were conducted in strict compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23 Rev. 1985) and approved by the Institutional Animal Care and Use Committee of Henan Institute of Science and Technology (Xinxiang, China). In order to obtain xenograft H22 tumor xenograft mice, approximately 107 of the H22 cells (murine liver cancer carcinoma cells, a gift from Dr Pengfei Cui in China Pharmaceutical University) were incubated subcutaneously to the flank region of the mice. Tumor volume (V) was determined by measuring length (L) and width (W) and calculated by the formula: V = L × W2/2.2.4. Synthesis and characterization of PTX–PEG
The carboxylated PEG was first synthesized according to the previous report with some modifications.16 Briefly, succinic anhydride (0.5 mmol, 50 mg) and PEG (0.25 mmol, 500 mg) were dissolved in 2 mL of anhydrous dimethylformamide (DMF) in a round bottom flask. Subsequently, DMAP (0.5 mmol, 61 mg) and triethylamine (TEA, 0.5 mmol, 70 mL) were homogeneously added to the solution under stirring.16 The reaction was carried out at 35 °C with N2 protection for 24 h. At the end of the reaction, iced ethyl ether was added and carboxylated PEG was precipitated. The white product was obtained by filtering ethyl ether, followed by vacuum desiccation for 24 h.The PTX–PEG was synthesized by conjugation of PTX and carboxylated PEG. Briefly, carboxylated PEG (0.1 mmol, ∼220 mg), NHS (0.8 mmol, 92.1 mg), and DCC (0.2 mmol, 38.3 mg) were dissolved in 5 mL of anhydrous dichloromethane (DCM). The reaction proceeded under the protection of N2 at room temperature for 24 h to activate the carboxyl of carboxylated PEG. In addition, PTX (0.2 mmol, 170.8 mg) was dissolved in anhydrous DCM in another round bottom flask. The activated PEG fluid was added dropwise to the PTX solution under stirring. Subsequently, DMAP (0.2 mmol, 24.4 mg) was added to the mixture. The reaction proceeded under the protection of N2 at room temperature for 48 h. The mixture was diluted to 40 mL (with DCM) and washed repeatedly with 1% HCl (40 mL) and water (40 mL). The DCM layer, obtained by a separating funnel, was further dried over anhydrous Na2SO4 and concentrated to approximately 1 mL under vacuum at 45 °C. Subsequently, iced ethyl ether was added to precipitate PTX–PEG. The white product was obtained by filtering ethyl ether followed by vacuum desiccation for 24 h.
The 1H NMR spectra of PTX–PEG were recorded using a nuclear magnetic resonance spectrometer (Avance™ 600, Bruker, Germany) that was operated at 300 MHz using CDCl3 as a solvent.
The mean molecular weights (MMW) of the copolymers were determined using gel permeation chromatography (GPC). The separation of the polymer was carried out on a PLgel MIXED-C column (pore size: 5 μm; dimensions: 7.5 mm × 300 mm) with DMSO as an eluent at the flow rate of 0.6 mL min−1. The standard curve was calibrated with narrow dextran monodisperse standards using a differential refractive index detector.
2.5. Preparation of micelles
Initially, 10.0 mg PTX–PEG with or without 2.0 mg DSPE–PEG–FA were dissolved in 2 mL dimethyl sulfoxide (DMSO). The mixture was stirred for 2 h at room temperature to ensure the complete solubilization of the polymers. Subsequently, 1 mL of distilled water was added to the polymer solution under stirring with a rate of 5 s a drop. The resultant solution was then placed into a dialysis bag (MWCO 1000 Da, 2L × 6) and dialyzed against distilled water for 2 days to remove the organic solvents. The final solution in the dialysis bag was ultrasonicated for 30 minutes in an ice bath by a probe-type ultrasonicator (JY92-2D, Ningbo Scientz Biotechnology Co., Ltd, Nanjing, People's Republic of China), followed by filtration through a 0.45 μm pore-sized microporous membrane and lyophilization. The resultant white powder was stored at −20 °C until further use.The critical micelle concentration (CMC) of SMs and MMs was determined by fluorescence spectroscopy (F-2500, Hitachi Co., Japan) similar to the previous report.23
2.6. Particle size and morphology analysis
The particle size and polydispersity index were assessed by dynamic light scattering (DLS) method using Zetasizer Nano ZS90 (Malvern Instruments Ltd., UK) at 25.0 °C ± 0.1 °C at 90°. The morphologies and size distributions were observed by transmission electron microscopy (TEM, Hitachi 7700, Japan) at an accelerating voltage of 80 kV.2.7. Drug loading content
The drug loading contents (DLs) of PTX in the MMs were estimated by ultraviolet (UV) spectrophotometer (TU-1810, Purkinje, China). The maximum absorbance peak of PTX was determined at 227 nm. Briefly, the micelle solution was diluted to a constant volume. A specific amount of acetonitrile was added to the solution to destroy the micelles. The absorbance of the solution at 227 nm was measured.24 In addition, a standard curve of PTX was established to determine the PTX content in the micelles.2.8. Stability and hemolysis assay
For the colloidal stability test, the freshly prepared MMs were diluted with a phosphate buffer (PBS, pH 7.4) and 50% FBS at the volume ratio of 1:10. The change in particle size was recorded at the predetermined time intervals. For the hemolysis assay, the red blood cells (RBCs) were first obtained from New Zealand rabbits and diluted to 2% suspension with a saline solution. SMs and MMs were added to the 2% RBCs suspension with the same volume to achieve the designated concentrations (0.1, 0.25, 0.5, 0.75, and 1 mg mL−1) and incubated at 37 °C for 1 h. In addition, the RBCs suspension was also incubated with saline and distilled water under the same condition as the negative (0% hemolysis) and positive controls (100% hemolysis), respectively. Following this, all the samples were centrifuged at 3000 rpm for 10 min and the absorption values of the same volume of the supernatants, which represented the counts of released hemoglobin, were measured by UV spectrophotometer.
2.9. In vitro cellular uptake
Fluorescent probe C6 was encapsulated into the nanoparticles after the formation of the micelles. Briefly, C6 (0.1 mg) in 0.5 mL of DMSO was added dropwise to the SMs or MMs solution (PBS, pH 7.4) under stirring at 150 rpm at room temperature. Subsequently, the final solution was ultrasonicated for 30 min in an ice bath. The resultant solution was dialyzed against an excess amount of PBS for 12 h using a dialysis bag (MWCO: 1000 Da, 2L × 4). The final solution was filtered through a 0.45 μm pore-sized microporous membrane to remove the large particles (if any), lyophilized, and stored at −20 °C until further use. To evaluate the cellular uptake of the SMs and MMs in Hela cell line, the cells were seeded in 24-well plates at a density of 1 × 105 cells per well and incubated for 12 h at 37 °C to allow their attachment. To investigate whether the nanoparticles were taken up through FA receptor-mediated endocytosis, the cells were incubated with 10 mg L−1 of free FA for 1 h prior to the addition of the nanoparticles. The medium was replaced by a serum-free cell culture medium containing 0.25 mg mL−1 of the C6 loaded SMs or MMs. After 2 and 4 h of incubation, the culture media were removed and the cells were rinsed with PBS thrice. Subsequently, the cells were fixed with 4% formaldehyde (10 min) and observed under an inverted fluorescence microscope (Axio Observer A1, Zeiss, Germany). The quantitative measurement of the C6 fluorescence intensity in the cells at each time point was conducted using flow cytometry (FCM, BD FACS Calibur, USA). Briefly, at the designated time intervals, the culture medium was removed and all cell samples were washed with PBS for three times. The cells were harvested by trypsinization and collected by centrifugation. Following supernatant aspiration, the cells were washed with PBS twice and then resuspended in 0.5 mL of PBS prior to being subjected to the FCM analysis.2.10. In vivo tumor targetability imaging
When the tumor volume reached about 300 mm3, the xenograft H22 tumor xenograft mice were randomly divided into two groups (n = 3) and intravenously administered with the DiR loaded SMs and MMs via the tail vein. At the prearranged time interval after injection (12 h), the NIR fluorescent images were captured by Maestro in vivo Imaging System (CRI Inc., USA). After in vivo imaging, the mice were sacrificed and their main organs (hearts, livers, spleens, lungs, and kidneys) as well as tumors were excised to measure their individual fluorescence intensities.2.11. In vitro cytotoxicity studies
The Hela cells were used to determine the comparative cytotoxicity of a commercial preparation of PTX (Taxol), SMs, and MMs by the MTT assay as reported previously.25 The cells were first seeded into 96-well plates at the density of 3.0 × 103 cells per well. After 12 h of incubation, the primary culture medium was discarded, following with the addition of a fresh serum-free medium containing different formulations at various PTX concentrations. After 48 h of incubation, the medium was replaced with an equal volume of the fresh medium containing 20 μL of 5 mg mL−1 MTT and incubated for 4 h at 37 °C. Then, MTT was removed and the cells were lysed with dimethyl sulfoxide (DMSO) under stirring for 15 min on a microtiter plate shaker. The viability of the cells was estimated according to the optical density (OD) values determined by a microplate reader at an absorption wavelength of 570 nm (Bio-Rad, model 680, USA).2.12. In vivo antitumor activity
The H22 murine liver cells overexpressing FA receptor26 was employed to test the in vivo antitumor activity of the nanoparticles. When the tumor volume of the H22 tumor-bearing BALB/c mice reached approximately 100 mm3, the mice were randomly divided into four groups (n = 3 for each group) as follows: (1) saline, (2) Taxol, (3) SMs, and (4) MMs. All groups were injected intravenously via the tail vein (noted as day 0). The therapy was repeated 7 times every 2 days. Tumor volume and body weight of each mouse were measured before the injection. At 2 days after the final injections, two mice in each group were randomly chosen and sacrificed to prepare the tumor sections. The tumors were extirpated, washed thrice with saline, weighed, and then fixed in 10% formalin. The formalin-fixed tumors were embedded in the paraffin blocks to prepare the hematoxylin and eosin (HE)-stained tumor sections. The sections were then visualized under an inverted fluorescence microscope.3. Results and discussion
3.1. Characteristics of PTX–PEG
Fig. 1 shows the 1H NMR spectra of PTX–PEG. The methyl (a) and methylene (b) peaks of PEG at around 3.37 ppm can be observed. According to the literature,27 among all hydroxyl groups in PTX, the most suitable position for the structural modification is the C-2′–OH. Therefore, the activated carboxylated PEG prefers to react with a hydroxyl group linked with C-2′ of PTX by forming an ester bond, which would lead to a shift of the neighboring double peak of CH-2′ hydrogen from d 4.79 ppm to d 6.0 ppm. The clearly observed peak at 6.0 ppm (c) indicated the successful connection of succinic anhydride with PTX. In addition, the characteristic peaks of PTX with aromatic protons at 7.0–8.0 ppm (d) also confirmed the conjugation of PTX. The presence of all characteristic peaks of PTX and PEG (Fig. 1) indicated the successful synthesis of PTX–PEG. The GPC data (Fig. S1†) also revealed that compared with PEG (MMW: 2000), PTX–PEG has an increased molecular weight of around 3500, which further confirmed that PTX–PEG was successfully synthesized.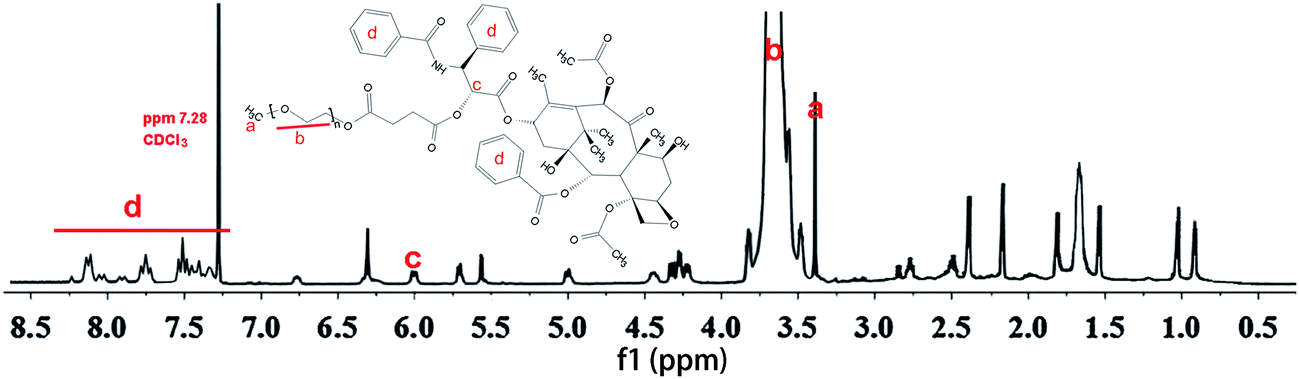 | ||
| Fig. 1 1H NMR spectra of PTX–PEG. | ||
3.2. Morphology and size distributions
The critical micelle concentration (CMC) of SMs and MMs was determined by fluorescence spectroscopy using pyrene as a hydrophobic probe (Fig. S2†). The CMC of SMs and MMs are 0.00700 and 0.00139 mg mL−1, respectively. The low CMC value ensures their stability when being diluted in vivo. The morphology and size of SMs and MMs are shown in Fig. 2. It can be observed from the TEM images that the uniform spherical micelles are formed in the two samples. The size and size distribution of the micelles were measured by DLS. The average size of the SMs is 56.9 nm with the polydispersion index (PDI) of 0.173 (Fig. 2A), while the average size of the MMs is 78.0 nm with the PDI of 0.146 (Fig. 2C), indicating that the introduction of DSPE–PEG–FA resulted in a slightly increased size without changing the micelle-forming ability of PTX–PEG. It is reported that the nanoparticles with a size of 10–200 nm can passively accumulate in the tumor cells via the enhanced penetration and retention effect.28 The grafted PEG chain could also increase the hydrophilicity of the micelles and serve as a shielding polymer to avoid the potential recognition by the reticuloendothelial system that is primarily responsible for the capture of ectogenic nanoparticles. Moreover, FA as a widely adopted targeting moiety was capable of specifically targeting the FA receptor that overexpressed in various types of cancer. The introduction of FA offers the possibility to active targeting cancers with an enhanced antitumor efficacy that has been employed and confirmed by many previous reports.29,30 Herein, the MMs formed by a rational mixture of DSPE–PEG–FA with PTX–PEG were expected to combine both passive and active targeting abilities to elevate the antitumor efficacy as compared to the SMs.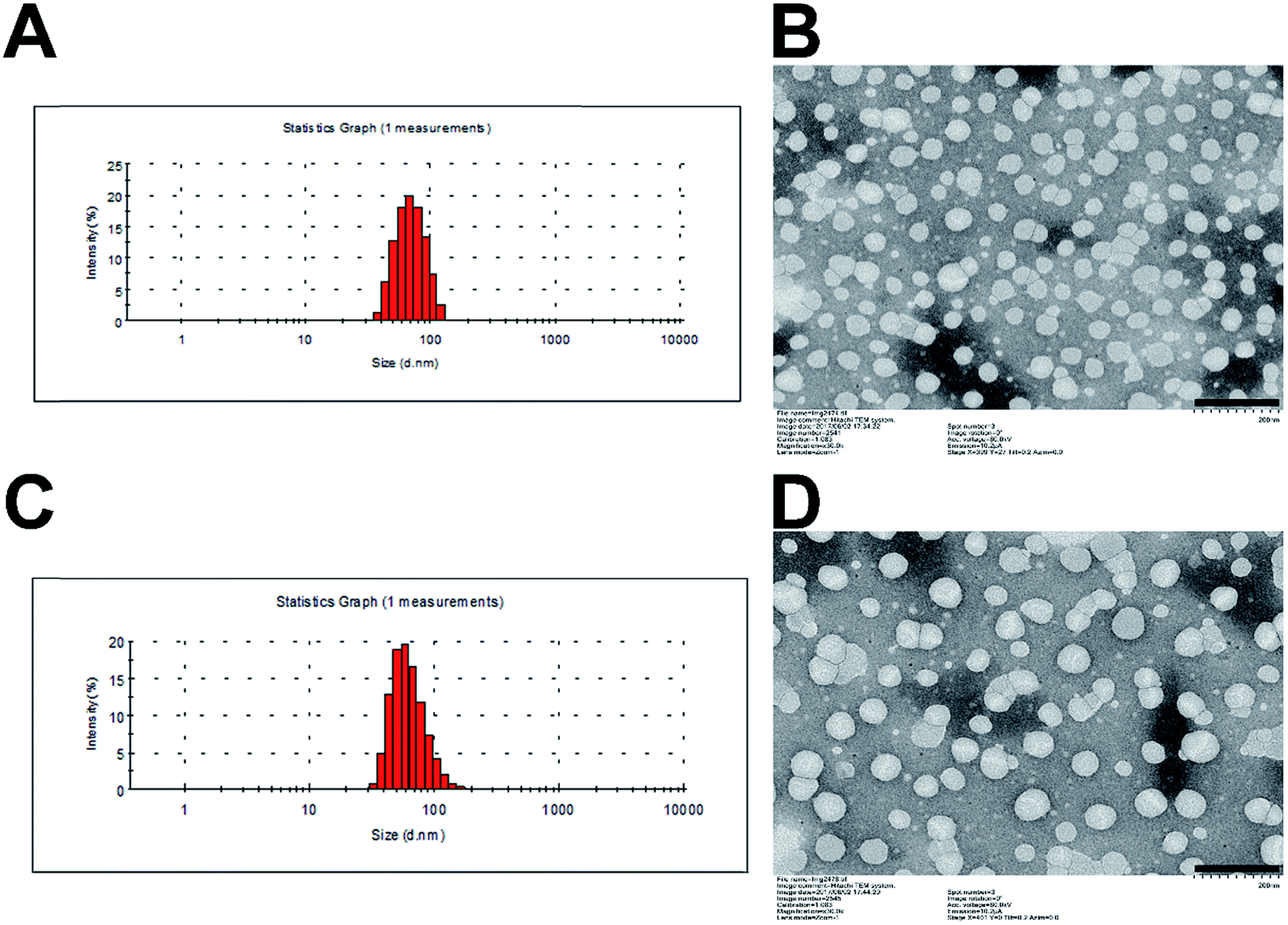 | ||
| Fig. 2 Particle size distribution of SMs (A) and MMs (C). TEM image of SMs (B) and MMs (D). Scale bar: 200 nm. | ||
3.3. Drug loading content
We prepared the micelles by a dialysis method. The maximum absorbance peak of PTX was determined at 227 nm (Fig. S3†). The drug loading content (DLC) of PTX in the SMs and the MMs was calculated by the following formula: DLC (%) = weight in PTX–PEG/weight of PTX–PEG × 100%, which was 20.3% and 16.4%, respectively.3.4. Stability and hemolysis assay
There are some basic requirements for an ideal drug delivery system intended to safely deliver the encapsulated drug molecules to bypass multiple extracellular barriers and locate specifically to the targeted site. It has been generally recognized that the nanoparticles should be able to maintain their morphology in the physiological conditions without a significant size change for at least a certain period of time, since the particle size is a critical parameter to determine the in vivo fate of the drug delivery system. Moreover, the drug delivery system should not induce any adverse reactions, such as hemolysis, during the entire process. As a result, the MMs were studied regarding their stability as well as hemolysis on time and concentration-dependent manner. To estimate the colloidal stability of the described nanoparticles, the change in particle size of the MMs, which serves as an indicator of the potential instability under the physiological conditions, was monitored in PBS (pH 7.4) and 50% FBS. As shown in Fig. 3A and S4,† the size of the MMs remained constant during the entire period with only slight fluctuation, which indicated that the as-prepared delivery system was capable of maintaining its size without being affected by the environment for a long time. This result offers the potential for controllable drug delivery for cancer therapy and is beneficial for its further development as a stable platform with a predictable behavior to fit more extensive applications.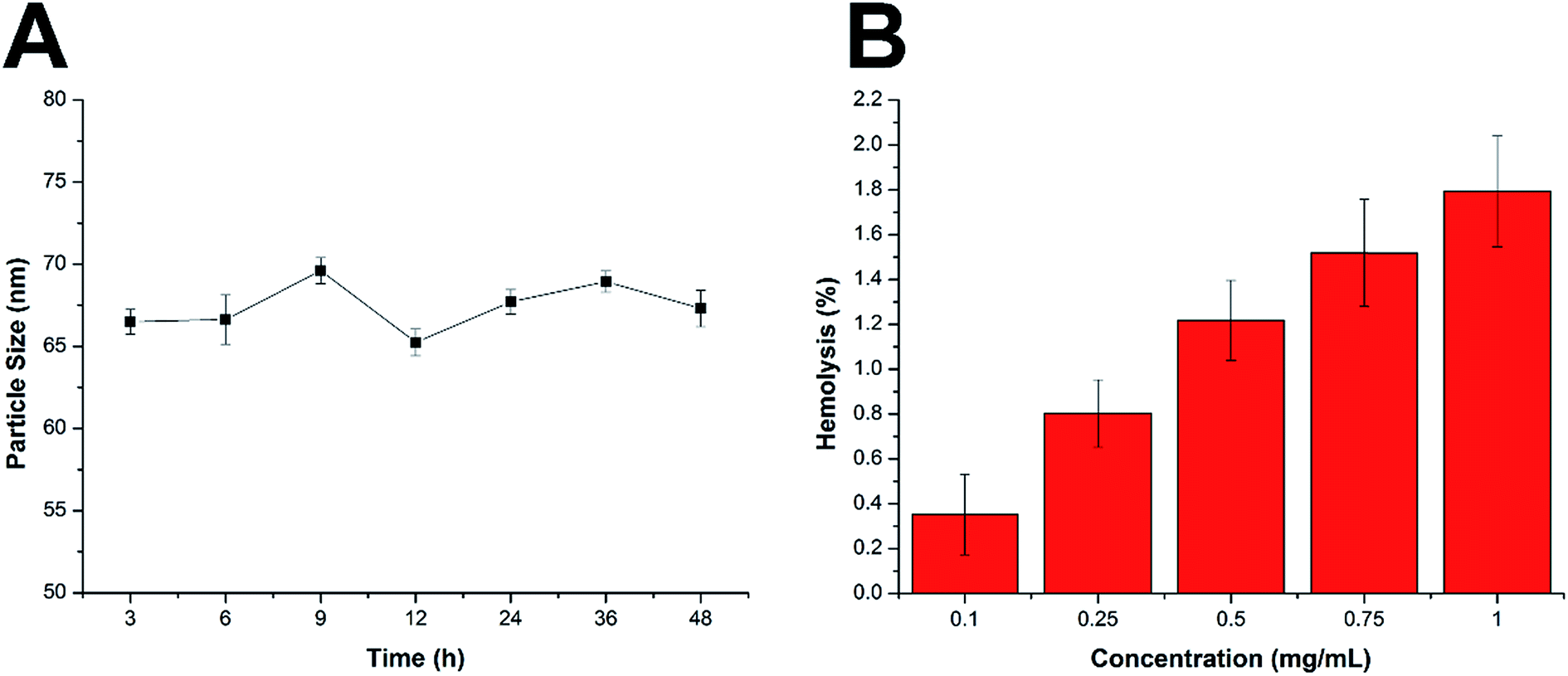 | ||
| Fig. 3 (A) Colloidal stability of MMs in PBS (pH 7.4) at 37 °C for up to 48 h. (B) Hemolysis of MMs at various concentrations. The data are shown as mean ± S.D. (n = 3). | ||
Apart from the steadily delivering and controlled release properties, hemolysis of the drug-loaded MMs was also investigated as a safety guide for intravenous administration and medicinal application.31 As shown in Fig. 3B, the drug-loaded MMs displayed no or neglectable hemolysis (merely 1.79%) on RBCs at the highest concentration of 1 mg mL−1. Considering that when applied in vivo, the MMs would be diluted to a much lower concentration compared with those being tested in our study, we therefore suggest that the as-prepared MMs could be relatively safe nanoparticles without inducing noticeable hemolysis. Collectively, we demonstrated that the MMs exerted a stable profile with a high biocompatibility, which holds a great promise to benefit the therapeutic effect in vivo.
3.5. Cellular uptake
PTX–PEG and DSPE–PEG–FA could automatically form the core–shell structured micelles in an aqueous solution on reaching their CMC. The advantages of this structure offer a preferable protection to the drugs encapsulated in the core, while the shell composed of PEG can shield the nanoparticles from the attack of plasma proteins. Moreover, the FA segment exposed towards the surface of the nanoparticles can target the folate receptors that are excessively expressed in the tumor cells. C6 as a fluorescent molecule capable of indicating the location and the endocytosis kinetics of nanoparticles was encapsulated in the micelles.32 The cells with or without FA pretreatment were incubated with the C6-loaded SMs and MMs and observed by fluorescence microscopy. The C6 fluorescence intensity was further quantified by FCM at different time points. The Hela cells overexpressing FA receptors were used for the cellular uptake evaluation.33As shown in Fig. 4A, the fluorescence microscopy images from both systems revealed the bright green fluorescence signals in the cytoplasm of the Hela cells, which was attributed to C6, indicating that the C6-incorporated nanoparticles successfully entered the cells. It could be also observed that an extended incubation time from 2 h to 4 h resulted in more intense fluorescence in the cell. This suggested that the cellular uptake of both SMs and MMs was positively related to the incubation time. However, under the same conditions, the C6-loaded MMs displayed a remarkably higher intensity compared with that for the SMs, which was also confirmed by the quantitative analysis by FCM as the fluorescence signals in the MMs treated cells was 2.15-fold to that of the SMs treated cells (Fig. 4B). To address the concept of FA receptor-targeting, the cells were pretreated with an excess FA solution and then incubated with the SMs and the MMs for 4 h. The FA untreated cells subjected to the same procedures were employed as a blank control. As displayed in Fig. 4C and D, the FA pretreatment resulted in a significant decrease in the cellular uptake in the MMs treated cells, but no visible changes were observed in the SMs treated cells. The FCM results revealed that the fluorescence signals in the MMs treated Hela cells dropped to 50% of that of the untreated cells. These results strongly suggested that the cellular uptake of the MMs was via the FA receptor-mediated pathways that contributed significantly to enhance the uptake of the nanoparticles as compared with the SMs.
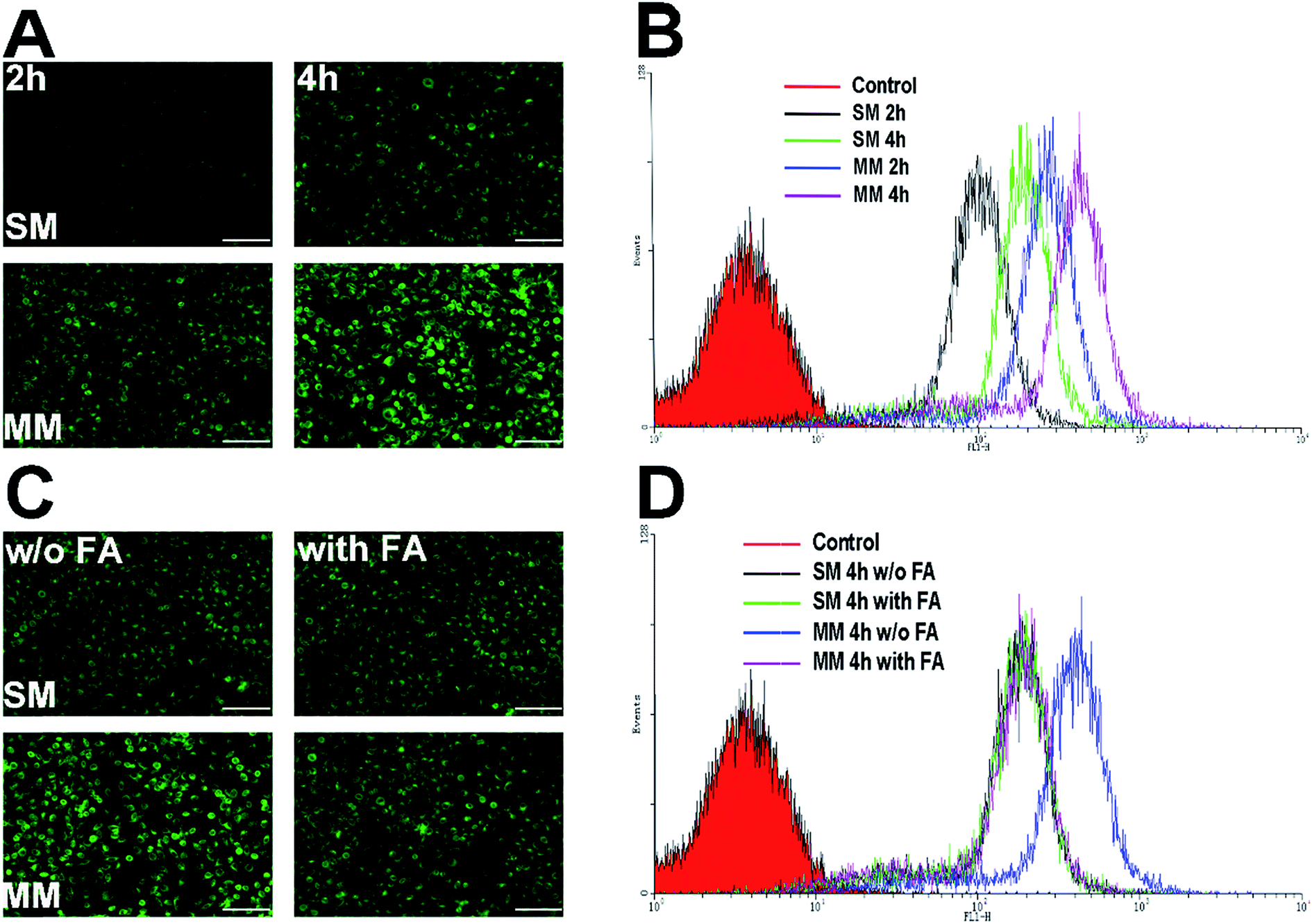 | ||
| Fig. 4 The in vitro cellular uptake analysis of SMs and MMs in the Hela cells. (A) The inverted fluorescence microscope images of the cells incubated with the C6-loaded SMs and MMs for 2 and 4 h. Scale bar: 200 μm. (B) Flow cytometric analysis of mean fluorescence intensity in the cells treated with the C6-loaded SMs and MMs for 2 and 4 h. (C) Inverted fluorescence microscope images of the cells incubated with the C6-loaded SMs and MMs with or without the FA pretreatment 4 h. Scale bar: 200 μm. The data are shown as mean ± S.D. (n = 3). (B) Flow cytometric analysis of mean fluorescence intensity in the cells treated with the C6-loaded SMs and MMs with or without the FA pretreatment 4 h. | ||
3.6. In vivo imaging
The introduction of DSPE–PEG–FA to the MMs was expected to increase the accumulation of the micelles at the tumor site. Herein, the fluorescence intensity of the DiR loaded MMs was monitored in comparison with the unmodified DiR-loaded SMs using a NIR fluorescence imaging system for 12 h to evaluate the targeting ability of different nanoparticles in the H22 tumor-bearing mice.34 Fig. 5A shows the in vivo images at the tumor site after the intravenous injection of the DiR loaded SMs and MMs at different time points. Significant differences in the targeting efficacy between SMs and MMs are observed. In detail, the fluorescence intensity of the MMs at the tumor site is markedly stronger than that of the SMs at 12 h post-injection, indicating the preferential accumulation of the MMs at the tumor site. This conclusion was further verified by the ex vivo imaging of the tumor and major organs (Fig. 5B), demonstrating that the SMs, whose distribution in the tumor tissue was inferior to that of the MMs, have only passive targetability. The powerful tumor targetability of the MMs could be ascribed to a combination of the EPR effect and the FA-mediated endocytosis mechanism.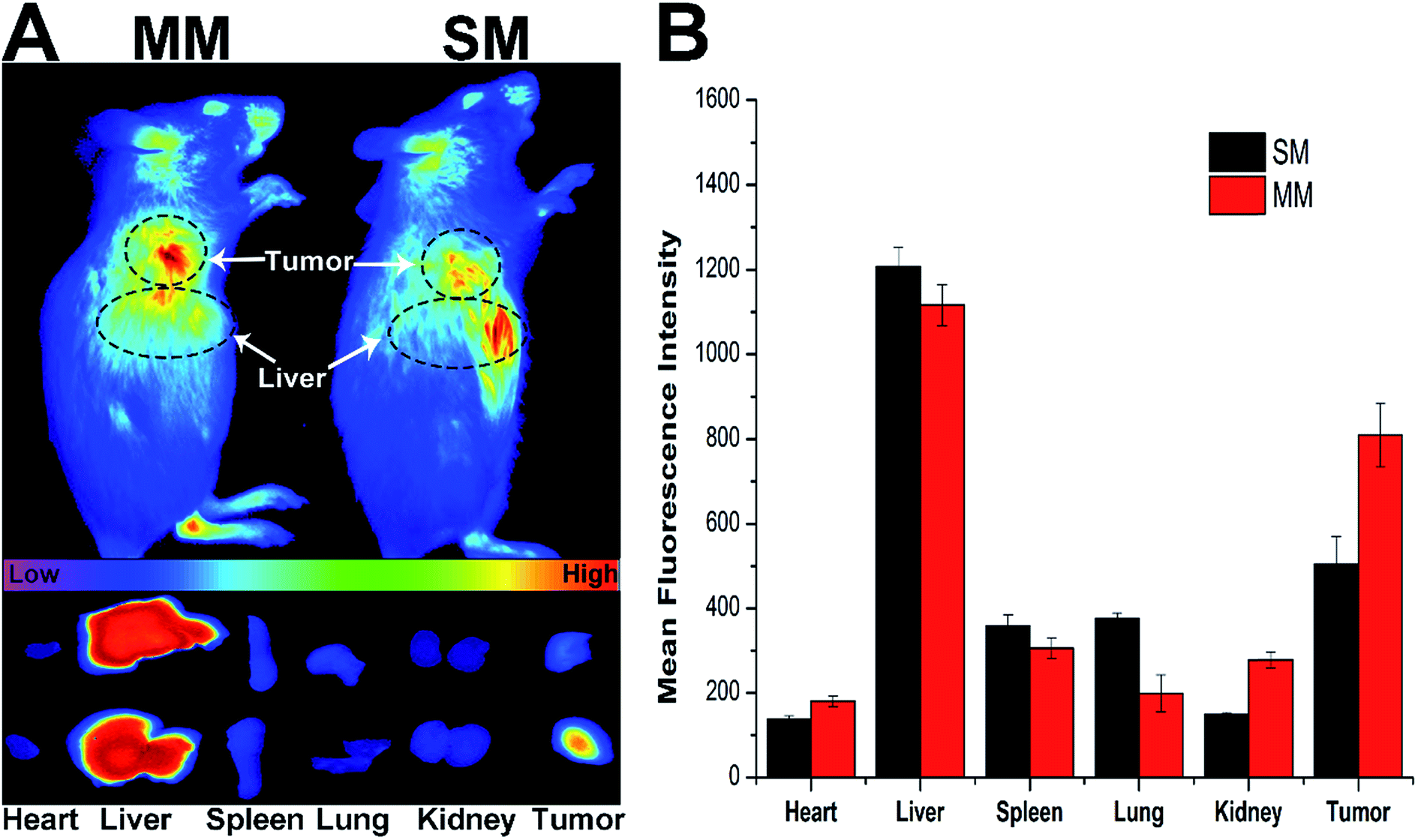 | ||
| Fig. 5 In vivo imaging and biodistribution of the H22 tumor xenograft mice after injection of DiR-loaded SMs and MMs. (A) NIR fluorescence images of nanoparticle localization in the mice (upper) as well as their main organs and tumor (lower) after the intravenous injection of the DiR-loaded SMs and MMs for 12 h. (B) Semiquantitative analysis of biodistribution of the DiR-loaded SMs and MMs in the mice determined by MFI in the tumor or tissues. The data are shown as mean ± S.D. (n = 3). | ||
3.7. In vitro cytotoxicity
The comparative in vitro cytotoxicity of Taxol, SMs, and MMs were investigated in the Hela cells for 24 h and 48 h. As shown in Fig. 6A and B, all three formulations exerted cytotoxicity on the Hela cells to some extent. The cytotoxicity effect was positively related to the PTX concentration and the exposure time. It was observed that the MMs exerted the best cytotoxicity effect with comparable cell viability to the cells after 24 h of incubation, which is superior to that of the SMs at the same condition. Moreover, after incubation for 48 h, the MMs showed more potent cytotoxicity than Taxol at all tested concentrations. These results suggested that the MMs could have an enhanced cytotoxicity to the Hela cells, which would be beneficial for the in vivo antitumor applications.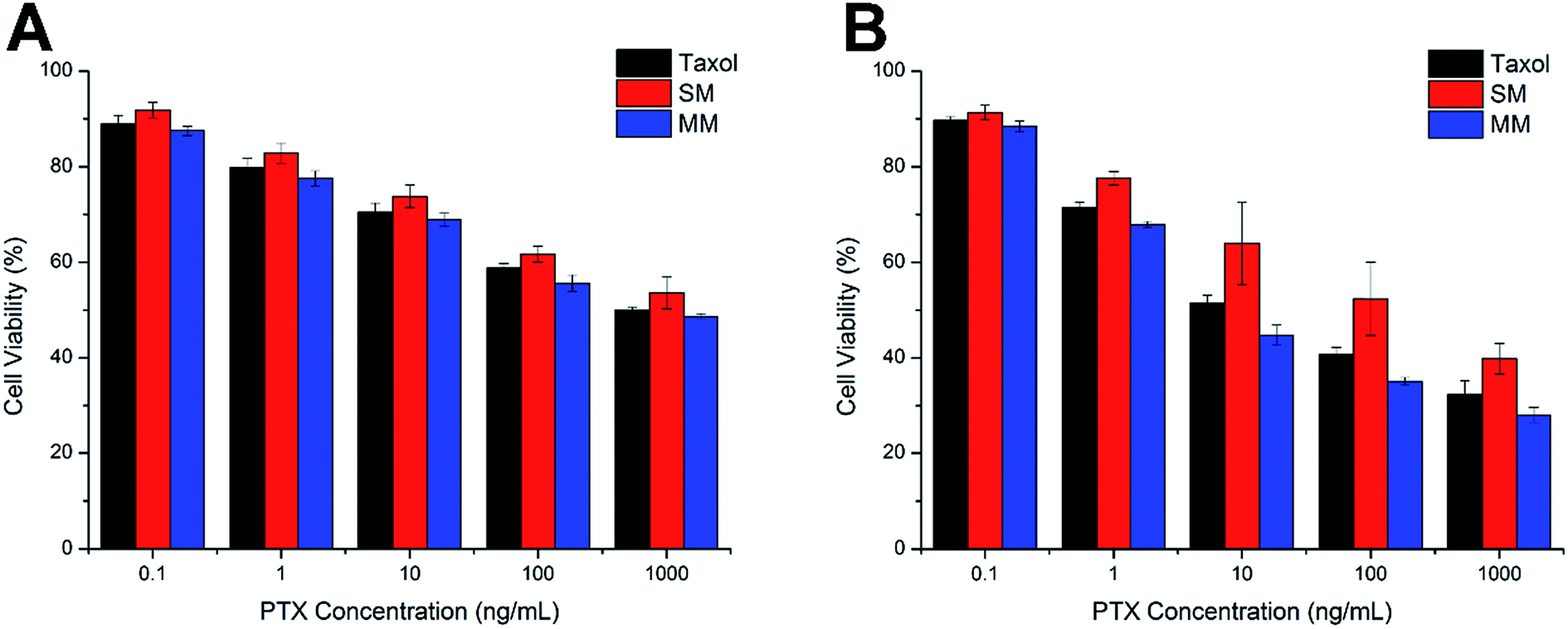 | ||
| Fig. 6 Cell viabilities of the Hela cells incubated with Taxol, SMs, and MMs at various PTX concentrations for 24 h (A) and 48 h (B). The data are expressed as mean ± S.D. (n = 3). | ||
3.8. In vivo anti-tumor
To confirm the antitumor potential of the MMs in vivo, the antitumor efficacy was evaluated in a subcutaneous H22 tumor xenograft model. As shown in Fig. 7A, the H22 tumor-bearing mice in the saline-treated group showed a rapid increase in the tumor growth over the entire period of the experiment. Moreover, the groups treated with Taxol and SMs exhibited a higher growth delay in comparison with the saline control group, which confirmed the antitumor effect of PTX. Interestingly, more elevated suppression of the tumor growth during the treatment with the MMs than other PTX-containing groups was observed. Fig. 5B shows the variation in the body weight of the mice with time. The body weight of the mice in the saline and Taxol groups began to steadily decrease after a period of time after administration, indicating that the living quality of the mice was compromised by the tumor burden and/or the side effects. In contrast, no noticeable body weight loss was observed in both SMs and MMs groups. In particular, in the MMs treated groups, the body weight of the mice was increased as compared to that at the initial assessment, implying that the tumor-homing property of the MMs not only increases their anti-cancer efficacy, but also reduces their safety risks. The representative HE-stained tumor sections from different experimental groups of the mice are displayed in Fig. 5C. The group treated with saline displayed the typical pathological characteristics of the tumor, including the closely packed tumor cells and the heavily stained cytoplasm. The images from other PTX-containing groups showed a cancer cell remission to some extent, while the group administered with the MMs displayed a massive remission with the most powerful antitumor ability. These observations presented substantial evidence to the stronger antitumor effect of the MMs. Overall, the MMs constructed in this study had a promising potential as a preferable tumor-targeting DDS for an efficient cancer therapy.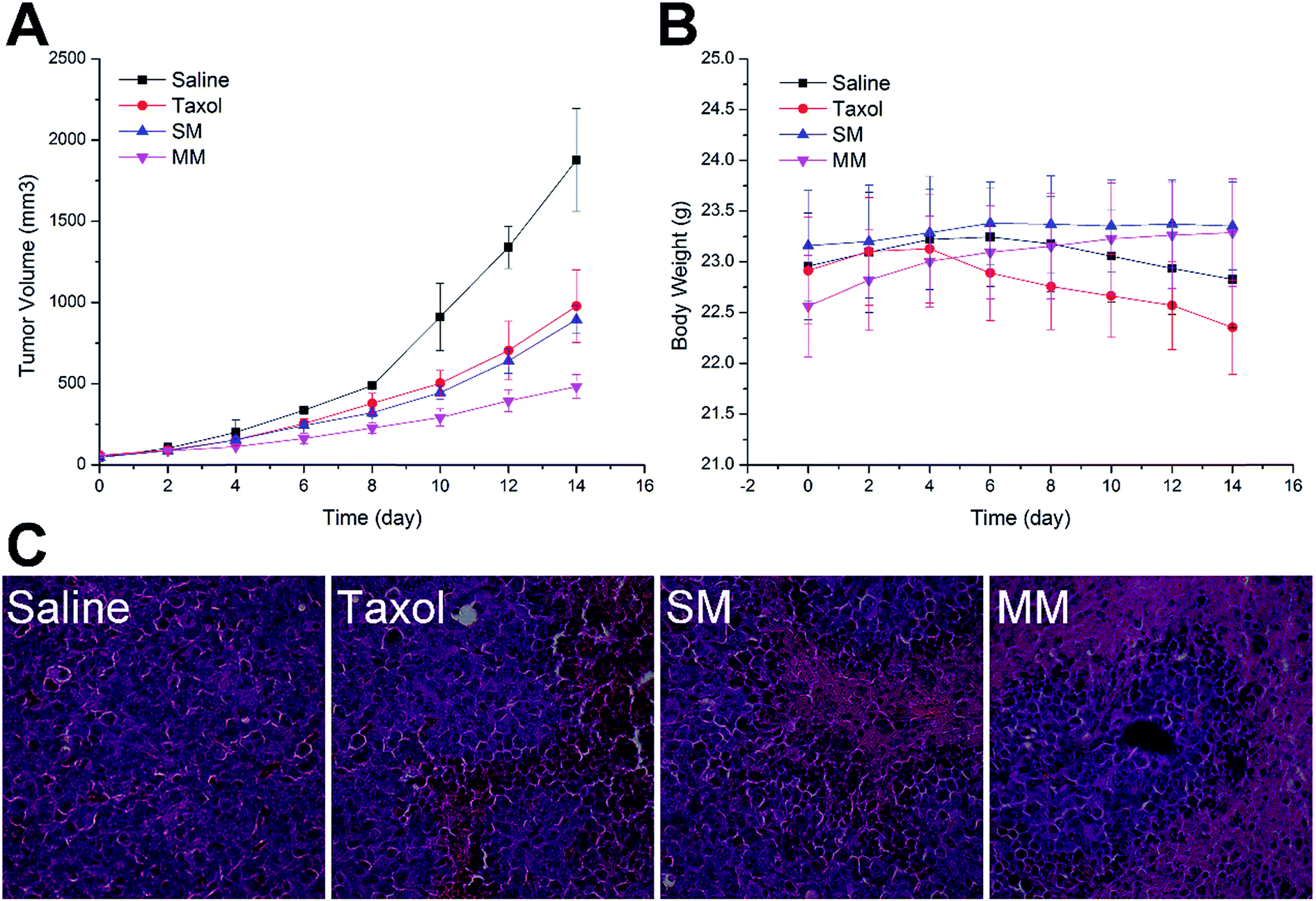 | ||
| Fig. 7 The tumor volume (A), body weight (B), and HE staining of tumor tissue (C) analysis of the H22 tumor-bearing BALB/c mice after the intravenous injection administration of different formulations (saline, Taxol, SMs, and MMs). The measurement of tumor volumes and the injection of formulations were repeated every 2 days for two weeks. Dose: 10 mg kg−1 PTX per mouse. The data are expressed as mean ± S.D. (n = 3). | ||
4. Conclusion
In summary, the paclitaxel prodrug mixed micelles (MMs) were successfully developed for tumor-targeted chemotherapy. The experimental results indicated that drug loading of PTX into the paclitaxel prodrug MMs was as high as 16.4%, which is superior to many currently available carriers. Furthermore, the paclitaxel prodrug MMs showed high biocompatibility with enduring colloidal stability and negligible hemolysis. Moreover, the paclitaxel prodrug MMs increased the accumulation of PTX in the Hela cancer cells via FA receptor-mediated endocytosis with an enhanced tumor-homing capability in the H22 tumor-bearing mice. In vitro and in vivo anti-cancer efficacy assays revealed that the MMs possessed a preferable anti-cancer ability, which exhibited the minimized toxic side effects of PTX and a strong tumor-suppression potential in the clinical application.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Shijun Chen (Henan Institute of Science and Technology) for the technical assistance on using the transmission electron microscope.References
- S. Biswas, P. Kumari, P. M. Lakhani and B. Ghosh, Eur. J. Pharm. Sci., 2016, 83, 184 CrossRef CAS PubMed.
- G. Liu, Q. Luo, H. Gao, Y. Chen, X. Wei, H. Dai, Z. Zhang and J. Ji, Biomater. Sci., 2015, 3, 490 RSC.
- D. Chiappetta and A. Sosnik, Eur. J. Pharm. Biopharm., 2007, 66, 303–317 CrossRef CAS PubMed.
- E. Kahraman, E. Karagö, S. Dinçer and Y. Ozsoy, J. Biomed. Nanotechnol., 2015, 11, 890 CrossRef CAS PubMed.
- A. B. E. Attia, Y. O. Zhan, J. L. Hedrick, P. P. Lee, P. L. R. Ee, P. T. Hammond and Y. Y. Yang, Curr. Opin. Colloid Interface Sci., 2011, 16, 182–194 CrossRef.
- M. Cagel, F. C. Tesan, E. Bernabeu, M. J. Salgueiro, M. B. Zubillaga, M. A. Moretton and D. A. Chiappetta, Eur. J. Pharm. Biopharm., 2017, 113, 211–228 CrossRef CAS PubMed.
- C. M. Hu and L. Zhang, Curr. Drug Metab., 2009, 10, 836 CrossRef CAS PubMed.
- Y. Shen, E. Jin, B. Zhang, C. J. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan and E. V. Kirk, J. Am. Chem. Soc., 2010, 132, 4259–4265 CrossRef CAS PubMed.
- H. Peng, X. Huang, A. Oppermann, A. Melle, L. Weger, M. Karperien, D. Wöll and A. Pich, J. Mater. Chem. B, 2016, 4, 7572–7583 RSC.
- S. A. Senevirathne, K. E. Washington, M. C. Biewer and M. C. Stefan, J. Mater. Chem. B, 2016, 4, 360–370 RSC.
- F. Kratz, I. A. Müller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20–53 CrossRef CAS PubMed.
- Y. Mi, J. Zhao and S. S. Feng, Int. J. Pharm., 2012, 438, 98 CrossRef CAS PubMed.
- Y. Wang, H. Wang, Y. Chen, X. Liu, Q. Jin and J. Ji, Chem. Commun., 2013, 49, 7123 RSC.
- N. T. Zaman, Y. Y. Yang and J. Y. Ying, Nano Today, 2010, 5, 9–14 CrossRef CAS.
- C. L. Lo, S. J. Lin, H. C. Tsai, W. H. Chan, C. H. Tsai, C. H. Cheng and G. H. Hsiue, Biomaterials, 2009, 30, 3961–3970 CrossRef CAS PubMed.
- D. Zhao, H. Zhang, S. Yang, W. He and Y. Luan, Int. J. Pharm., 2016, 515, 281–292 CrossRef CAS PubMed.
- L. Mu, T. A. Elbayoumi and V. P. Torchilin, Int. J. Pharm., 2006, 306, 142–149 CrossRef PubMed.
- T. Li, J. Lin, T. Chen and S. Zhang, Polymer, 2006, 47, 4485–4489 CrossRef CAS.
- J. Lu, X. Chuan, H. Zhang, W. Dai, X. Wang, X. Wang and Q. Zhang, Int. J. Pharm., 2014, 471, 525–535 CrossRef CAS PubMed.
- H. Zhang, H. Hu, H. Zhang, W. Dai, X. Wang, X. Wang and Q. Zhang, Nanoscale, 2015, 7, 10790–10800 RSC.
- L. Dai, J. Liu, Z. Luo, M. Li and K. Cai, J. Mater. Chem. B, 2016, 4, 6758–6772 RSC.
- J. Li, S. Xue and Z.-W. Mao, J. Mater. Chem. B, 2016, 4, 6620–6639 RSC.
- S. Abbad, C. Wang, A. Y. Waddad, H. Lv and J. Zhou, Int. J. Nanomed., 2015, 10, 305 Search PubMed.
- F. Chen, J. Wu, C. Zheng, J. Zhu, Y. Zhang, X. You, F. Cai, V. Shah, J. Liu and L. Ge, J. Mater. Chem. B, 2016, 4, 3959–3968 RSC.
- D. Han, W. Fang, R. Zhang, J. Wei, N. D. Kodithuwakku, L. Sha, W. Ma, L. Liu, F. Li and Y. Li, Brain, Behav., Immun., 2016, 51, 56–69 CrossRef CAS PubMed.
- Y. Zhang, H. Zhang, W. Wu, F. Zhang, S. Liu, R. Wang, Y. Sun, T. Tong and X. Jing, Int. J. Nanomed., 2014, 9, 2019–2030 Search PubMed.
- Z. Xie, H. Guan, X. Chen, C. Lu, L. Chen, X. Hu, Q. Shi and X. Jing, J. Controlled Release, 2007, 117, 210–216 CrossRef CAS PubMed.
- A. Hervault, A. E. Dunn, M. Lim, C. Boyer, D. Mott, S. Maenosono and N. T. Thanh, Nanoscale, 2016, 8, 12152 RSC.
- J. Liu, W. E. Hennink, M. J. van Steenbergen, R. Zhuo and X. Jiang, J. Mater. Chem. B, 2016, 4, 7022–7030 RSC.
- D. Zhou, C. Li, M. He, M. Ma, P. Li, Y. Gong, H. Ran, Z. Wang, Z. Wang, Y. Zheng and Y. Sun, J. Mater. Chem. B, 2016, 4, 4164–4181 RSC.
- P. Huang, B. Zeng, Z. Mai, J. Deng, Y. Fang, W. Huang, H. Zhang, J. Yuan, Y. Wei and W. Zhou, J. Mater. Chem. B, 2016, 4, 46–56 RSC.
- Y. Guo, B. Niu, Q. Song, Y. Zhao, Y. Bao, S. Tan, L. Si and Z. Zhang, J. Mater. Chem. B, 2016, 4, 2338–2350 RSC.
- B. Yu, H. Li, J. Zhang, W. J. Zheng and T. Chen, J. Mater. Chem. B, 2015, 3, 2497–2504 RSC.
- Q. Han, X. Jia, Y. Qian, Z. Wang, S. Yang, Y. Jia, W. Wang and Z. Hu, J. Mater. Chem. B, 2016, 4, 7087–7091 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07796c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |