
Functionalization of wet-spun graphene films using aminophenol molecules for high performance supercapacitors†
Muhammad
Salman
,
Xingyuan
Chu
,
Tieqi
Huang
,
Shengying
Cai
,
Qiuyan
Yang
,
Xiaozhong
Dong
,
Karthikeyan
Gopalsamy
and
Chao
Gao
 *
*
MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Key Laboratory of Adsorption and Separation Materials & Technologies of Zhejiang Province, Zhejiang University, 38 Zheda Road, Hangzhou 310027, P. R. China. E-mail: chaogao@zju.edu.cn
First published on 12th October 2018
Abstract
Aminophenol isomer functionalized graphene film electrodes are fabricated with wet-spinning technology for supercapacitors. According to the different electrochemical mechanisms of aminophenol isomers on graphene, we optimize the conditions of o-aminophenol combined graphene, achieving a high specific capacitance of 636 F g−1 and excellent cycling stability of 97% after 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.
000 cycles.
Introduction
Supercapacitors (SCs) are an important class of electrochemical energy storage devices, owing to its significant benefits, such as fast charging–discharging rates, high power density, long cycle lifetime and safe operation.1–3Graphene has been used as a favorable supercapacitor electrode material for the last few decades due to its fascinating properties such as low weight, chemical inertness, high specific surface area, good electrical conductivity and electrochemical properties.4–7 Recently, graphene-based SCs have been widely used in electrochemical applications due to their high specific area and good conductivity.8–10 However, like other carbon materials which store energy based on the electric double layer adsorption mechanism, graphene also suffers from less satisfactory capacitance and energy density.11 To overcome this problem many researches have been conducted including into electrochemical materials like metal oxides and conducting polymers to introduce extra pesudo-capacitance contribution. Nevertheless, their practical applications are still limited because of the decreased conductivity induced by these low conductive materials and poor cycling stability resulting from the structural breakdown during multiple redox processes of polymers.12,13
The insertion of a heteroatoms (B, N, O, S and P) into the graphene sheet is also an efficient approach to change the electronic properties of the graphene structure and thus to increase the overall energy density of SCs.14–18 In particular, the nitrogen atom is believed to be an ideal dopant for graphene because it can be easily doped onto graphene sheets owing to its comparable atomic size to carbon as well as strong valence bonds with respect to carbon atoms.19,20 The introduction of nitrogen not only increases the pseudo-capacitance by reversible redox changes but also improves the wettability of electrolyte.21 For this purpose, several methods have been reported for N-doping into graphene sheets, for example, chemical vapor deposition (CVD), the nitrogen plasma process, arc-discharge, thermal annealing of GO with ammonia, and segregation growth.22–24 Recently, small molecule precursors containing nitrogen have been introduced to achieve high-performance nitrogen-doped graphene electrodes, including ammonia,25,26 urea,27,28 pyrrole,29 amino acids,30,31 aromatic amines,32–37 aliphatic amines38,39 and ammonium salts.40,41 Because of their high cost, rigorous conditions and complexity, these approaches are not worthy of scale-up processes. Therefore, achieving convenient preparation of scalable nitrogen-doped graphene electrodes is still needed.
Herein, we present a simple and mild protocol for the chemical functionalization of a wet-spun graphene oxide film using with three common isomers of aminophenol, namely o-aminophenol (o-AP), p-aminophenol (p-AP) and m-aminophenol (m-AP), using a one-pot hydrothermal method. The capacitance achieved by the OAP/rGO electrodes can reach up to 636 F g−1 with excellent cycling stability of 97% after 16000 cycles. The mechanisms of the doping of these three molecules into graphene have been analyzed in detail. Accordingly, OAP molecules are covalently coupled with GO via a condensation reaction to form a benzoxazole ring, while PAP molecules reacted with GO via a nucleophilic addition reaction and MAP displayed lower reactivity toward the nucleophilic reaction due to electronic effects. This strategy affords a new methodology to prepare high-performance AP functionalized graphene electrodes for supercapacitor applications. Consequently, the synthesized materials were directly used as an electrode for supercapacitors without adding any binder or active materials. The flexibility of the OAP/rGO film was good and enough for it to be free standing (Scheme 1).
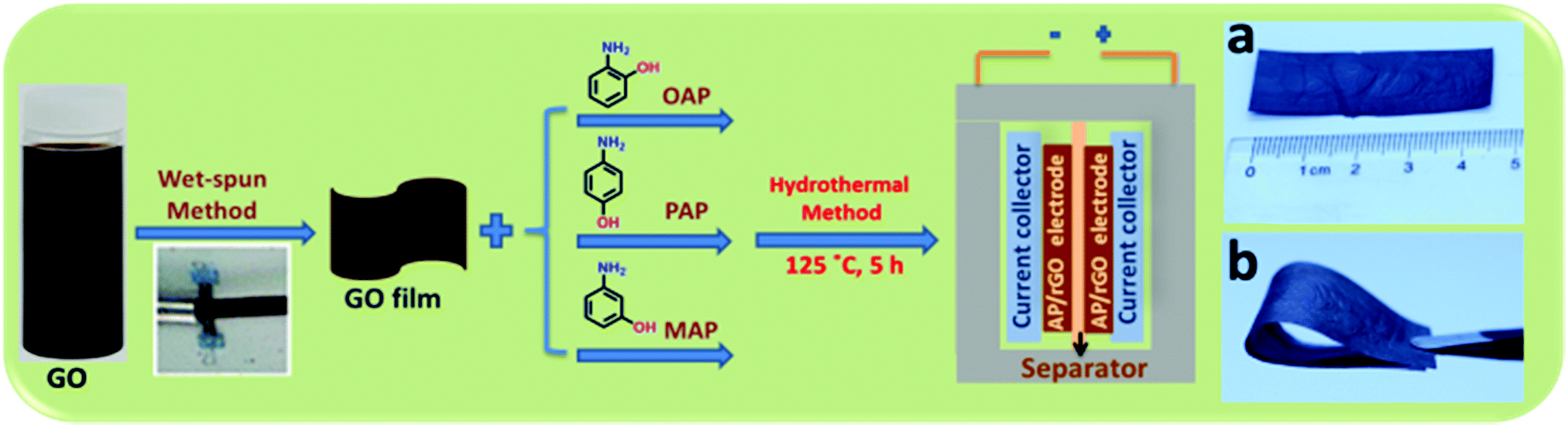 | ||
| Scheme 1 Illustration of the preparation processes of AP/rGO composites. | ||
In addition, the OAP/rGO film can be bent from a straight rectangular shape to a folded shape without cracking, showing its good flexibility and high toughness (Scheme 1b).
Results and discussion
The N-doped graphene films were achieved using a wet-spun method followed by a hydrothermal treatment of OAP/PAP/MAP aqueous solution with the wet-spun GO film at 125 °C for 5 h, as demonstrated in Scheme 1. More experimental details can be found in the ESI.† As we know, the electron mobility is an important factor for supercapacitors. First of all, we determined the mobility of the AP/rGO composites as a function of temperature (Fig. S1, ESI†). The results revealed that all composites, particularly OAP/rGO, exhibited a maximum electron mobility of 3.475 × 1019 cm2 V−1 s−1 at 125 °C. When the temperature increased from 125 °C to 180 °C, the formation of the benzoxazole ring would not occur, considering that most oxygen-containing groups were reduced at higher temperature,42 and therefore the possibility of benzoxazole ring formation decreased. Furthermore, we investigated the mass percentage of the AP/rGO composites by thermal gravimetric analysis (TGA) in an N2 atmosphere (Fig. S2, ESI†). TGA analysis was performed at a temperature ranging from 25 °C to 800 °C. The weight percent of AP was 12% in OAP/rGO, higher than those of the PAP/rGO and MAP/rGO composites, which are 6% and 4.8% respectively. In addition, in the OAP/rGO composites the sharp weight loss at 460–520 °C was attributed to the benzoxazole ring43 that covalently attached to the graphene sheets. However, the weight loss of the PAP/rGO composite lies in the range of 350–420 °C due to decomposition of PAP molecules.44 In contrast, the weight loss of the MAP/rGO sample lies in the range of 200 °C to 300 °C because of the MAP molecules.45 This indicated that the OAP/rGO composite was considerably more stable at the synthesis temperature.The cross-section structures of rGO, OAP/rGO, PAP/rGO and MAP/rGO were examined by SEM (Fig. 1). The SEM image of OAP/rGO and PAP/rGO (Fig. 1b) showed wrinkled and crumbled graphene sheets similar to rGO, which contributed to the fast adsorption and diffusion of the ions during hydrothermal treatment.30,46 However, the SEM image of MAP/rGO displayed severely stacked graphene sheets and a tightly packed structure. This stacked configuration suggested serious agglomeration during the hydrothermal reaction between m-aminophenol and GO. The difference in the microscopic structures of the AP/rGO composites was due to different electrostatic interactions between the aminophenol molecules and GO.
 | ||
| Fig. 1 SEM images of (a) rGO, (b) OAP/rGO, (c) PAP/rGO and (d) MAP/rGO films. | ||
In addition, the mechanical properties are quite important in energy storage devices. Therefore, the mechanical properties of the AP/rGO composites are shown in Fig. S3 (ESI†). In the OAP/rGO composite, the observed maximum tensile strength was due to strong covalent bonding between OAP and the GO sheets. This directly enhances the interlayer shear strength up to 20.17 MPa. However, the PAP/RGO composite displayed lower tensile strength (19.49 MPa) as compared to the OAP/rGO composite. In contrast, the tensile strength of MAP/rGO was 7.66 MPa, lower than that of OAP/rGO and PAP/rGO. The weakened mechanical property was due to weak interaction which leads to the destruction of the stacking of the interlayered structure of MAP molecules in the GO sheets.47 Similarly, the strain of OAP/rGO (1.03%) and PAP/rGO (0.87%) at breakage is higher than that of MAP/rGO, 0.63%, as a result of the unfolding process of the wrinkled and crumbled graphene sheets during stretching.48
The chemical interaction of GO films with aminophenol isomers and the presence of surface functional groups were identified by FTIR analysis (Fig. 2a). The GO precursor showed a series of peaks at 3392, 1731, 1253, and 1058 cm−1 in the FTIR spectrum, which can be allocated to hydroxyl (O–H), carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), epoxy (C–O–C), and alkoxyl (C–O) groups, respectively.49 In contrast, a broad peak appeared at 3544–3343 cm−1 in the OAP/rGO composite, while in the PAP/rGO composite a peak appeared at 3540–3437 cm−1 with a sharp medium intensity. Two clear overlapping peaks at 3536–3408 cm−1 were shown in the case of the MAP/rGO composite, which indicated primary amines and –OH groups, while the intensity of the peak at 1731 cm−1 of CO groups on the GO sheets significantly reduced in the presence of AP molecules. However, the emergence of two new peaks for C–N and N–H was considered as evidence for the functionalization of AP molecules on the GO surface via covalent bonds. Furthermore, the peaks which appeared at 1169–1163 cm−1 were attributed to C–N vibrational modes in N-graphene sheets contributed by AP molecules. Besides, the vibrations around 856–730 cm−1 correspond to –NH group wagging modes in the C–NH chemical bonds.50,51
O), epoxy (C–O–C), and alkoxyl (C–O) groups, respectively.49 In contrast, a broad peak appeared at 3544–3343 cm−1 in the OAP/rGO composite, while in the PAP/rGO composite a peak appeared at 3540–3437 cm−1 with a sharp medium intensity. Two clear overlapping peaks at 3536–3408 cm−1 were shown in the case of the MAP/rGO composite, which indicated primary amines and –OH groups, while the intensity of the peak at 1731 cm−1 of CO groups on the GO sheets significantly reduced in the presence of AP molecules. However, the emergence of two new peaks for C–N and N–H was considered as evidence for the functionalization of AP molecules on the GO surface via covalent bonds. Furthermore, the peaks which appeared at 1169–1163 cm−1 were attributed to C–N vibrational modes in N-graphene sheets contributed by AP molecules. Besides, the vibrations around 856–730 cm−1 correspond to –NH group wagging modes in the C–NH chemical bonds.50,51
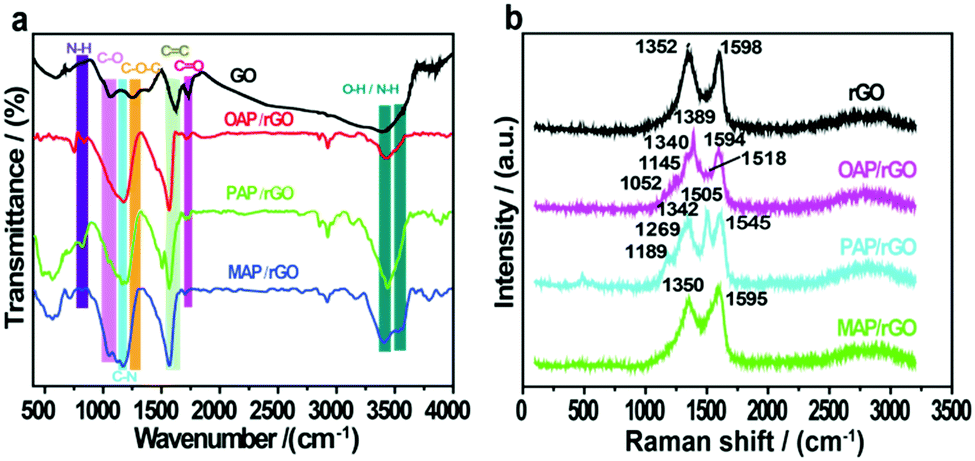 | ||
| Fig. 2 (a) FTIR and (b) Raman characterization of rGO, OAP/rGO, PAP/rGO and MAP/rGO. | ||
Furthermore, to explore the structural properties of the as-prepared materials, we analyzed the Raman spectra of rGO, OAP/rGO, PAP/rGO and MAP/rGO as shown in Fig. 2b. In the rGO sample, the broad peak near 1352 cm−1 (D band) was related with structural defects and partially disordered structures of the sp2 domains.52 Correspondingly, the peak which appeared at 1598 cm−1 (G band) was associated with the E2g vibration mode of sp2-C areas which can be used to explain the degree of graphitization.52–54 The Raman spectra of the OAP/rGO composite displayed values of 1389 cm−1 and 1594 cm−1 for the D and G bands respectively. The peak which appeared at 1518 cm−1 showed CN stretching of the benzoxazole ring,55 while the peak observed around 1340 cm−1 is assigned to the phenyl carbon–nitrogen bond (C–N). The C–O–C stretching vibration of the benzoxazole ring appeared at 1145 cm−1.56 The C–H bonding vibration occurred at 1052 cm−1 which was consistent with the previous reports.57 In the case of the PAP/rGO sample, the ratio of ID/IG is 0.84, which is a bit different to that of rGO (0.86), because of the PAP molecules attached on the basal plane of graphene. The peak which occurred at 1189 cm−1 of PAP/rGO was related to the C–H bending of quinoid type rings.58 The peaks at 1269 cm−1 and 1505 cm−1 can be attributed to the C–N stretching vibration of the benzenoid ring and the CN stretching vibration of the quinonoid ring in PAP, respectively.59 The results disclosed that the surface of rGO was successfully modified with PAP molecules. In Raman spectra of the MAP/rGO sample, two major peaks appeared at 1350 cm−1 and 1595 cm−1 corresponding to the D and G bands respectively. The results revealed that the intensity of the G band increased as compared to the G band of rGO. This indicated that the graphitic structure was highly disordered. The incorporation of the N atom into the conjugated system of graphene resulted in the observed downshift of the G band value.60
The elemental composition of AP/rGO composites was also characterized using XPS measurements. The typical survey spectra of rGO show peaks at 284.3 eV and 531.3 eV which corresponded to the C 1s and O 1s peaks respectively. However, for all AP/rGO samples, an additional peak was observed around 400 eV which was assigned to the N 1s peak (Fig. 3a). This indicated that AP molecules were successfully attached to graphene oxide with the overall N contents more than 5% (Table S1, ESI†) for all three types of AP/rGO samples. To investigate the nitrogen configurations in all AP/rGO samples, a high-resolution N 1s characterization was carried out (Fig. 3b–d). All the N 1s spectra were mainly composed of four peaks. The peak at 398.4 eV was attributed to pyridinic-N species, which donated a π-electron to the hexagonal-unsaturated system. The peak that appeared at 399.5 eV corresponded to the amine species attached via a covalent bond to sp3-C and sp2-C of graphene. The third peak located at 400.2 eV was due to pyrrolic-N species, which donated two π-electrons to the five membered rings of the conjugated system. The fourth peak that appeared at 401.5 eV was associated with quaternary-N, which corresponds to the C atoms, replaced inside the graphene layers by the N atoms.32,61 These nitrogen configurations can change the efficiency of charge transfer due to multiple electronic states,62 which is also evident from Raman spectra, showing that benzoid and quinoid type formation occurred.
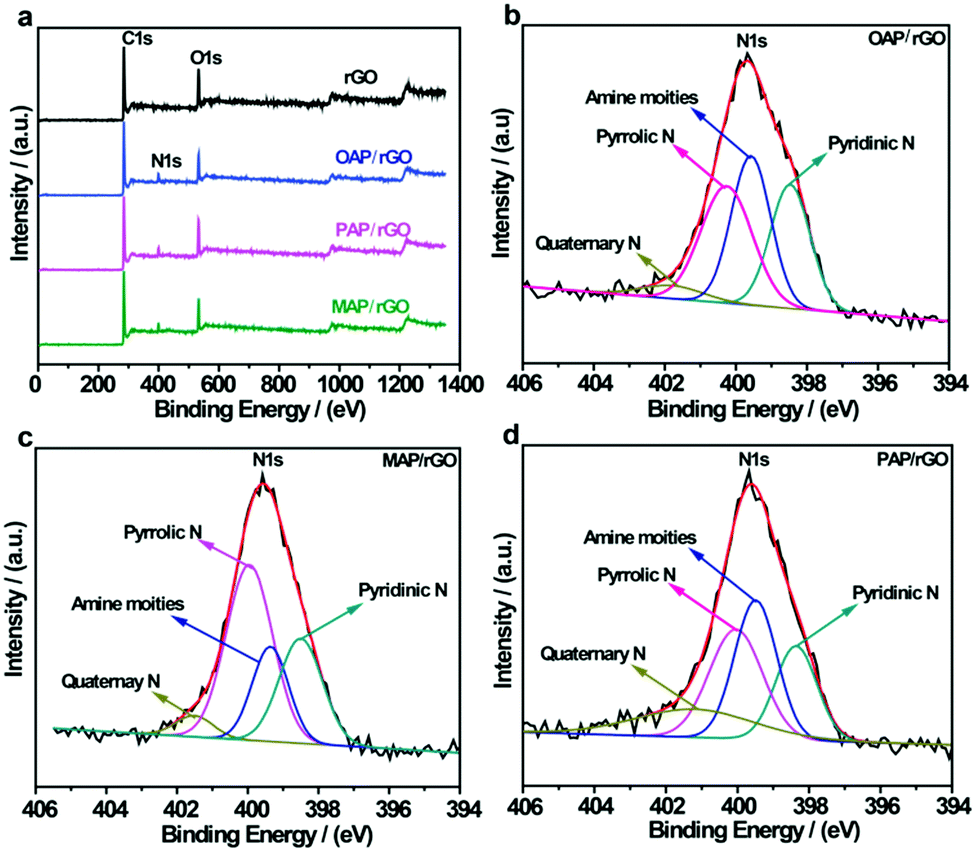 | ||
| Fig. 3 XPS analysis of rGO, OAP/rGO, PAP/rGO and MAP/rGO; (a) survey spectra of rGO and AP/rGO; and (b–d) N 1s spectra of OAP/rGO, PAP/rGO and MAP/rGO graphene films. | ||
The high-resolution C 1s spectra of the rGO and AP/rGO composites consist of four peaks (Fig. S4, ESI†). The main peak at 284.4 eV was connected to the C–C/CC bond, indicating that the C atoms in the AP/rGO samples are organized in a conjugated honeycomb lattice pattern.39 Three peaks which appeared at higher binding energy show the presence of oxygen groups, including C–OH at 285.4 eV, CO at 288.2 eV, and O–CO at 289.2 eV, respectively.28,39,63 After functionalization, the intensities of the sp3-C peak and the C–O peak, especially the peak of C–O (epoxy and alkoxy), reduced intensely confirming that numerous oxygen-containing functional groups were fruitfully involved in N-doping. A new peak appeared at 285.3 eV for the AP/rGO samples which originated from C–N functional groups.32 The results obtained from XPS and FTIR analysis confirmed the existence of C–N bonds and the decreased intensity of C–O bonds, implying the covalent linkage of amino groups on the graphene films.
Electrochemical performance of the AP/rGO materials
To investigate the electrochemical performance of the AP/rGO composites for supercapacitor applications, cyclic voltammetry (CV) and galvanostatic charge/discharge (GCD) techniques were conducted within the potential window of 0–0.8 V in 1 M H2SO4 with two electrode systems. First of all, to evaluate the effect of different molar ratios on supercapacitor performance, we used various amounts of aminophenol during the hydrothermal process (Fig. S5, ESI†). The results revealed that the concentration of aminophenol had an obvious effect on capacitance. Typically the capacitance increased gradually as the molar ratio increased from 0.5 mmol to 1 mmol, whereas a further increase in the concentration from 1 mmol to 1.5 mmol resulted in the decrease of capacitance. This phenomenon may be caused by the residual OAP molecules stacked within the graphene sheets via π–π interactions and thus poor rate performance due to hindrance of the charge transfer pathway.64–66 Therefore, 1 mmol of aminophenol was selected to evaluate the supercapacitor performance.The CV curves of OAP/rGO, PAP/rGO, MAP/rGO and rGO are shown in Fig. 4a and Fig. S6 (ESI†). The CV shape of OAP/rGO and PAP/rGO showed a pear-like shape with two prominent redox peaks due to the redox transition of OAP and PAP between the oxidation and reduction state.67 Hence, it indicated that both the electrical double-layer capacitance and pseudo-capacitance are co-existent. The maximum capacitance of OAP/rGO and PAP/rGO was found to be 590 F g−1 at 10 mV s−1, while MAP/rGO and rGO samples showed typical nearly rectangular curves which indicated the electric double-layer capacitance behavior. However, the MAP/rGO composite represented a maximum capacitance of 216 F g−1 at 10 mV s−1, much lower than those of OAP and PAP composites and even that of rGO. In addition, the CV curves started to tilt when the scan rate increased and at a low scan rate it appeared as quasi-rectangular. This behavior was representative of small aromatic amine molecules because of anodic oxidation reactions in the amino groups.68
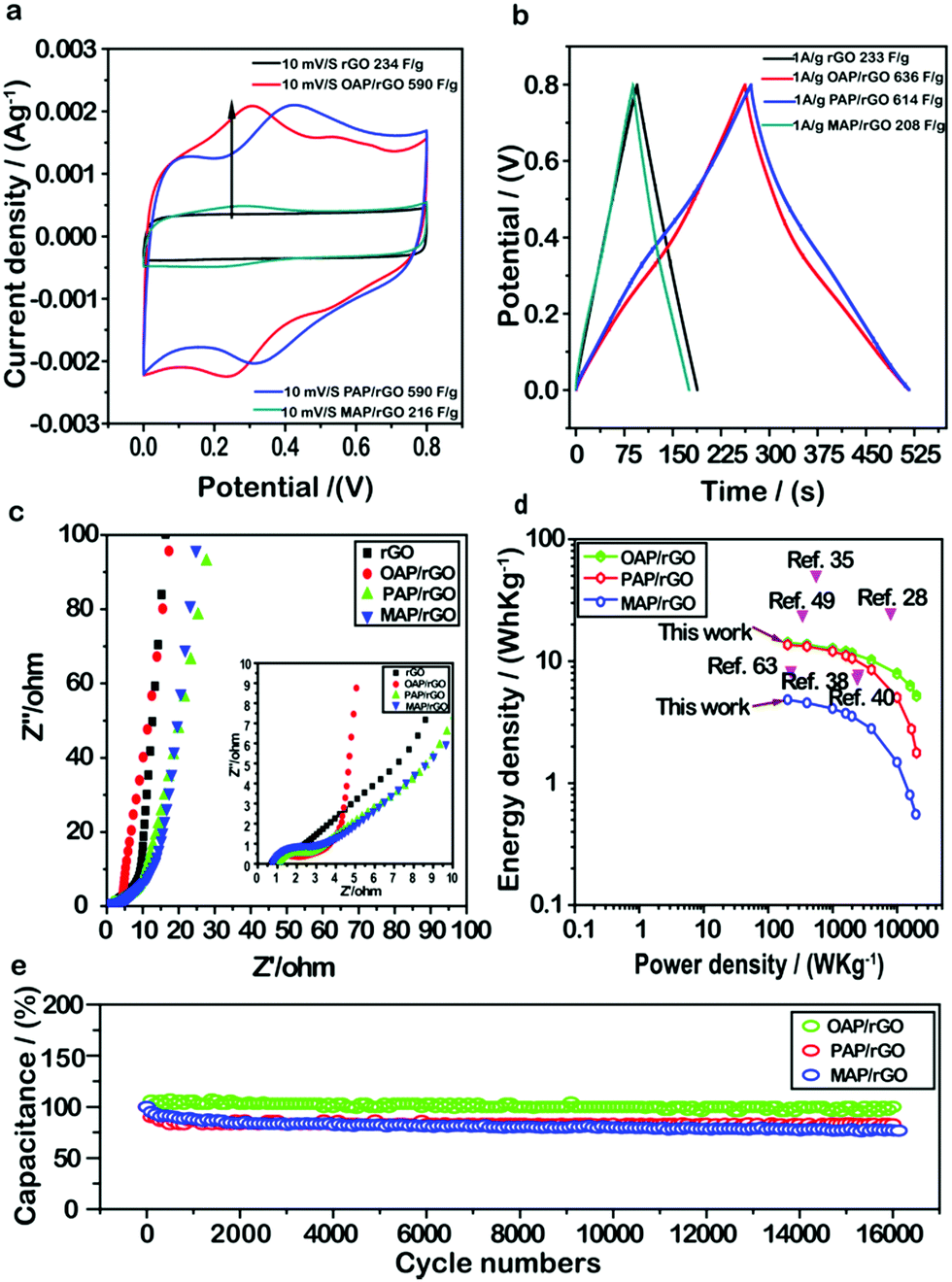 | ||
| Fig. 4 (a) CV curve of rGO/, OAP/rGO, PAP/rGO and MAP/rGO at 10 mV s−1; (b) GCD curve of rGO, OAP/rGO, PAP/rGO and MAP/rGO at 1 A g−1; (c) Nyquist plots of rGO and AP/rGO composites; (d) Ragone plots of AP/rGO composites; and (e) long cycle test of AP/rGO composites. | ||
Similar conclusions can also be drawn from the GCD curves of AP/rGO and rGO samples (Fig. 4b). Even though the as-prepared electrode materials OAP/rGO and PAP/rGO showed slightly distorted symmetry curves due to electric double-layer capacitance and pseudo-capacitance,43 the specific capacitances of OAP/rGO and PAP/rGO were computed to be as high as 637 F g−1 and 614 F g−1 at a current density of 1 A g−1 respectively, which were much higher than that of rGO (231 F g−1) and MAP/rGO (208 F g−1). This indicated the significant role of OAP and PAP molecules in increasing the capacitance of graphene sheets. In contrast, the MAP/rGO composite showed poor electrochemical activity of MAP molecules and the compact structural morphology (as depicted by SEM images in Fig. 1d) became the main obstacle for high energy storage capability. The specific capacitance of the rGO and as-prepared AP/rGO electrode materials against the scan rate and current densities were also calculated from CV (Fig. S6, ESI†) and GCD (Fig. S7, ESI†) measurements, as shown in Fig. S8 (ESI†). The specific capacitance increased with decreasing scan rate and current density; this phenomenon was observed because the limitation of ion diffusion decreases.69 An electrochemical impedance spectroscopy (EIS) study was further conducted to deeply understand the electrochemical behaviors of the as-prepared electrode materials. As shown in Fig. 4c, in the low-frequency region, all of the AP/rGO samples showed a nearly steep line, indicating that all composites show good EDLC behaviour.5,70 From Nyquist plots, the equivalent series resistance (ESR) values were found to be 8.9 Ω, 3.2 Ω, 6.3 Ω and 12.1 Ω for rGO, OAP/rGO, PAP/rGO, and MAP/rGO, respectively, which somehow explained the high capacitance of OAP/rGO and PAP/rGO.
Ragone plots of the AP/rGO materials are shown in Fig. 4d. The OAP/rGO composite showed an energy density of 14.1 W h kg−1 with a power density of 199.9 W kg−1, and PAP/rGO of 13.64 W h kg−1 with a power density of 199.8 W kg−1. The electrochemical stability of the OAP/rGO, PAP/rGO and MAP/rGO composites was tested upto 16000 cycles at a current density of 5 A g−1 with the capacitance retention of 97%, 80% and 76%, respectively. This suggested that the OAP/rGO and PAP/rGO electrode materials showed excellent cycling stability (Fig. 4e), as compared to previous reports.31 Moreover, the PAP/rGO sample attained high cycling stability after 16000 cycles, which displayed better performance compared with GN/PAP composites.44 The excellent cycling stability of OAP/rGO and PAP/rGO was assigned to superior structural stability and reversible redox reactions.71 On the contrary, the relatively low cycling stability of MAP/rGO may be due to its high Rct value, low reversible faradaic reactions, and limited propagation of ions resulting from the disordered graphene nanostructure.
Mechanism
According to the above characterization results, we proposed a reaction mechanism between AP molecules and GO as schemed in Fig. 5. When OAP molecules were treated hydrothermally with GO, a benzoxazole ring was formed by covalent linkage through condensation reactions (Fig. 5a), as confirmed by Raman spectroscopy.51,55 The FTIR spectra of OAP/rGO and PAP/rGO also showed that carbonyl peaks around 1731 cm−1 reduced significantly and a new peak was observed at 1623 cm−1, due to a typical reaction which occurred between the carboxylic groups and amino groups of OAP and PAP molecules via condensation or the CO stretching of untreated carboxylate groups. Therefore, PAP molecules also reacted with GO through a nucleophilic addition reaction (Fig. 5b). In the MAP/rGO composite, no peak appeared in this region and there was also less reduction of the CO and C–O peaks than that of OAP/rGO and PAP/rGO.
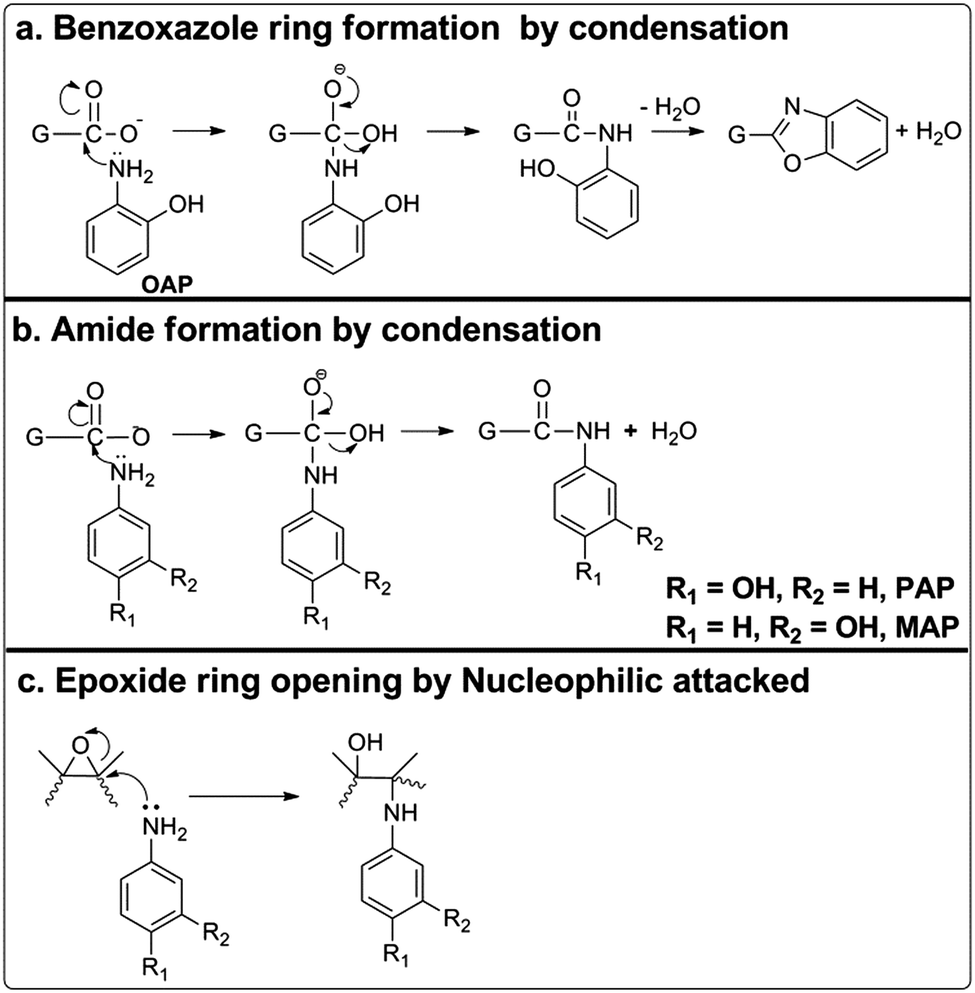 | ||
| Fig. 5 Possible mechanism of AP molecules with GO. | ||
The difference in reactivity of AP molecules due to a different position of –NH2 groups on the benzene ring and thus difference of electrochemical performance can be explained by their resonance structures (Fig. 6). The OAP and PAP molecules favored the nucleophilic addition reaction due to more electron density in the amine group which is beneficial to the stable intermediates and multiple redox transitions (Fig. 6a and b). In contrast, reduced electron density on MAP molecules because of electronic as well as resonance effects decreased the redox reaction by the weaker redox transition state (Fig. 6c).
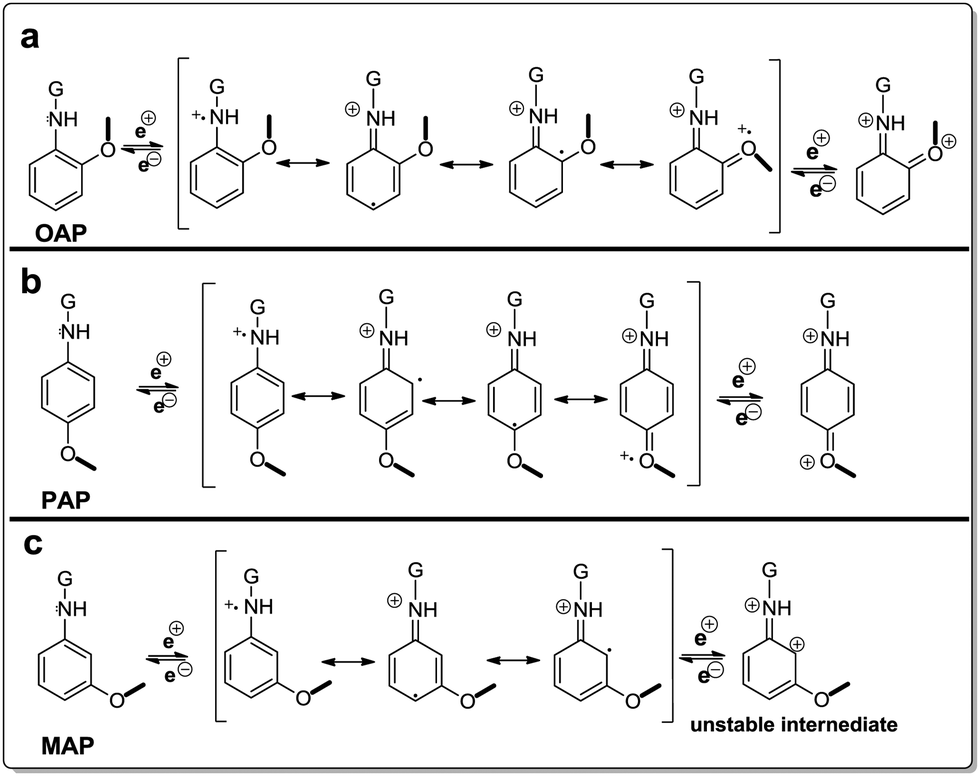 | ||
| Fig. 6 Resonance structures of AP molecules. | ||
The energy storage mechanisms of AP molecules were proposed based on the component characterizations of the charged and discharged electrodes (Scheme S1, ESI†). The CV curve of the OAP/rGO composite displayed an oxidation peak at 0.30 V and a reduction peak at 0.25 V, which are attributed to benzoxazole rings embedded on graphene sheets72,73 (Scheme S1, eqn (S1), ESI†). In the case of the PAP/rGO composite, it displayed a quasi-reversible redox peak which is assigned to p-benzoquinoneimine, formed by oxidation and reduction of PAP molecules74 (Scheme S1, eqn (S2), ESI†), while in the MAP/rGO composite, the CV curve exhibited a redox peak at 0.28 V and 0.21 V, which appeared due to the poly(m-aminophenol) block polymer formed at the surface of the electrode by the crosslinked structure (Scheme S1, eqn (S3), ESI†). In an acidic medium amino groups of MAP molecules are protonated and easily coupled with monomers through electrophilic substitution reactions due to high electron density on the para position with respect to the –NH2 group. Therefore, such a structure is more likely to result in low conductivity.75
Conclusions
In conclusion, we reported the functionalization of wet-spun GO films with three AP isomers (OAP, PAP and MAP) using a one-pot hydrothermal method. The synthesized OAP/rGO, PAP/rGO and MAP/rGO composites showed wrinkled, crumbled and stacked structures. The chemical bonding between the GO films and AP precursors was confirmed by FTIR, Raman spectroscopy and XPS analysis. Among these three N-rGO isomers, the highest capacitance observed for OAP and PAP was 590 F g−1 in CV measurements, while for the MAP composite it was only 218 F g−1 at 10 mV s−1. In GCD measurements, the specific capacitances were 636 F g−1, 614 F g−1 and 207 F g−1 for OAP, PAP and MAP at 1 A g−1, respectively. OAP/rGO and PAP/rGO showed excellent cycling stability after 16000 CD cycles with 97% and 80% retention respectively. The rGO/MAP materials only showed 76% capacitance retention after similar CD cycles. The mechanism discussion demonstrated that the as-prepared AP/rGO composites with different microstructures and electrochemical performance were due to reactivity differences of the AP isomers as well as different positions of the –NH2 groups on the benzene rings. The OAP molecules formed benzoxazole rings through a condensation reaction with GO, while on the other hand, PAP molecules coupled via a nucleophilic addition reaction and MAP displayed lower reactivity toward nucleophilic reactions due to electronic effects. These electrochemical results manifested that OAP functionalized GO composites improved the supercapacitor performance and can be utilized as excellent electrodes for practical application in energy storage devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 21325417, 51533008, 51703194 and 51603183), the National Key R&D Program of China (No. 2016YFA0200200), and the Fundamental Research Funds for the Central Universities (No. 2017QNA4036 and 2017XZZX008-06).References
- D. Yu, Q. Qian, L. Wei, W. Jiang, K. Goh, J. Wei, J. Zhang and Y. Chen, Chem. Soc. Rev., 2015, 44, 647–662 RSC.
- G. Yu, X. Xie, L. Pan, Z. Bao and Y. Cui, Nano Energy, 2013, 2, 213–234 CrossRef CAS.
- P. Sharma and T. S. Bhatti, Energy Convers. Manage., 2010, 51, 2901–2912 CrossRef CAS.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279 CrossRef CAS PubMed.
- L. Zhang and G. Shi, J. Phys. Chem. C, 2011, 115, 17206–17212 CrossRef CAS.
- L. Wang, D. Wang, J. Zhu and X. Liang, Ionics, 2012, 19, 215–220 CrossRef.
- T. Liu, G. Shao, M. Ji and Z. Ma, Ionics, 2013, 20, 145–149 CrossRef.
- Y. B. Tan and J.-M. Lee, J. Mater. Chem. A, 2013, 1, 14814–14843 RSC.
- J. Mao, J. Iocozzia, J. Huang, K. Meng, Y. Lai and Z. Lin, Energy Environ. Sci., 2018, 11, 772–799 RSC.
- J. Mao, M. Ge, J. Huang, Y. Lai, C. Lin, K. Zhang, K. Meng and Y. Tang, J. Mater. Chem. A, 2017, 5, 11873–11881 RSC.
- P. J. Hall, M. Mirzaeian, S. I. Fletcher, F. B. Sillars, A. J. R. Rennie, G. O. Shitta-Bey, G. Wilson, A. Cruden and R. Carter, Energy Environ. Sci., 2010, 3, 1238 RSC.
- C. Zheng, X. Zhou, H. Cao, G. Wang and Z. Liu, J. Power Sources, 2014, 258, 290–296 CrossRef CAS.
- S. L. Candelaria, Y. Shao, W. Zhou, X. Li, J. Xiao, J.-G. Zhang, Y. Wang, J. Liu, J. Li and G. Cao, Nano Energy, 2012, 1, 195–220 CrossRef CAS.
- C. N. R. Rao, K. Gopalakrishnan and A. Govindaraj, Nano Today, 2014, 9, 324–343 CrossRef CAS.
- D. Wang, Y. Min, Y. Yu and B. Peng, J. Colloid Interface Sci., 2014, 417, 270–277 CrossRef CAS PubMed.
- W. S. V. Lee, M. Leng, M. Li, X. L. Huang and J. M. Xue, Nano Energy, 2015, 12, 250–257 CrossRef CAS.
- Z. Wen, X. Wang, S. Mao, Z. Bo, H. Kim, S. Cui, G. Lu, X. Feng and J. Chen, Adv. Mater., 2012, 24, 5610–5616 CrossRef CAS PubMed.
- J. Han, L. L. Zhang, S. Lee, J. Oh, K.-S. Lee, J. R. Potts, J. Ji, R. S. Ruoff, X. Zhao and S. Park, ACS Nano, 2013, 7, 19–26 CrossRef CAS PubMed.
- S. U. Lee, R. V. Belosludov, H. Mizuseki and Y. Kawazoe, Small, 2009, 5, 1769–1775 CrossRef CAS PubMed.
- M. Fan, Z.-Q. Feng, C. Zhu, X. Chen, C. Chen, J. Yang and D. Sun, J. Mater. Sci., 2016, 51, 10323–10349 CrossRef CAS.
- X. Wei, X. Jiang, J. Wei and S. Gao, Chem. Mater., 2016, 28, 445–458 CrossRef CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- R. Yadav and C. K. Dixit, J. Sci.: Adv. Mater. Devices, 2017, 2, 141–149 Search PubMed.
- S. Zhuang, B. B. Nunna, D. Mandal and E. S. Lee, Nano-Struct. Nano-Objects, 2018, 15, 180–185 CrossRef.
- D. Li, C. Yu, M. Wang, Y. Zhang and C. Pan, RSC Adv., 2014, 4, 55394–55399 RSC.
- V. H. Pham, S. H. Hur, E. J. Kim, B. S. Kim and J. S. Chung, Chem. Commun., 2013, 49, 6665–6667 RSC.
- H.-L. Guo, P. Su, X. Kang and S.-K. Ning, J. Mater. Chem. A, 2013, 1, 2248–2255 RSC.
- L. Sun, L. Wang, C. Tian, T. Tan, Y. Xie, K. Shi, M. Li and H. Fu, RSC Adv., 2012, 2, 4498 RSC.
- C. H. Yang Zhao, Y. Hu, H. Cheng, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2012, 51, 11371–11375 CrossRef PubMed.
- T. Wang, L. Wang, D. Wu, W. Xia, H. Zhao and D. Jia, J. Mater. Chem. A, 2014, 2, 8352–8361 RSC.
- Z. Yang, G. Xing, P. Hou and D. Han, Mater. Sci. Eng., B, 2018, 228, 198–205 CrossRef CAS.
- Y. Lu, F. Zhang, T. Zhang, K. Leng, L. Zhang, X. Yang, Y. Ma, Y. Huang, M. Zhang and Y. Chen, Carbon, 2013, 63, 508–516 CrossRef CAS.
- Y. Zou, W. Zhong, S. Li, J. Luo, C. Xiong and W. Yang, Electrochim. Acta, 2016, 212, 828–838 CrossRef CAS.
- Y. Cui, Q.-Y. Cheng, H. Wu, Z. Wei and B.-H. Han, Nanoscale, 2013, 5, 8367–8374 RSC.
- B. Song, J. Zhao, M. Wang, J. Mullavey, Y. Zhu, Z. Geng, D. Chen, Y. Ding, K.-s. Moon, M. Liu and C.-P. Wong, Nano Energy, 2017, 31, 183–193 CrossRef CAS.
- X. Lu, L. Li, B. Song, K.-s. Moon, N. Hu, G. Liao, T. Shi and C. Wong, Nano Energy, 2015, 17, 160–170 CrossRef CAS.
- A. Śliwak, B. Grzyb, N. Díez and G. Gryglewicz, Appl. Surf. Sci., 2017, 399, 265–271 CrossRef.
- Y. Zhang, G. Wen, P. Gao, S. Bi, X. Tang and D. Wang, Electrochim. Acta, 2016, 221, 167–176 CrossRef CAS.
- J. W. Lee, J. M. Ko and J.-D. Kim, Electrochim. Acta, 2012, 85, 459–466 CrossRef CAS.
- H. Zhang, T. Kuila, N. H. Kim, D. S. Yu and J. H. Lee, Carbon, 2014, 69, 66–78 CrossRef CAS.
- D. Wang, Y. Min, Y. Yu and B. Peng, J. Colloid Interface Sci., 2014, 417, 270–277 CrossRef CAS PubMed.
- P. Wiench, Z. González, R. Menéndez, B. Grzyb and G. Gryglewicz, Sens. Actuators, B, 2018, 257, 143–153 CrossRef CAS.
- W. Ai, W. Zhou, Z. Du, Y. Du, H. Zhang, X. Jia, L. Xie, M. Yi, T. Yu and W. Huang, J. Mater. Chem., 2012, 22, 23439 RSC.
- C. Chen, W. Fan, Q. Zhang, T. Ma and Z. Wang, Ionics, 2015, 21, 2639–2645 CrossRef CAS.
- T. Gopalasamy, M. Gopalswamy, M. Gopichand and J. Raj, J. Polym., 2014, 2014, 1–11 Search PubMed.
- X. Zhang, Y. Lai, M. Ge, Y. Zheng, K.-Q. Zhang and Z. Lin, J. Mater. Chem. A, 2015, 3, 12761–12768 RSC.
- T. Huang, S. Cai, H. Chen, Y. Jiang, S. Wang and C. Gao, J. Mater. Chem. A, 2017, 5, 8255–8260 RSC.
- T. Huang, B. Zheng, Z. Liu, L. Kou and C. Gao, J. Mater. Chem. A, 2015, 3, 1890–1895 RSC.
- M. Acik, G. Lee, C. Mattevi, M. Chhowalla, K. Cho and Y. J. Chabal, Nat. Mater., 2010, 9, 840–845 CrossRef CAS PubMed.
- W.-S. Hung, C.-H. Tsou, M. De Guzman, Q.-F. An, Y.-L. Liu, Y.-M. Zhang, C.-C. Hu, K.-R. Lee and J.-Y. Lai, Chem. Mater., 2014, 26, 2983–2990 CrossRef CAS.
- H. L. Ma, H. B. Zhang, Q. H. Hu, W. J. Li, Z. G. Jiang, Z. Z. Yu and A. Dasari, ACS Appl. Mater. Interfaces, 2012, 4, 1948–1953 CrossRef CAS PubMed.
- S. Li, C. Yu, J. Yang, C. Zhao, M. Zhang, H. Huang, Z. Liu, W. Guo and J. Qiu, Energy Environ. Sci., 2017, 10, 1958–1965 RSC.
- W. Hu, D. Xu, X. N. Sun, Z. H. Xiao, X. Y. Chen and Z. J. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 8630–8640 CrossRef CAS.
- A. Sanchez-Sanchez, M. T. Izquierdo, S. Mathieu, J. González-Álvarez, A. Celzard and V. Fierro, Green Chem., 2017, 19, 2653–2665 RSC.
- T. D. Klots and W. B. Collier, Spectrochim. Acta, Part A, 1995, 51, 1291–1316 CrossRef.
- B. Adriano and P. Barbara, J. Raman Spectrosc., 2001, 32, 953–959 CrossRef.
- M. Muniz-Miranda, Vib. Spectrosc., 1999, 19, 227–232 CrossRef CAS.
- A.-u.-H. A. Shah and R. Holze, Electrochim. Acta, 2008, 53, 4642–4653 CrossRef CAS.
- M. Tagowska, B. Pałys and K. Jackowska, Synth. Met., 2004, 142, 223–229 CrossRef CAS.
- D. Deng, X. Pan, L. Yu, Y. Cui, Y. Jiang, J. Qi, W.-X. Li, Q. Fu, X. Ma, Q. Xue, G. Sun and X. Bao, Chem. Mater., 2011, 23, 1188–1193 CrossRef CAS.
- X. Fan, C. Yu, J. Yang, Z. Ling and J. Qiu, Carbon, 2014, 70, 130–141 CrossRef CAS.
- B. Song, J. I. Choi, Y. Zhu, Z. Geng, L. Zhang, Z. Lin, C.-c. Tuan, K.-s. Moon and C.-P. Wong, Chem. Mater., 2016, 28, 9110–9121 CrossRef CAS.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS.
- Y. Wang, X. Yang, L. Qiu and D. Li, Energy Environ. Sci., 2013, 6, 477–481 RSC.
- K. Zhang, L. Mao, L. L. Zhang, H. S. On Chan, X. S. Zhao and J. Wu, J. Mater. Chem., 2011, 21, 7302–7307 RSC.
- H. Bai, Y. Xu, L. Zhao, C. Li and G. Shi, Chem. Commun., 2009, 1667–1669, 10.1039/B821805F.
- Y. F. Nie, Q. Wang, H. T. Yi, X. Y. Chen and Z. J. Zhang, RSC Adv., 2015, 5, 65100–65109 RSC.
- D. N. Futaba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura and S. Iijima, Nat. Mater., 2006, 5, 987–994 CrossRef CAS PubMed.
- M. R. Nateghi, M. H. Mosslemin and H. Hadjimohammadi, React. Funct. Polym., 2005, 64, 103–109 CrossRef CAS.
- B. Hsia, J. Marschewski, S. Wang, J. B. In, C. Carraro, D. Poulikakos, C. P. Grigoropoulos and R. Maboudian, Nanotechnology, 2014, 25, 055401 CrossRef PubMed.
- Z. Yu, L. Tetard, L. Zhai and J. Thomas, Energy Environ. Sci., 2015, 8, 702–730 RSC.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- Z. Wang, X. Li, Y. Wu, Y. Tang and S. Ma, J. Electroanal. Chem., 1999, 464, 181–186 CrossRef CAS.
- H. J. Salavagione, J. Arias, P. Garcés, E. Morallón, C. Barbero and J. L. Vázquez, J. Electroanal. Chem., 2004, 565, 375–383 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00260f |
| This journal is © the Partner Organisations 2018 |