
Hydrogen-bonded-assisted supramolecular microwires for pure violet lasers: benefits of preventing intermolecular π–π stacking and aggregation in single crystals†
Man
Xu‡
a,
Chang-Jin
Ou‡
a,
Chao
Gao‡
a,
Jin-Yi
Lin
*abc,
Wei
Xu
d,
Meng-Na
Yu
b,
Wen-Sai
Zhu
a,
Zong-Qiong
Lin
c,
Lu-Bin
Bai
a,
Ya-Min
Han
a,
Ling-Hai
Xie
 *b,
Ling
Huang
a,
Chun-Xiang
Xu
d,
Jian-Feng
Zhao
a,
Jian-Pu
Wang
a and
Wei
Huang
*abc
*b,
Ling
Huang
a,
Chun-Xiang
Xu
d,
Jian-Feng
Zhao
a,
Jian-Pu
Wang
a and
Wei
Huang
*abc
aKey Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University, Nanjing 211816, China. E-mail: iamjylin@njtech.edu.cn; wei-huang@njtech.edu.cn
bCenter for Molecular Systems and Organic Devices (CMSOD), Key Laboratory for Organic Electronics and Information Displays (KLOEID) & Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing University of Posts & Telecommunications, Nanjing 210046, China. E-mail: iamlhxie@njupt.edu.cn
cShaanxi Institute of Flexible Electronics (SIFE), Northwestern Polytechnical University (NPU), 127 West Youyi Road, Xi’an, Shaanxi 710072, China
dState Key Laboratory of Bioelectronics, School of Electronic Science and Medical Engineering, Southeast University, Nanjing 210096, China
First published on 26th September 2018
Abstract
Deep-violet lasing at the microscale is crucial for the realization of highly integrated nano-photonic and communication devices. In the work reported herein, we fabricated a type of deep-violet organic microwire laser by suppressing the intermolecular π–π stacking interaction and aggregation based on a hydrogen-bonded-assisted molecular encapsulation mechanism. Interestingly, a regular supramolecular encapsulation layer can be constructed by controlling S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–C hydrogen-bonded superstructures in crystalline frameworks and simultaneously effectively inhibiting the intermolecular π–π stacking interactions, allowing for single-molecular excitonic behavior. For the synergistic effects of shorter effective conjugated-length and negligible intermolecular excitonic coupling, the microwire therein can act as a high-quality whispering-gallery-mode microcavity for low threshold deep-violet lasing (41 W cm−2, 6 tims lower than those of controlled materials) with an emission peak of 405 ± 2 nm, attributed to 0–1 band emission of a single molecule in diluted solution. Therefore, our work provides a proof-of-concept trial for developing a highly stable and easily processed single crystal via a molecular encapsulation strategy in a supramolecular crystalline framework.
O⋯H–C hydrogen-bonded superstructures in crystalline frameworks and simultaneously effectively inhibiting the intermolecular π–π stacking interactions, allowing for single-molecular excitonic behavior. For the synergistic effects of shorter effective conjugated-length and negligible intermolecular excitonic coupling, the microwire therein can act as a high-quality whispering-gallery-mode microcavity for low threshold deep-violet lasing (41 W cm−2, 6 tims lower than those of controlled materials) with an emission peak of 405 ± 2 nm, attributed to 0–1 band emission of a single molecule in diluted solution. Therefore, our work provides a proof-of-concept trial for developing a highly stable and easily processed single crystal via a molecular encapsulation strategy in a supramolecular crystalline framework.
Introduction
Organic solid-state lasers (OSLs) have attracted significant attention for their tunability, light weight, flexibility, and ability to fabricate cheap optoelectronic devices with high absorption cross-sections and tunable emission.1–3 Although recent progress and significant improvements in designing and fabricating OSLs with distributed feedback (DBF) resonator structures4–6 and possessing light-emitting field effect transistors have been achieved,7–9 the critical issues related to low charge-carrier mobility and high lasing threshold in the gain material still must be addressed. In this regard, organic single crystals (SCs) with excellent molecular alignment and regular packing hold great promise to be employed as a gain medium in electrically driven organic lasers as they are virtually free from light scattering, critical carrier densities, and large refractive indices.10–15 However, compared to red, green, and blue emissive SCs for lasers, easily processed deep-violet SCs with a lasing peak of <410 nm have been quite rare and expensive15–19 in the last several decades owing to their complex aggregation, excitonic coupling and energy transfer under extremely concentrated conditions, resulting in larger Stokes shift, red-shifted emission, or lower luminescence efficiency.1,3,15,20 More importantly, emission peaks of approximately 405 ± 2 nm show unique applications in photo-assisted information storage, laser printing, undersea communication, projection displays, sensors, and optical detectors.1–3,15 Therefore, the development of a novel molecular design strategy to construct highly stable and easily processed deep-violet organic fluorophores for low-threshold SC lasers is urgently required.Compared to the common amorphous/polycrystalline organic fluorophores in solid states, small molecules always show long-range and highly molecular order in SCs.15 In this regard, molecules exhibiting stronger π–π interaction and dense aromatic stacking with regular molecular aggregation in SCs may result in stronger intermolecular electron delocalization, hybridization and coupling.15,21–23 The strong excitonic coupling in both H- and J-type aggregates is detrimental for lasing properties: the former affects excitonic coupling cases and enhanced reabsorption losses, while the latter always induces a low radiative-decay rate and decreased photoluminescence quantum yield (PLQY),10,12,20,24 which are undesirable, especially for the fabrication of deep-violet lasers. In this regard, a weaker coupling regime can suppress exciton diffusion and thus reduce the impact of exciton annihilation at high exciton densities required for lasing action.3,6,18,19,22 Besides, the laser threshold values are directly related to the radiative decay rate; therefore, molecules with short excited-state lifetimes and higher PLQY are preferred for lasers.8,19,22 Thus, a rational molecular design for encapsulation of fluorophores in SCs of organic lasers (Scheme 1a) is essential for suppressing π–π stacking interactions and avoiding excitonic coupling in condensed states. In these designed SCs, multi-dimensional fluorophores can be seperated into several “cages” and physically isolated by a self-assembled encapsulation layer (substituent groups, represented by pink and green layers in Scheme 1) to obtain single-molecular excitonic behavior [Scheme 1(a)]. To confirm the effectiveness of our hypothesis above [Scheme 1(a)], we designed and constructed a highly stable steric supramolecule, 2′,7′-bis(9,9-dioctyl-9H-fluoren-2-yl)spiro[fluorene-9,9′-thioxanthene]10′,10′-dioxide (DOF-SFSO), for deep-violet microlasers with single-molecular excitonic behavior via synergistic effects of steric- and hydrogen-bonding interactions [Scheme 1(b)]. Steric and flexible alkyl units can act as encapsulated layers to inhibit intermolecular π–π stacking interactions, and simultaneously improve material stability, processing ability, and PLQY in SCs. Impressively, the lasing peak of our SCs is approximately 405–408 nm, similar to the 0–1 vibronic transition of a single DOF-SFSO molecule, indicating intramolecular photophysical behavior in SCs. The lasing threshold is 6 times lower than those of controlled terfluorene without steric units, 2,2′:7′,2′′-ter(9,9′-dimethylfluorene) (TDMeF).19
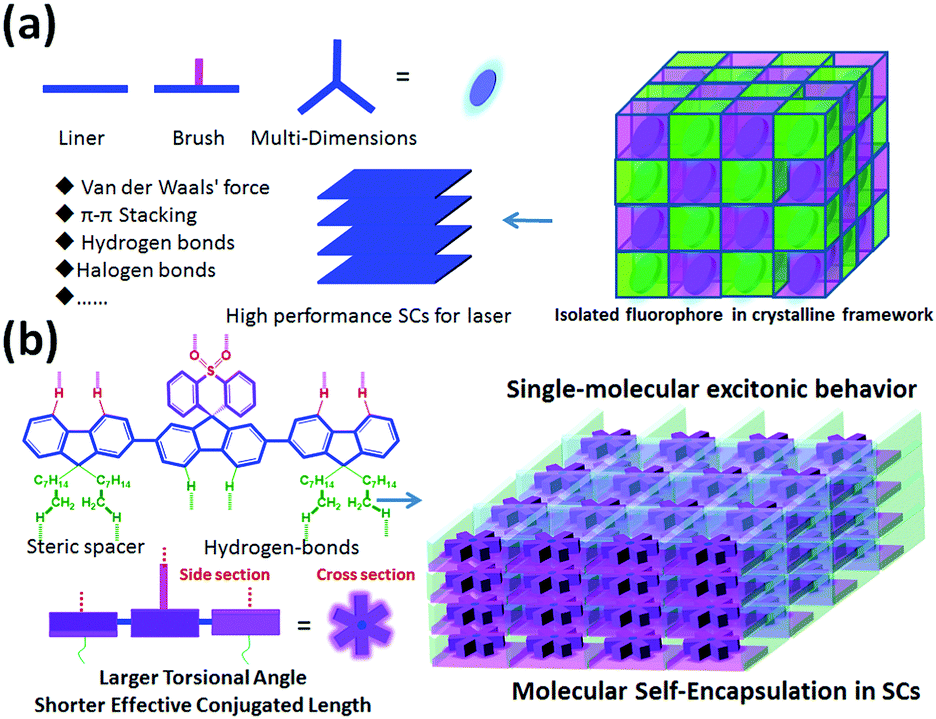 | ||
| Scheme 1 Schematic representation of (a) rational design for high performance SCs for a low-threshold organic laser by controlling intermolecular π–π stacking interaction and aggregation. (b) Designed blueprint of regular aromatic crystalline packing based on DOF-SFSO molecules to isolate fluorophores in the SC framework for violet microlasers. DOF-SFSO molecules show a non-planar conformation with a larger adjacent angle of 38.5° and 145° in SCs, indicating that the fluorene units show dislocated arrangement with a larger adjacent angle in our DOF-SFSO molecules. Subsequently, we set a purple rectangle to present the fluorene unit from the cross-section. | ||
Results and discussion
DOF-SFSO was readily synthesized by a one-step reaction, shown in Scheme S1 (see the ESI†), and its chemical structure and purity were confirmed by 1H nuclear-magnetic-resonance (NMR) and 13C NMR spectroscopy and single crystal X-ray diffraction measurements (Fig. S1 and S2, ESI†). For the substitution of longer, flexible alkyl groups at the 9-position, DOF-SFSO has excellent solubility in organic solvents (>20 mg mL−1), such as dichloromethane, chloroform, and tetrahydrofuran, allowing for excellent solution-processed ability for the construction of optoelectronic devices. In addition, DOF-SFSO displays a high thermal decomposition temperature (Td) of 387 °C (Fig. S3, ESI†). Furthermore, there is no obvious phase-transition temperature observed from 30 °C to 300 °C (Fig. S4, ESI†), indicating that our terfluorene has excellent thermal stability for laser applications.Subsequently, wire-type DOF-SFSO SCs (∼10 μm) were obtained by a traditional mixed-solvent method. Molecular packing modes are clearly screened by SC X-ray diffraction analysis (Fig. 1 and Fig. S5, ESI†). As expected, the molecules show non-planar conformation with larger adjacent angles of 38.5° and 145° in SCs [Fig. 1(a) and Scheme 1(b)], allowing for shorter effective conjugated length and lower π-electron delocalization to obtain deep and stable violet emission. More interestingly, a centered terfluorene backbone structure can pack into zigzag single regular layers (width ∼8.0 Å) in SCs, which can be effectively isolated by the steric spacer SO aromatic rigid layer [pink lines in Fig. 1(b)], consistent with our hypothesis as illustrated in Scheme 1(b). As shown in Fig. 1(c), from the crystallographic structure of a DOF-SFSO SC, dual supramolecular hydrogen-bonding interactions SO⋯H–C (side fluorene: 4,5-position) form between DOF-SFSO molecules, with a characteristic distance of 2.586 or 2.678 Å, and further enable the molecules to self-assemble into a hydrogen-bonded-assisted superstructure. In addition, in our designed blueprint, the flexible units at the 9-position can also act as a soft “insulated” layer to isolate the fluorophore in a self-assembled crystalline framework (green line in Fig. 1b). Interestingly, weaker noncovalent interactions between C–H (flexible chain)⋯H–C (centered fluorene: 4,5-position) are also observed with a lower distance of 2.355 Å, which induced the formation of encapsulated layers to physically separate emissive molecules in SCs. In this regard, the fluorophore can be divided into regular “cages” in SCs (Scheme 1b), enabling them to obtain single-molecular excitonic behavior. Similar to previous terfluorene TDMeF SCs,19 DOF-SFSO molecules show a larger torsion angle of nearly 40° between adjacent fluorene moieties. What's more, a nearly complete dislocated π–π stacked interaction with a larger distance of 3.786 Å is also observed in Fig. 1(e), suggesting the extremely weaker, and even negligible, intermolecular π–π stacked interaction in SCs. In short, there is no obvious intermolecular π–π stacked interaction and aggregation, further avoiding intermolecular excitonic coupling in DOF-SFSO hydrogen-bonded-assisted supramolecular SCs.
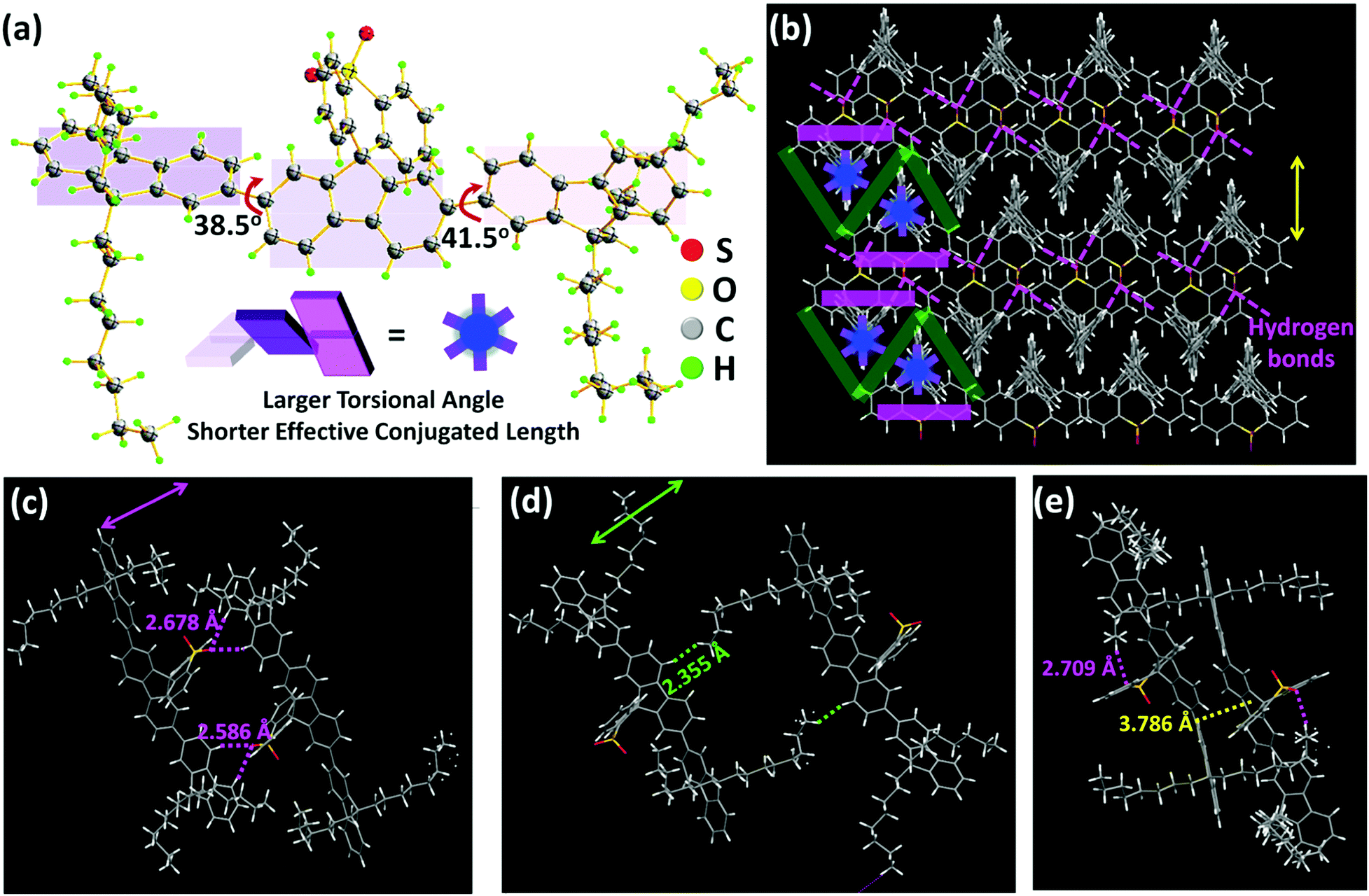 | ||
| Fig. 1 (a) DOF-SFSO crystallographic structure. (b) Molecular packing modes for encapsulated DOF-SFSO SC. Pink lines denote hydrogen-bonds. (c–e) Molecular packing of the DOF-SFSO dimer in different directions. | ||
Among all organic semiconducting materials, terfluorene compounds have proved to be some of the most successful wide-band-gap and stable-gain media in violet-blue organic lasers.1,3,15 However, a terfluorene-based amplified spontaneous emission (ASE)/laser always shows red-shifted deep blue emission (0–1, >420 nm) owing to its longer effective conjugated length induced by quasi-planar conformation or intermolecular aggregation in solid states.3,25 In fact, according to our previous studies,19,25 our TDMeF exhibited a large torsion conformation and weaker intermolecular electronic coupling in SCs, allowing for keeping violet blue emission with high fluorescent efficiency for violet microlasers. As expected, for the same conjugated backbone structure, similar absorption and fluorescence (PL) spectra of our terfluorene compounds (DOF-SFSO and TDMeF) were observed in diluted solution with a maximum absorption peak of 348 nm and three emission peaks at 394 nm (0–0), 415 nm (0–1), and 438 nm (0–2) (Fig. 2).19 However, both controlled TDMeF neat film and crystals exhibited slight green-band emission at 530 nm with a long spectral tail to 600 nm, indicating weaker intermolecular π–π interaction or aggregation, which resulted in excimer behavior in these solid states.17,25–27 Similar to abundant steric-fluorene-based materials, DOF-SFSO shows higher stable emission in the violet-blue region without long-wavelength emission in the film states, even under thermal annealing at 200 °C.5,28 The unstructured bands are most likely caused by non-rigid conjugated backbones giving rise to the absorption arising from a variety of differently twisted conformers in the solution or film states [Fig. 1(a)].12 However, a well-resolved vibronic structure in the emission spectra is a result of an ultrafast torsional relaxation to a defined lower-energy excited state, known to occur in single bonded molecular structures.29 Emission spectra of isolated molecules in the diluted solution dominant 0–0 vibronic bands. Conversely, the 0–1 vibronic replicas prevail in the spectra of the neat films and SCs. The rather slightly bathochromic shift of the PL spectra of our terfluorene neat spin-coated films (15 nm) compared to diluted solution is attributed to enhanced intermolecular interaction, more quasi-planar conformation and longer effective conjugated length. Obviously, the ASE spectra of DOF-SFSO neat spin-coated film have a maximum peak at 428 nm, attributed to 0–1 vibronic band emission [Fig. 2, bottom], consistent with the findings of previous studies.1,3,15 In this regard, DOF-SFSO molecules may show longer effective conjugated length, weaker intermolecular aggregation, or π–π interaction in spun-film states.29 In general, a sign of the resulting reduced intermolecular excitonic behavior, energy transfer and quenching processes is the very small red-shift, and even blue-shift, of the PL spectrum in the solid state compared to that in diluted solution.30,31 Amazingly, compared to diluted solution, a blue-shift of approximately ∼8 nm of 0–1 vibronic band emission was observed in the PL spectra of DOF-SFSO SCs. This distinctive blue spectral shift in emission of SCs may originate from the increase of the dihedral angles between adjacent fluorenes, confirming the non-planar conformation with a larger adjacent angle. Furthermore, emission peaks of the diluted solutions match those of the SCs; however, their intensity distribution is different. Significantly lower intensity of the higher 0–0 energy vibronic replicas in SCs is due to the larger reabsorption in the bulk of the crystal for the thick sample (∼5 μm).10 These intrachain emission properties of our SCs were further investigated by time-resolved transient PL measurements. Our diluted solution and SCs show one lifetime as fitted by the single-exponential decay. The PL decay times (τ) of diluted solution and SCs are 0.50 and 0.59 ns, respectively, but the film had two lifetimes, 0.67 and 0.89 ns, respectively. In addition, the PLQYs of our DOF-SFSO diluted solution, films and SCs were 89% ± 2%, 36% ± 2%, and 58% ± 2%, respectively, which are attributed to the suppression of aggregation-induced quenching in SC states. Correspondingly, the rate constants of radiative fluorescence decay (kr) are calculated with kr = ΦPLQY/τ. All the kr values are on the order of ≈108 and 109 s−1 for our DOF-SFSO pristine film and SCs, respectively, which are high compared with those of conventional organic lasing dyes. Compared to neat films, blue-shifted PL spectra and higher kr values for DOF-SFSO SCs indicate single-molecular excitonic behavior in these condensed SC states.
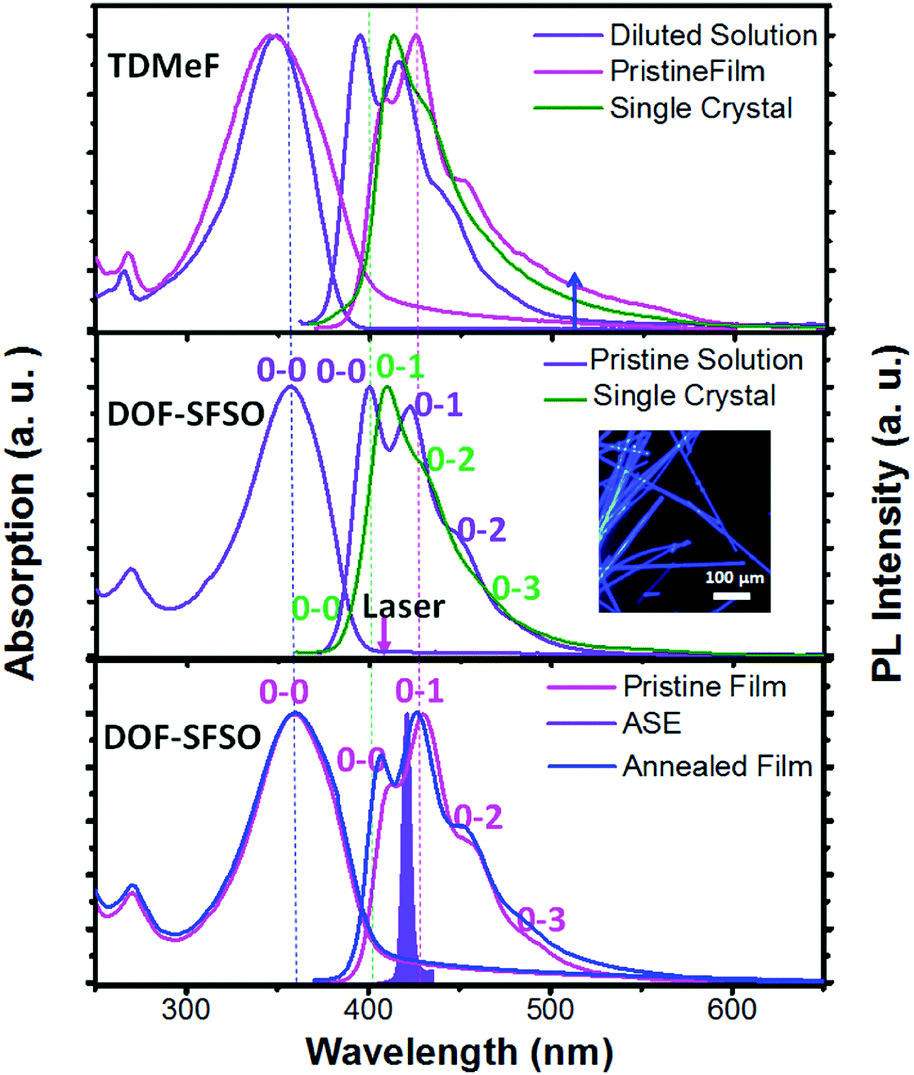 | ||
| Fig. 2 Absorption, emission, or ASE spectra of DOF-SFSO in diluted solution, pristine and annealed film (200 °C), and microcrystals, together with absorption and emission spectra of the controlled TDMeF spin-coated film and crystal.19 | ||
In order to further confirm our assumptions and explore the intrinsic potential of the gain medium, its lasing properties were explored by utilizing the thin excitation stripe technique (Fig. 3). Measurements were performed by exciting microwire SCs with a pulsed laser stripe at the edge and recording the emission spectra. Evidently, broad spontaneous emission bands observed at low excitation density become narrower with the increasing pump density above the onset of stimulated optical transitions. As shown in Fig. 3, the lasing peak appears at the DOF-SFSO SCs 0–1 vibronic replicas (also the 0–1 vibronic band of single molecules), effectively confirming the single-excitonic behavior in SCs. A set of sharp lasing peaks appears on the top of the 0–1 transition with linewidth Δλ = 0.18 nm (0.52 nm for TDMeF) and centered at 406 nm for DOF-SFSO (409 nm for TDMeF) microcrystals.19 Therefore, the cavity quality factor (Q = λ/Δλ) can be estimated to be as high as ∼2255 and 786 for DOF-SFSO and TDMeF SCs, respectively. Spectral narrowing with increasing excitation power density is accompanied by an abrupt change in the emission intensity profile from linear to superlinear at ILaserth. Owing to the weaker excitonic coupling and high PLQY, the ILaserth value estimated for the studied DOF-SFSO SCs is 41.5 W cm−2 compared to that for controlled TDMeF (256 W cm−2, 1 order of magnitude higher at similar lasing peak, 6 times lower) was obtained. Such differences in ILaserth values of the terfluorene SCs studied can have several causes, namely different radiative-decay rates, exciton annihilation impeding attainment of critical exciton density, excited state absorption overlapping with the gain region, etc. More importantly, it is difficult to simultaneously construct two of the same optical resonators that meet the conditions that can produce laser oscillation by solution self-assembly. In this regard, molecular encapsulation in a supramolecular SC framework is an effective strategy with which to suppress excitonic coupling to obtain stable and low-threshold organic laser.
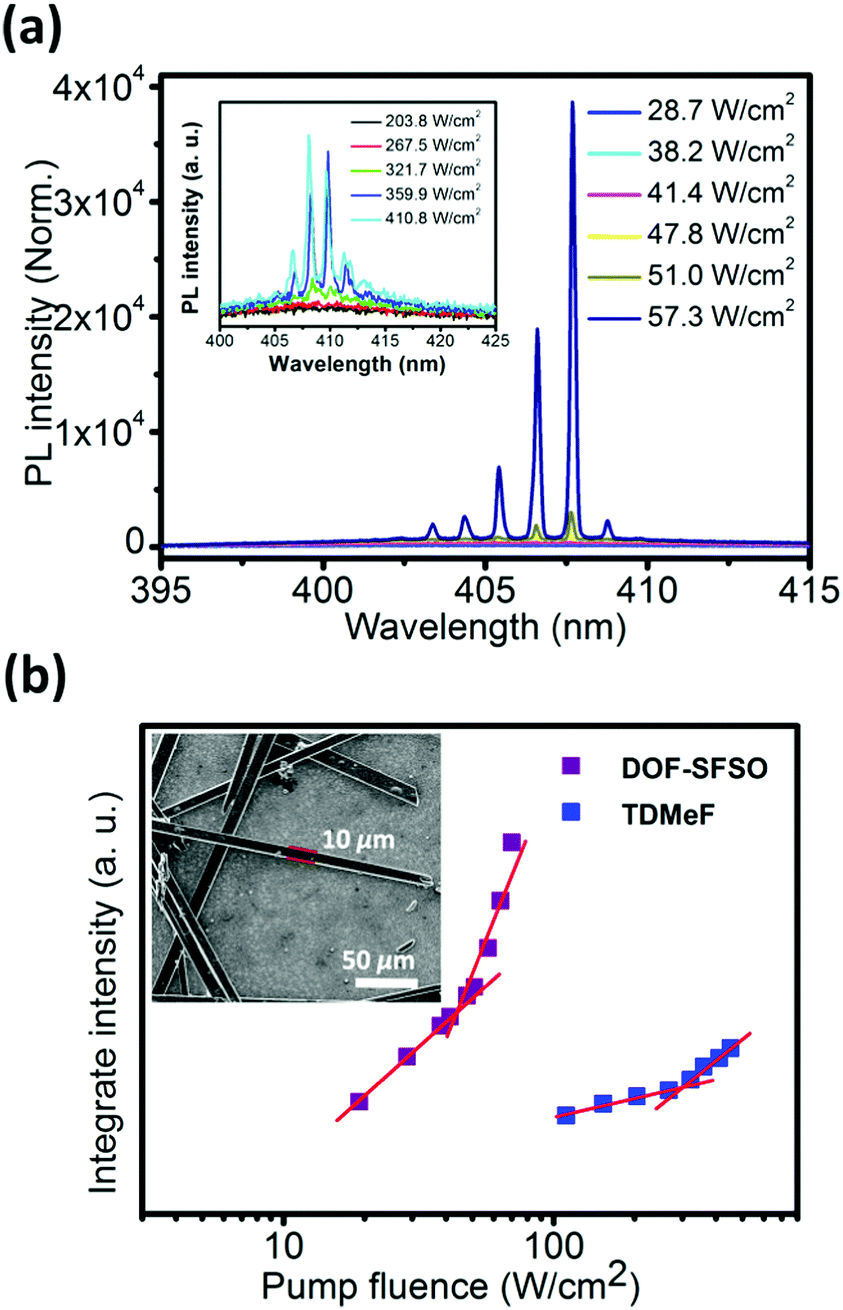 | ||
| Fig. 3 (a) PL spectra obtained from DOF-SFSO SCs excited at different energies. PL integrated area of the 0–1 peak (at approximately 405 nm) as a function of pump densities for DOF-SFSO microwires. (b) PL integrated area of the 0–1 peak (at approximately 405 nm) as a function of pump densities for DOF-SFSO and TDMeF SCs. The lasing threshold is identified as the intersection between the sublinear and superlinear regions.19 The inset shows the SEM images of DOF-SFSO microwires. The microcrytal had a width of 10 μm and a length of 300 μm. | ||
Conclusions
In summary, we have proposed an effective strategy to construct a lower-threshold deep-violet microlaser by isolating fluorophores and obtaining single-molecular excitonic behavior in SCs. Intermolecular SO⋯H–C hydrogen-bonds in our supramolecular framework enable DOF-SFSO molecules to show a non-planar conformation with larger adjacent angles of 38.5 and 145° in SCs. Apart from good thermal stability, the DOF-SFSO micro-SCs matched the emission behavior of single molecules in diluted solution, further confirming their intramolecular photophysical properties. Impressively, further evidence for the effectiveness of our strategy was obtained by demonstrating the violet-laser action, which peaked at 405 ± 2 nm, similar to the 0-1 vibronic band for a single DOF-SFSO molecule. What's more, the corresponding threshold is rather lower, 6 times lower in fact, than those of controlled materials without steric units (TDMeF). This encapsulation strategy approach in a crystalline framework is thus shown to be a promising route for the fabrication of low-cost optoelectronic nano-devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (61874053, 21504041, 21502091, 21502092, and 21774061), the National Key Basic Research Program of China (973) (2015CB932200), the Natural Science Funds of the Education Committee of Jiangsu Province (18KJA430009), the Natural Science Foundation of Jiangsu Province (BK20171470), “High-Level Talents in Six Industries” of Jiangsu Province (XYDXX-019), the open research fund from Key Laboratory for Organic Electronics and Information Displays and State Key Laboratory of Supramolecular Structure and Materials at Jilin University (sklssm201808).References
- H.-H. Fang, J. Yang, J. Feng, T. Yamao, S. Hotta and H.-B. Sun, Laser Photonics Rev., 2014, 8, 687–715 CrossRef.
- H. Dong, C. Zhang and Y. S. Zhao, J. Mater. Chem. C, 2017, 5, 5600–5609 RSC.
- A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed.
- B. K. Yap, R. Xia, M. Campoy-Quiles, P. N. Stavrinou and D. D. C. Bradley, Nat. Mater., 2008, 7, 376–380 CrossRef CAS PubMed.
- J.-Y. Lin, W.-S. Zhu, F. Liu, L.-H. Xie, L. Zhang, R. Xia, G.-C. Xing and W. Huang, Macromolecules, 2014, 47, 1001–1007 CrossRef CAS.
- Q. Zhang, J. Liu, Q. Wei, X. Guo, Y. Xu, R. Xia, L. Xie, Y. Qian, C. Sun, L. Lüer, J. Cabanillas-Gonzalez, D. D. C. Bradley and W. Huang, Adv. Funct. Mater., 2018, 0, 1705824 CrossRef.
- C. Zhang, P. Chen and W. Hu, Small, 2016, 12, 1252–1294 CrossRef CAS PubMed.
- H. Nakanotani, S. Akiyama, D. Ohnishi, M. Moriwake, M. Yahiro, T. Yoshihara, S. Tobita and C. Adachi, Adv. Funct. Mater., 2007, 17, 2328–2335 CrossRef CAS.
- T. Takenobu, S. Z. Bisri, T. Takahashi, M. Yahiro, C. Adachi and Y. Iwasa, Phys. Rev. Lett., 2008, 100, 066601 CrossRef PubMed.
- Z. Xu, Q. Liao, Q. Shi, H. Zhang, J. Yao and H. Fu, Adv. Mater., 2012, 24, OP216–OP220 CAS.
- S. Han, W. Zhang, B. Qiu, H. Dong, W. Chen, M. Chu, Y. Liu, X. Yang, F. Hu and Y. S. Zhao, Adv. Opt. Mater., 2018, 6, 1701077 CrossRef.
- P. Baronas, G. Kreiza, P. Adomenas, O. Adomeniene, K. Kazlauskas, J.-C. Ribierre, C. Adachi and S. Jursenas, ACS Appl. Mater. Interfaces, 2018, 10, 2768–2775 CrossRef CAS PubMed.
- X. Wang, Q. Liao, H. Li, S. Bai, Y. Wu, X. Lu, H. Hu, Q. Shi and H. Fu, J. Am. Chem. Soc., 2015, 137, 9289–9295 CrossRef CAS PubMed.
- X. Wang, H. Li, Y. Wu, Z. Xu and H. Fu, J. Am. Chem. Soc., 2014, 136, 16602–16608 CrossRef CAS PubMed.
- J. Gierschner, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2016, 4, 348–364 CrossRef CAS.
- X. H. Zhu, D. Gindre, N. Mercier, P. Frere and J. M. Nunzi, Adv. Mater., 2003, 15, 906–909 CrossRef CAS.
- J.-Y. Lin, G.-Y. Zhu, B. Liu, M.-N. Yu, X.-H. Wang, L. Wang, W.-S. Zhu, L.-H. Xie, C.-X. Xu, J.-P. Wang, P. N. Stavrinou, D. D. C. Bradley and W. Huang, ACS Macro Lett., 2016, 5, 967–971 CrossRef CAS.
- C.-J. Ou, C. Zhu, X.-H. Ding, L. Yang, J.-Y. Lin, L.-H. Xie, Y. Qian, C.-X. Xu, J.-F. Zhao and W. Huang, J. Mater. Chem. C, 2017, 5, 5345–5355 RSC.
- C.-J. Ou, X.-H. Ding, Y.-X. Li, C. Zhu, M.-N. Yu, L.-H. Xie, J.-Y. Lin, C.-X. Xu and W. Huang, J. Phys. Chem. C, 2017, 121, 14803–14810 CrossRef CAS.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- H.-H. Fang, J. Yang, R. Ding, Q.-D. Chen, L. Wang, H. Xia, J. Feng, Y.-G. Ma and H.-B. Sun, Appl. Phys. Lett., 2010, 97, 101101 CrossRef.
- J. Gierschner, H. G. Mack, L. Luer and D. Oelkrug, J. Chem. Phys., 2002, 116, 8596–8609 CrossRef CAS.
- S. Cai, H. Shi, Z. Zhang, X. Wang, H. Ma, N. Gan, Q. Wu, Z. Cheng, K. Ling, M. Gu, C. Ma, L. Gu, Z. An and W. Huang, Angew. Chem., Int. Ed., 2018, 130, 4069–4073 CrossRef.
- P.-Z. Chen, H. Zhang, L.-Y. Niu, Y. Zhang, Y.-Z. Chen, H.-B. Fu and Q.-Z. Yang, Adv. Funct. Mater., 2017, 27, 1700332 CrossRef.
- E. Y. Choi, L. Mazur, L. Mager, M. Gwon, D. Pitrat, J. C. Mulatier, C. Monnereau, A. Fort, A. J. Attias, K. Dorkenoo, J. E. Kwon, Y. Xiao, K. Matczyszyn, M. Samoc, D. W. Kim, A. Nakao, B. Heinrich, D. Hashizume, M. Uchiyama, S. Y. Park, F. Mathevet, T. Aoyama, C. Andraud, J. W. Wu, A. Barsella and J. C. Ribierre, Phys. Chem. Chem. Phys., 2014, 16, 16941–16956 RSC.
- J. Lin, Z. Yu, W. Zhu, G. Xing, Z. Lin, S. Yang, L. Xie, C. Niu and W. Huang, Polym. Chem., 2013, 4, 477–483 RSC.
- J. Y. Lin, J. Wong, L. H. Xie, X. C. Dong, H. Y. Yang and W. Huang, Macromol. Rapid Commun., 2014, 35, 895–900 CrossRef CAS PubMed.
- B. Liu, J. Lin, F. Liu, M. Yu, X. Zhang, R. Xia, T. Yang, Y. Fang, L. Xie and W. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 21648–21655 CrossRef CAS PubMed.
- J. K. Gallaher, K. Chen, G. S. Huff, S. K. K. Prasad, K. C. Gordon and J. M. Hodgkiss, J. Phys. Chem. Lett., 2016, 7, 3307–3312 CrossRef CAS PubMed.
- C. Pan, K. Sugiyasu, Y. Wakayama, A. Sato and M. Takeuchi, Angew. Chem., Int. Ed., 2013, 52, 10775–10779 CrossRef CAS PubMed.
- F. Cacialli, J. S. Wilson, J. J. Michels, C. Daniel, C. Silva, R. H. Friend, N. Severin, P. Samorì, J. P. Rabe, M. J. O'Connell, P. N. Taylor and H. L. Anderson, Nat. Mater., 2002, 1, 160 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00397a |
| ‡ Man Xu, Changjin Ou and Chao Gao contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |