DOI:
10.1039/C8QM00170G
(Research Article)
Mater. Chem. Front., 2018,
2, 1523-1528
Pearson's principle-inspired strategy for the synthesis of amorphous transition metal hydroxide hollow nanocubes for electrocatalytic oxygen evolution†
Received
16th April 2018
, Accepted 6th June 2018
First published on 6th June 2018
Abstract
Hollow nanostructures with higher surface area offer great advantages for electrocatalytic water splitting. Here, we demonstrate the fabrication of amorphous hollow M(OH)x (M = Fe, Co, Ni) nanocubes through a template-assisted route inspired by Pearson's hard and soft acid–base (HSAB) principle with Cu2O nanocubes with different sizes (50 nm, 500 nm) as the sacrificial templates. A comparative study of the electrocatalytic oxygen evolution reaction (OER) of the hollow M(OH)x nanocubes with a similar size indicates that Ni(OH)2 has better OER catalytic activity. It has been revealed that the metal oxyhydroxides formed at the surface are actually the real active species for the OER electrocatalysis. In particular, Ni(OH)2 nanocubes obtained by the Cu2O (50 nm) template provide the best OER activity, with a low overpotential of 349 mV vs. RHE to achieve a current density of 10 mA cm−2 and a low Tafel slope of 63 mV dec−1. The hollow metal hydroxide nanostructures through the Pearson's principle-inspired strategy can be highly efficient electrocatalysts for OER applications.
Introduction
The increasing environmental crisis and global energy demand have accelerated the search for clean and renewable energy carrier alternatives to fossil fuels. The electrolysis of water has been considered as a promising method for the production of hydrogen and oxygen.1,2 However, with sluggish kinetics due to the multistep proton-coupled electron transfer, the oxygen evolution reaction (OER) is always more challenging and thermodynamically and kinetically demanding.3–8 Currently, the most active OER catalysts remain the noble metal oxides (RuO2 and IrO2), but the high cost and scarcity of such catalysts have impeded their scale-up applications in energy conversion systems.9–11 Therefore, cost-effective and earth-abundant catalysts as possible alternatives to Ru/Ir-based compounds for OER are highly desired. Recent advances indicate that transition metal sulfides,12 phosphides,13,14 oxides,4,15,16 oxyhydroxides,17,18 and hydroxides19–21 are efficient in OER catalysis.
Among these catalysts, 3d metal hydroxides exhibit good OER performances because of their miscellaneous chemical compositions.22–25 In particular, Fe/Co/Ni-based hydroxides are promising OER electrocatalysts owing to their environmental friendliness, earth-abundance, and excellent catalytic activity.26 A great deal of effort has been devoted to the design and synthesis of cost-effective hydroxides (Fe/Co/Ni-based) through adjusting the morphology and exposed facets.27 Hollow materials have received tremendous interest due to their structural feature that more catalytically active sites may be exposed to accomplish excellent performance.28–30 It has been demonstrated that hollow cubes, when compared with hollow spheres, have an anisotropic shape with more sides and facets and thus better photocatalytic properties.27,31
Template-assisted synthesis plays an important role in the fabrication of hollow catalysts.32,33 Among these, a route inspired by Pearson's hard and soft acid–base (HSAB) principle has received more and more attention in preparing metal hydroxides and metal oxides with controllable size, morphology and properties, because the morphology and size are important for the catalytic activity.34–40 Inspired by the Pearson's HSAB principle, Guo et al. synthesized uniform amorphous Ni(OH)2 nanoboxes with undamaged shell structures and various sizes by selecting S2O32− as the coordinating etchant toward Cu2O templates, and the as-prepared Ni(OH)2 nanoboxes demonstrated an improved electrochemical sensing ability for glucose.41 Recently, hollow Co(OH)2 cubes were prepared via a template-assisted route with Cu2O as the sacrificial template for further conversion into hollow CdS/Co9S8 cubes for photocatalytic water splitting.42 These studies offer a new route to prepare amorphous transition metal hydroxides with enhanced ionic conductivity as well as robust electrochemical and mechanical stability,43 which may be more feasible for electrocatalytic applications.36
In this work, we demonstrate the synthesis of M(OH)x (M = Fe, Co, Ni) hollow nanocubes using a template-assisted process with Cu2O nanocubes as the sacrificial templates (Fig. 1). With Cu2O nanocubes of different sizes, hollow M(OH)x cubic structures are obtained and their electrocatalytic activity towards the OER is systematically compared. It is found that hollow Ni(OH)2 nanocubes prepared from 50 nm Cu2O display preferable OER activity, with a low overpotential of 349 mV vs. RHE to achieve a current density of 10 mA cm−2. This work provides a new avenue for the synthesis of hollow transition metal hydroxides for energy conversion applications.
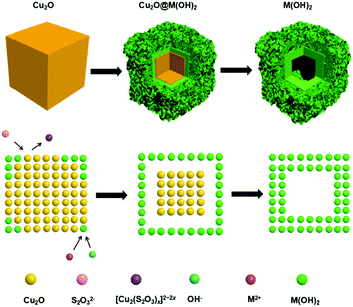 |
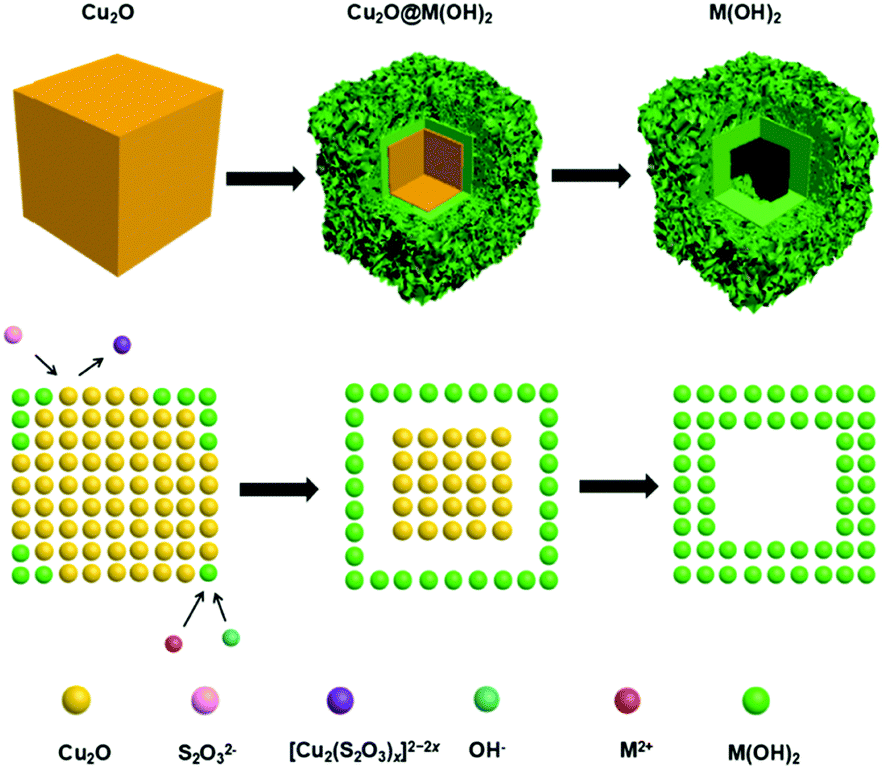
| | Fig. 1 Schematic illustration of the morphology evolution of M(OH)x (M = Fe, Co, Ni) hollow nanocubes prepared through the Pearson's principle-inspired strategy and using Cu2O nanocubes as the sacrificial templates. | |
Experimental
Preparation of Cu2O nanocubes (50 nm)
Cu2O nanocubes with an average size of 50 nm were synthesized using a modified reductive solution chemistry route.44 In a typical procedure, 0.5 g of poly(ethylene glycol) (PEG, MW: 2000) was first dissolved in 10 mL of Cu(Ac)2 aqueous solution (0.1 mM) under magnetic stirring. Once PEG was completely dissolved, 50 μL of NaOH solution (6.0 M) was added dropwise. Upon addition, the solution immediately changed to blue color, indicating the formation of Cu(OH)2 precursors. After 10 min, 0.2 mL of ascorbic acid (AA) solution (1 M) was added dropwise to the solution, where the solution slowly turned into orange color. The products were collected by centrifugation after a reaction time of 30 min by repeatedly rinsing with deionized water and ethanol in order to minimize the surface adsorbed PEG molecules.
Preparation of Cu2O nanocubes (500 nm)
500 nm Cu2O nanocubes were prepared following a reported procedure with slight modifications.45,46 In a typical procedure, 0.087 g of sodium dodecyl sulfate (SDS) surfactant was first dissolved in 9.6 mL of 0.1 mM CuCl2 solution. Then, 0.25 mL of 1 M NaOH solution was added and kept stirring for 10 min. Blue Cu(OH)2 colloidal particles were formed. Next, 0.15 mL of 0.2 M NH2OH·HCl solution was added and the vial was stirred for 20 min. The solution gradually turned from light blue to orange, indicating the formation of Cu2O. After ageing for 2 h, Cu2O nanocrystals were collected by centrifugation and repeatedly rinsed with deionized water and ethanol.
Preparation of hollow M(OH)x (M = Fe, Co, Ni) nanocubes
Hollow transition metal hydroxides, M(OH)x (M = Fe, Co, Ni), were synthesized using a template-assisted process.41 In a typical procedure, the as-prepared Cu2O nanocubes (5 mg) and MCl2·yH2O (1.7 mg) were ultrasonically dissolved in 10 mL of ethanol/water mixture solvent (volume ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with the existence of PVP (0.33 g, Mw = 40000). Then, 4.0 mL of Na2S2O3 aqueous solution (1 M) was added dropwise to the above solution. After continuous stirring for 10 min, M(OH)x was collected by centrifuging and rinsing with deionized water and ethanol. Zn(OH)2 and Mn(OH)2 hollow nanocubes were also prepared using the same procedure. Notably, hollow Fe(OH)3 nanocubes were synthesized with FeCl2·4H2O (2.1 mg) and 1 mL of Na2S2O3 aqueous solution via a similar process using only deionized water as a solvent.
:1) with the existence of PVP (0.33 g, Mw = 40000). Then, 4.0 mL of Na2S2O3 aqueous solution (1 M) was added dropwise to the above solution. After continuous stirring for 10 min, M(OH)x was collected by centrifuging and rinsing with deionized water and ethanol. Zn(OH)2 and Mn(OH)2 hollow nanocubes were also prepared using the same procedure. Notably, hollow Fe(OH)3 nanocubes were synthesized with FeCl2·4H2O (2.1 mg) and 1 mL of Na2S2O3 aqueous solution via a similar process using only deionized water as a solvent.
Characterization
Powder X-ray diffraction (XRD) patterns were recorded on a Rigaku D/max 2500 X-ray diffractometer using Cu Kα radiation (λ = 1.5406 Å). X-ray photoelectron spectra (XPS) were obtained on a PHI 5700 ESCA System spectrometer using an Al Kα excitation source. Raman spectra were collected on a Renishaw inVia confocal micro-Raman spectroscopy system using a TE air-cooled 576 × 400 CCD array with a 532 nm excitation laser. Transmission electron microscopy (TEM, Tecnai G2 F20) and scanning electron microscopy (SEM, FEI Quanta 200F) were applied to observe the particle size and morphology.
Electrochemical measurements
All electrochemical measurements were performed on a CHI-660D electrochemical workstation in 1 M KOH solution employing a three-electrode configuration on a rotating disk electrode (RDE) operating at 800 rpm. A graphite rod was used as the counter electrode, an Hg/HgO (in 1 M KOH solution) electrode as the reference electrode, and a glassy carbon electrode (GCE, 5 mm in diameter) as the working electrode. In order to prepare catalyst ink, the hollow nanocubes (2 mg) were dispersed in an ethanol/water mixture solvent (0.4 mL) with 2.5 μL of Nafion and sonicated for about 20 min to obtain a uniform dispersion. Then, 7.5 μL of the ink was drop-cast on the GCE, with a catalyst loading of 0.19 ± 0.01 mg cm−2. Linear sweep voltammetry (LSV) was carried out with a scan rate of 5 mV s−1. Cyclic voltammetry (CV) was carried out at various scan rates in order to obtain the double layer capacitance (Cdl). Electrochemical impedance spectroscopy (EIS) was measured in the frequency ranging from 106 to 0.1 Hz. To better compare the true catalytic activity of the different catalysts, the series resistance determined from EIS experiments was used to correct the polarization measurements and subsequent Tafel analysis for the iR losses. All potentials were referenced to the reversible hydrogen electrode (RHE).
Results and discussion
The successful preparation of Cu2O nanocubes (50 nm) and Cu2O nanocubes (500 nm) can be reflected by the TEM and XRD studies (Fig. S1 and S2 in the ESI†). The reaction between Cu2O and M2+ (M = Fe, Co, Ni) involves a Pearson's hard and soft acid–base principle, namely hard Lewis acids form stable complexes with hard bases, whereas soft acids prefer soft bases.47 Hence, choosing a soft base ligand (S2O32−, CN−, SCN−, etc.) as the coordinating etchant should be more suitable than a hard base (Cl−, NH3, etc.) due to the soft acid feature of Cu+ within the Cu2O templates. In this work, we employ S2O32− as the coordinating etchant and the processes can be described as shown in Fig. 1. The general chemical processes could be elaborated as follows:| | | Cu2O + xS2O32− + H2O → [Cu2(S2O3)x]2−2x + 2OH− | (1) |
| | | S2O32− + H2O → HS2O3− + OH− | (2) |
In the above processes, S2O32− introduced in the reaction system plays miscellaneous roles for the synthesis of M(OH)x (M = Fe, Co, Ni) hollow nanocubes. On one hand, coordinating etching of Cu2O (eqn (1)) occurs, and a soluble [Cu2(S2O3)x]2−2x complex forms subsequently, since the soft–soft interaction of Cu+–S2O32− is stronger than the soft–hard interaction of Cu+–O2− within Cu2O. On the other hand, the OH− released from the etching of Cu2O (eqn (1)) and the hydrolysis of S2O32− (eqn (2)) can facilitate the formation of M(OH)x (eqn (3)), adhering to the surface of unreacted Cu2O nanocubes. As the reaction proceeds, M(OH)x starts forming synchronously and the shell structure prefers to form when the ion concentration reaches sedimentary conditions. Therefore, Na2S2O3 first etches the outside surface of Cu2O rather than the interior part of the Cu2O in this synthesis process,35,37,48 which can be proved by a time-dependent TEM study of the intermediates (Fig. S3 in the ESI†). When the shell is formed, further reaction will lead to a topographic transformation from solid to hollow nanocubes, which can be proven by SEM (Fig. S4 in the ESI†) and TEM studies (Fig. 2). It should be noted that the morphology of the hollow hydroxide nanocubes is dependent on the nature of the metal ions. The iron hydroxide nanocubes are composed of solid nanoframes with a hollow interior part (Fig. 2a and d). The shell of the hollow cobalt hydroxide nanocubes appears to be assembled by numerous loose nanosheets (Fig. 2b and e). While, the frame of the hollow nickel hydroxide nanocubes is decorated with small nanoparticles (Fig. 2c and f). A selected area electron diffraction (SAED) study (insets in Fig. 2) implies that the as-prepared M(OH)x hollow nanocubes are all in an amorphous structure.
 |
| | Fig. 2 TEM and SAED (inset) images of the as-prepared (a) Fe(OH)3 (500 nm), (b) Co(OH)2 (500 nm), (c) Ni(OH)2 (500 nm), (d) Fe(OH)3 (50 nm), (e) Co(OH)2 (50 nm), and (f) Ni(OH)2 (50 nm) hollow nanocubes. | |
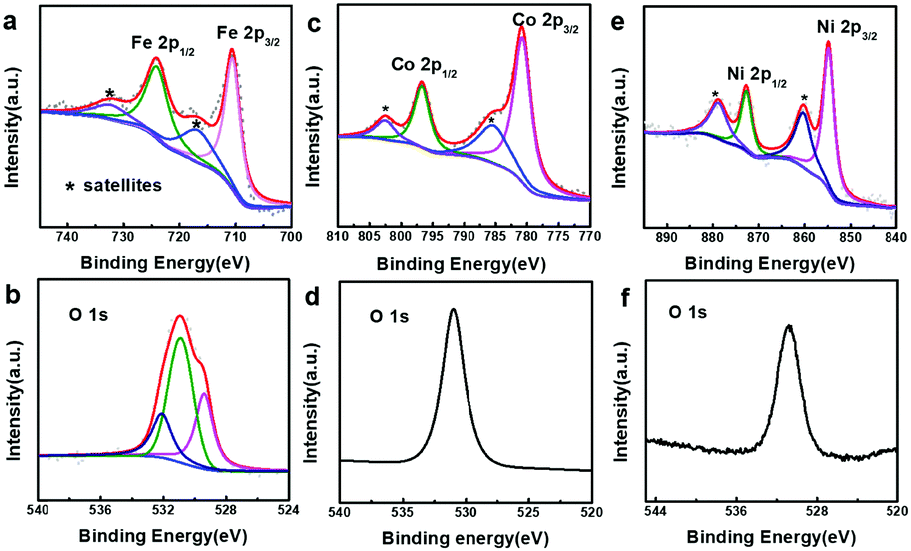
In order to better understand the composition of the products, X-ray photoelectron spectroscopy (XPS) was carried out (Fig. 3). The strong peaks at 712.4 and 726.1 eV in the Fe 2p XPS spectrum (Fig. 3a) correspond to Fe 2p3/2 and Fe 2p1/2, respectively, whereas the satellite peaks can also be observed around 717.9 and 734.0 eV, indicating the formation of Fe(OH)3.49–51 This reveals that the produced Fe(OH)2 can be oxidized into Fe(OH)3 under our experimental conditions.37 Actually, hollow Fe(OH)3 nanocubes could also be obtained by using FeCl3 (Fig. S5 in the ESI†). The Co 2p XPS spectrum (Fig. 3c) exhibits two obvious peaks centred at 780.7 and 796.8 eV associated with Co 2p3/2 and Co 2p1/2, respectively, whereas the binding energies at 785.5 and 802.6 eV can be assigned to satellite peaks, suggesting the presence of Co(OH)2.52–54 The strong peaks at 855.4 and 872.8 eV in the Ni 2p XPS spectrum (Fig. 3e) correspond to Ni 2p3/2 and Ni 2p1/2, respectively, and the satellite peaks can be observed around 860.6 and 879.0 eV, indicating the production of Ni(OH)2. The single peak (Fig. 3b, d and f) at ∼530.8 eV in the O 1s XPS spectra corresponds to OH−, confirming the formation of Fe(OH)3, Co(OH)2 and Ni(OH)2.55,56 The XPS data of the 500 nm hydroxides are similar to those of their 50 nm counterparts (Fig. S6 in the ESI†). A Raman study (Fig. S7 in the ESI†) of the as-prepared samples also indicates the formation of Fe(OH)3 (243, 384, 553 and 692 cm−1), Co(OH)2 (462 and 523 cm−1) and Ni(OH)2 (534 cm−1).57,58 However, an X-ray powder diffraction (XRD) study implies that the as-prepared M(OH)x hollow nanocubes are amorphous (Fig. S8 in the ESI†), agreeing well with the HR-TEM and SAED results (Fig. S9 in the ESI†). Full range XPS survey spectra reveal that pure M(OH)x materials are formed without any impurities under the present process with Cu2O as the sacrificial templates (Fig. S10 in the ESI†). Notably, hollow Mn(OH)2 and Zn(OH)2 nanocubes could also be synthesized using this template-assisted route (Fig. S11 and S12 in the ESI†), while they are excluded in the following electrochemical performance investigations as they are generally not active for OER applications.
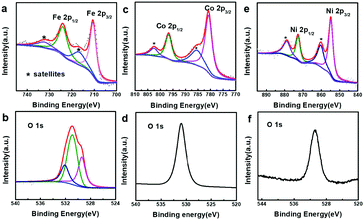 |
| | Fig. 3 XPS spectra of the as-prepared samples. (a) Fe 2p and (b) O 1s of iron hydroxide (50 nm), (c) Co 2p and (d) O 1s of cobalt hydroxide (50 nm), and (e) Ni 2p and (f) O 1s of nickel hydroxide (50 nm). | |
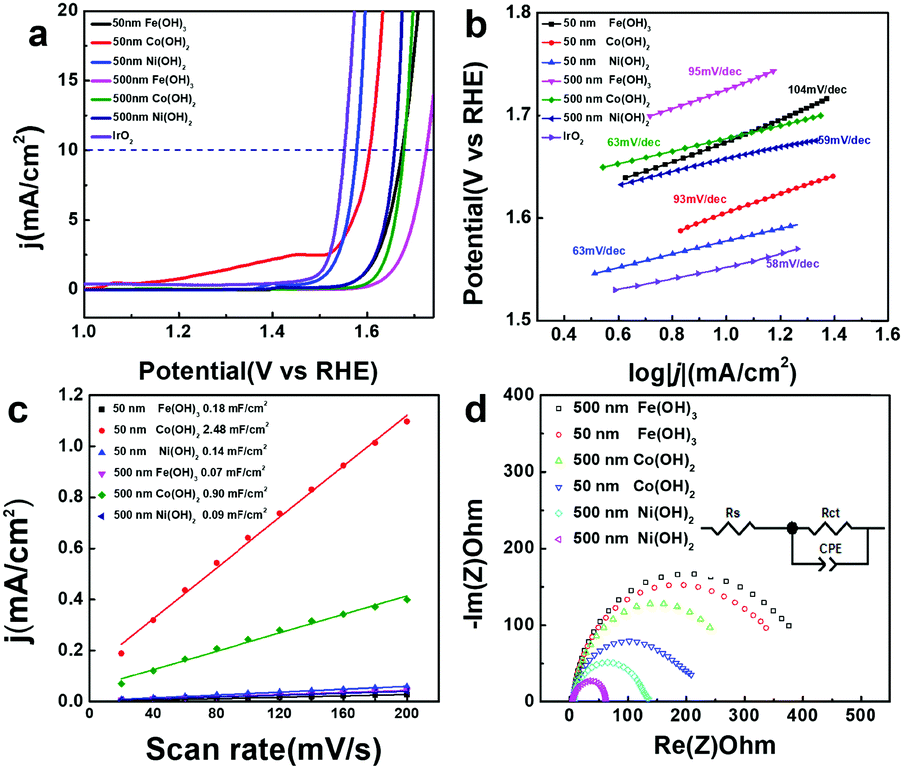
Transition metal hydroxides have been reported to be active in OER catalysis. Here, the electrocatalytic OER activity of all the M(OH)x materials and commercial IrO2 were compared in 1 M KOH using a rotating disk electrode (RDE) at a rate of 800 rpm. As shown in Fig. 4a, hollow Fe(OH)3 (500 nm), Co(OH)2 (500 nm) and Ni(OH)2 (500 nm) nanocubes achieve a current density of 10 mA cm−2 at high overpotentials of 495, 447 and 428 mV vs. RHE, respectively. In contrast, with smaller sizes, hollow Fe(OH)3 (50 nm), Co(OH)2 (50 nm) and Ni(OH)2 (50 nm) nanocubes display higher activity for the OER process, with lower overpotentials of 444, 375 and 349 mV vs. RHE at a current density of 10 mA cm−2, respectively. This suggests that the size of the catalysts has a huge impact on their OER electrocatalytic activity. With a similar size, the electrocatalytic activity of Ni(OH)2 is better than that of Co(OH)2 and Fe(OH)3, which is consistent with previous reports.21,59 A theoretical study also shows that for the various coordinated transition metal sites, the OER activity follows the order of Ni > Co > Fe when comparing the Gibbs free energy for the evolution of oxygen.60 We have compared the OER activity of Ni(OH)2 (50 nm) hollow nanocubes with recently reported transition metal-based OER electrocatalysts, and it can be seen that the hollow Ni(OH)2 (50 nm) has competitive activity in terms of overpotential and Tafel slope (Table S1 in the ESI†).61–63 From the extrapolation of the linear region of overpotential (η) vs. logj (Fig. 4b), Tafel slopes of 95, 63, 59, 104, 93, and 63 mV dec−1 (after iR correction) can be obtained for Fe(OH)3 (500 nm), Co(OH)2 (500 nm), Ni(OH)2 (500 nm), Fe(OH)3 (50 nm), Co(OH)2 (50 nm) and Ni(OH)2 (50 nm), respectively. A low Tafel slope value of 63 mV dec−1 for Ni(OH)2 (50 nm) suggests greatly efficient kinetics of O2 evolution. Double-layer capacitance (Cdl) is deduced from the cyclic voltammetry (CV) curves to evaluate the electrochemically active surface area (ECSA) during the OER catalysis (Fig. S13 in the ESI†). Capacitive current was plotted as a function of scan rate to extract the Cdl values (Fig. 4c) to be 0.07, 0.90, 0.09, 0.11, 2.49, and 0.14 mF cm−2 for Fe(OH)3 (500 nm), Co(OH)2 (500 nm), Ni(OH)2 (500 nm), Fe(OH)3 (50 nm), Co(OH)2 (50 nm) and Ni(OH)2 (50 nm), respectively. Though the hollow Co(OH)2 nanocubes assembled by numerous nanosheets have higher ECSA, it does not necessarily guarantee improved OER activity. Moreover, electrochemical impedance spectroscopy (EIS) is carried out to provide further insight into the electrode kinetics during the OER catalysis. The Nyquist plots (Fig. 4d) were fitted using an equivalent circuit (inset in Fig. 4c) to extract the charge transfer resistance (Rct) of 373.1, 332.0, 236.2, 202.9, 127.6 and 56.4 Ω for Fe(OH)3 (500 nm), Co(OH)2 (500 nm), Ni(OH)2 (500 nm), Fe(OH)3 (50 nm), Co(OH)2 (50 nm) and Ni(OH)2 (50 nm), respectively. Ni(OH)2 (50 nm) has a relatively low Rct of 56.4 Ω, implying a facilitated charge transfer process during the OER process. The above results indicate that for the same kind of hollow metal hydroxide, a smaller size can lead to higher ECSA and lower Rct, which may explain the better electrocatalytic OER activity obtained from the hollow metal hydroxide with a smaller size. Electrochemical stability is also important for the electrocatalysts in practical applications. All of the catalysts only exhibit slightly apparent degradation in terms of overpotential after 1000 CV cycles (Fig. S14 in the ESI†), and the hollow nanocube structures of these metal hydroxides can also be well maintained (Fig. S15 in the ESI†). Potentiostatic current–time curves display unobvious decay in the electrocatalytic current density for a time period of 12 h for all the samples (Fig. S16 in the ESI†). Therefore, the as-prepared amorphous hollow metal hydroxide nanocubes can be promising OER electrocatalysts with high durability.
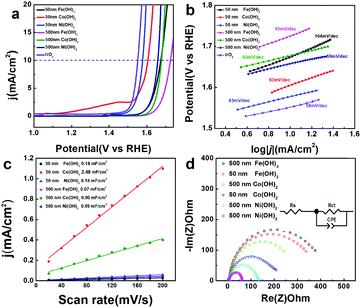 |
| | Fig. 4 Electrochemical properties of various hollow metal hydroxide nanocubes toward the OER in 1 M KOH solution. (a) J–V curves after iR correction in comparison to an IrO2 commercial catalyst, (b) Tafel plots, (c) plots showing the extraction of the Cdl values, and (d) Nyquist plots (inset shows the equivalent circuit). | |
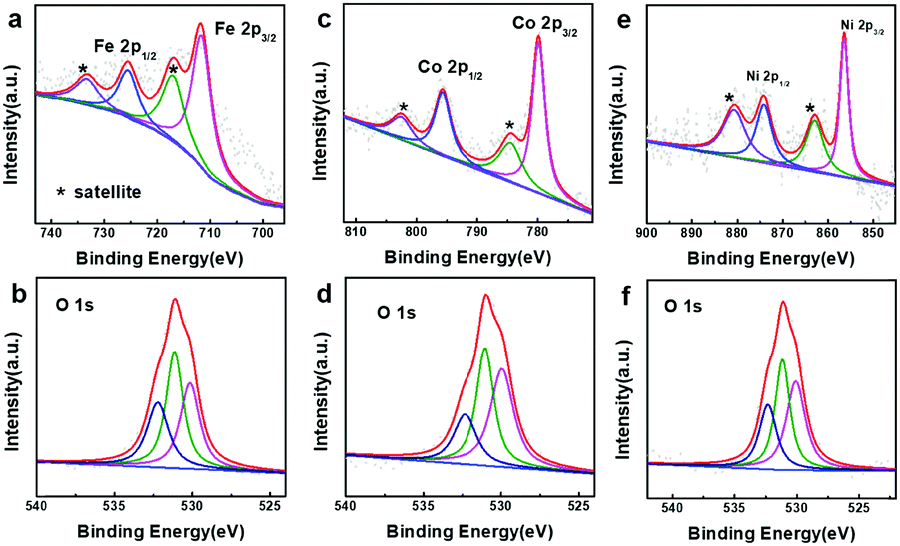
To probe the active species for OER catalysis, XPS spectra of all the samples after the OER measurements were collected (Fig. 5 and Fig. S17 in the ESI†). In Fig. 5a, main peaks at 711.2 and 725.1 eV together with satellite peaks at 718.4 and 733.3 eV in the Fe 2p XPS spectrum can be assigned to Fe3+.64,65 The main peaks at 779.9 and 796.1 eV and satellite peaks at 784.4 and 802.4 eV in the Co 2p XPS spectrum are ascribed to Co3+ (Fig. 5c),53,66 and the main peaks at 856.3 and 873.9 eV and satellite peaks at 862.5 and 880.7 eV are due to Ni3+ (Fig. 5e).67–69 The peaks at 530.0, 531.1 and 532.6 eV in the O 1s XPS spectra should be assigned to “O2−” and “OH−” (Fig. 5b, d and f).64,65 The results above clearly suggest that the surface of the metal hydroxides was transformed into oxyhydroxide layers, which are the real active species for the OER catalysis.
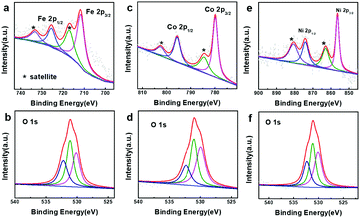 |
| | Fig. 5 XPS spectra of the as-prepared samples after the OER process. (a) Fe 2p and (b) O 1s of iron hydroxide (50 nm), (c) Co 2p and (d) O 1s cobalt hydroxide (50 nm), and (e) Ni 2p and (f) O 1s of nickel hydroxide (50 nm). | |
Conclusions
In conclusion, we demonstrate the Pearson's principle-inspired strategy for the fabrication of amorphous hollow M(OH)x (M = Fe, Co, Ni) nanocubes using Cu2O nanocubes with different sizes (50 nm, 500 nm) as the sacrificial template. Hollow M(OH)x nanocubes have a similar size to the applied Cu2O precursor, while the surface morphology is dependent on the nature of the metal ions. A comparative study of the electrocatalytic oxygen evolution reaction (OER) of the three hollow M(OH)x nanocubes with a similar size reveals that Ni(OH)2 has better activity in terms of overpotential and Tafel slope. Among all the samples, Ni(OH)2 nanocubes obtained by the template-assisted route with Cu2O (50 nm) show the best OER activity, with a low overpotential of 349 mV vs. RHE to achieve a current density of 10 mA cm−2 and a low Tafel slope of 63 mV dec−1. An XPS study after the OER process suggests the formation of a metal oxyhydroxide surface layer as the real active species for the OER catalysis. This template-assisted synthesis of hollow nanostructures may provide a new platform for fabricating highly efficient electrocatalysts for energy conversion and storage applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant No. 21471039, 21571043, and 21671047), the Fundamental Research Funds for the Central Universities (PIRS of HIT A201502 and HIT. BRETIII. 201223), the China Postdoctoral Science Foundation (2014M560253), the Postdoctoral Scientific Research Fund of Heilongjiang Province (LBH-Q14062, LBH-Z14076), and the Natural Science Foundation of Heilongjiang Province (B2015001).
Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef PubMed.
- P. Z. Chen, K. Xu, Z. W. Fang, Y. Tong, J. C. Wu, X. L. Lu, X. Peng, H. Ding, C. Z. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 14710–14714 CrossRef PubMed.
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. G. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- J. X. Wang, C. Y. Yan, G. F. Cai, M. Q. Cui, A. L. S. Eh and P. S. Lee, Adv. Mater., 2016, 28, 4490 CrossRef PubMed.
- L. S. Xie, C. Tang, K. Y. Wang, G. Du, A. M. Asiri and X. P. Sun, Small, 2017, 13, 1613–6810 Search PubMed.
- Q. S. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef PubMed.
- W. X. Zhu, R. Zhang, F. L. Qu, A. M. Asiri and X. P. Sun, ChemCatChem, 2017, 9, 1721–1743 CrossRef.
- J. Jiang, Q. X. Liu, C. M. Zeng and L. H. Ai, J. Mater. Chem. A, 2017, 5, 16929–16935 RSC.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef PubMed.
- Y. Zhang, G. Jia, H. Wang, B. Ouyang, R. S. Rawat and H. J. Fan, Mater. Chem. Front., 2017, 1, 709–715 RSC.
- L. L. Feng, G. T. Yu, Y. Y. Wu, G. D. Li, H. Li, Y. H. Sun, T. Asefa, W. Chen and X. X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef PubMed.
- G. Zhang, G. C. Wang, Y. Liu, H. J. Liu, J. H. Qu and J. H. Li, J. Am. Chem. Soc., 2016, 138, 14686–14693 CrossRef PubMed.
- H. H. Duan, D. G. Li, Y. Tang, Y. He, S. F. Ji, R. Y. Wang, H. F. Lv, P. P. Lopes, A. P. Paulikas, H. Y. Li, S. X. Mao, C. M. Wang, N. M. Markovic, J. Li, V. R. Stamenkovic and Y. D. Li, J. Am. Chem. Soc., 2017, 139, 5494–5502 CrossRef PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef PubMed.
- B. Li, S. W. Chien, X. Ge, J. Chai, X. Y. Goh, K. T. Nai, T. S. Andy Hor, Z. Liu and Y. Zong, Mater. Chem. Front., 2017, 1, 677–682 RSC.
- M. Asnavandi and C. Zhao, Mater. Chem. Front., 2017, 1, 2541–2546 RSC.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef PubMed.
- C. Tang, H. S. Wang, H. F. Wang, Q. Zhang, G. L. Tian, J. Q. Nie and F. Wei, Adv. Mater., 2015, 27, 4516–4522 CrossRef PubMed.
- H. D. Yang, Y. Long, Y. Zhu, Z. M. Zhao, P. Ma, J. Jin and J. T. Ma, Green Chem., 2017, 19, 5809–5817 RSC.
- A. Balram, H. F. Zhang and S. Santhanagopalan, Mater. Chem. Front., 2017, 1, 2376–2382 RSC.
- M. Chhowalla, Z. F. Liu and H. Zhang, Chem. Soc. Rev., 2015, 44, 2584–2586 RSC.
- Y. Zhan, G. J. Du, S. L. Yang, C. H. Xu, M. H. Lu, Z. L. Liu and J. Y. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 12930–12936 CrossRef PubMed.
- Q. Fu and X. H. Bao, Chem. Soc. Rev., 2017, 46, 1842–1874 RSC.
- C. K. Ranaweera, C. Zhang, S. Bhoyate, P. K. Kahol, M. Ghimire, S. R. Mishra, F. Perez, B. K. Gupta and R. K. Gupta, Mater. Chem. Front., 2017, 1, 1580–1584 RSC.
- D. U. Lee, J. Y. Choi, K. Feng, H. W. Park and Z. W. Chen, Adv. Energy Mater., 2014, 4, 1614–6832 Search PubMed.
- J. L. Zheng, W. Zhou, Y. R. Ma, W. Cao, C. B. Wang and L. Guo, Chem. Commun., 2015, 51, 12863–12866 RSC.
- Z. Y. Wang, D. Y. Luan, C. M. Li, F. B. Su, S. Madhavi, F. Y. C. Boey and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 16271–16277 CrossRef PubMed.
- Z. Y. Wang, Z. C. Wang, H. B. Wu and X. W. Lou, Sci. Rep., 2013, 3, 2045–2322 CrossRef PubMed.
- X. J. Wang, J. Feng, Y. C. Bai, Q. Zhang and Y. D. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef PubMed.
- K. Kato, F. Dang, K. Mimura, Y. Kinemuchi, H. Imai, S. Wada, M. Osada, H. Haneda and M. Kuwabara, Adv. Powder Technol., 2014, 25, 1401–1414 CrossRef.
- H. Wang, S. Zhuo, Y. Liang, X. Liang and B. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9055–9059 CrossRef PubMed.
- Y. Xin, Y. Huang, K. Lin, Y. Yu and B. Zhang, Sci. Bull., 2018, 63, 601–608 CrossRef.
- Z. Wang and X. W. Lou, Adv. Mater., 2012, 24, 4124–4129 CrossRef PubMed.
- Z. Wang, D. Luan, F. Y. Boey and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 4738–4741 CrossRef PubMed.
- J. Nai, H. Yin, T. You, L. Zheng, J. Zhang, P. Wang, Z. Jin, Y. Tian, J. Liu, Z. Tang and L. Guo, Adv. Energy Mater., 2015, 5, 1401880 CrossRef.
- J. Nai, Y. Tian, X. Guan and L. Guo, J. Am. Chem. Soc., 2013, 135, 16082–16091 CrossRef PubMed.
- M. Waqas, Y. Wei, D. Mao, J. Qi, Y. Yang, B. Wang and D. Wang, Nano Res., 2017, 10, 3920–3928 CrossRef.
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
- J. Qi, K. Zhao, G. Li, Y. Gao, H. Zhao, R. Yu and Z. Tang, Nanoscale, 2014, 6, 4072–4077 RSC.
- J. W. Nai, S. Q. Wang, Y. Bai and L. Guo, Small, 2013, 9, 3147–3152 CrossRef PubMed.
- B. C. Qiu, Q. H. Zhu, M. M. Du, L. G. Fan, M. Y. Xing and J. L. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2684–2688 CrossRef PubMed.
- F. Tian, M. D. Radin and D. J. Siegel, Chem. Mater., 2014, 26, 2952–2959 CrossRef.
- Q. Li, P. Xu, B. Zhang, G. Wu, H. Zhao, E. Fu and H. L. Wang, Nanoscale, 2013, 5, 7397–7402 RSC.
- C. H. Kuo and M. H. Huang, J. Phys. Chem. C, 2008, 112, 18355–18360 CrossRef.
- C. H. Kuo, Y. T. Chu, Y. F. Song and M. H. Huang, Adv. Funct. Mater., 2011, 21, 792–797 CrossRef.
- R. G. Pearson, J. Am. Chem. Soc., 1985, 107, 6801–6806 CrossRef.
- J. Nai, S. Wang, Y. Bai and L. Guo, Small, 2013, 9, 3147–3152 CrossRef PubMed.
- Y. Ma, Y. Wang, D. Xie, Y. Gu, H. Zhang, G. Wang, Y. Zhang, H. Zhao and P. K. Wong, ACS Appl. Mater. Interfaces, 2018, 10, 6541–6551 CrossRef PubMed.
- C. Long, L. Jiang, T. Wei, J. Yan and Z. Fan, J. Mater. Chem. A, 2014, 2, 16678–16686 RSC.
- Y. Liu, Z. Jin, P. Li, X. Tian, X. Chen and D. Xiao, ChemElectroChem, 2018, 5, 593–597 CrossRef.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef.
- J. Yang, H. W. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef.
- X. Guo, R. M. Kong, X. Zhang, H. Du and F. Qu, ACS Catal., 2017, 8, 651–655 CrossRef.
- M. A. Peck and M. A. Langell, Chem. Mater., 2012, 24, 4483–4490 CrossRef.
- H. Yang, Y. Long, Y. Zhu, Z. Zhao, P. Ma, J. Jin and J. Ma, Green Chem., 2017, 19, 5809–5817 RSC.
- T. Shinagawa, M. T. K. Ng and K. Takanabe, Angew. Chem., Int. Ed., 2017, 56, 5061–5065 CrossRef PubMed.
- B. Wehling, P. Vandenabeele, L. Moens, R. Klockenkamper, A. von Bohlen, G. Van Hooydonk and M. de Reu, Mikrochim. Acta, 1999, 130, 253–260 CrossRef.
- F. Z. Sun, L. B. Li, G. Wang and Y. Q. Lin, J. Mater. Chem. A, 2017, 5, 6849–6859 RSC.
- G. Gao, S. Bottle and A. Du, Catal. Sci. Technol., 2018, 8, 996–1001 RSC.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef PubMed.
- Y. Liang, Y. Yu, Y. Huang, Y. Shi and B. Zhang, J. Mater. Chem. A, 2017, 5, 13336–13340 RSC.
- J. Zhang, Y. Hu, D. Liu, Y. Yu and B. Zhang, Adv. Sci., 2017, 4, 1600343 CrossRef PubMed.
- J. Baltrusaitis, D. M. Cwiertny and V. H. Grassian, Phys. Chem. Chem. Phys., 2007, 9, 5542–5554 RSC.
- C. L. Long, L. L. Jiang, T. Wei, J. Yan and Z. J. Fan, J. Mater. Chem. A, 2014, 2, 16678–16686 RSC.
- Z. Chen, C. X. Kronawitter, Y. W. Yeh, X. F. Yang, P. Zhao, N. Yao and B. E. Koel, J. Mater. Chem. A, 2017, 5, 842–850 RSC.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef.
- A. P. Grosvenor, M. C. Biesinger, R. S. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef.
- B. Song, K. Li, Y. Yin, T. Wu, L. N. Dang, M. Caban-Acevedo, J. C. Han, T. L. Gao, X. J. Wang, Z. H. Zhang, J. R. Schmidt, P. Xu and S. Jin, ACS Catal., 2017, 7, 8549–8557 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S17 and Table S1. See DOI: 10.1039/c8qm00170g |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Xijiang
Han
and
Ping
Xu
,
Xijiang
Han
and
Ping
Xu