
Directionally assembled MoS2 with significantly expanded interlayer spacing: a superior anode material for high-rate lithium-ion batteries†
Qilin
Wei‡
a,
Min-Rui
Gao‡
 b,
Yan
Li‡
c,
Dongtang
Zhang
a,
Siyu
Wu
a,
Zonghai
Chen
c and
Yugang
Sun
*a
b,
Yan
Li‡
c,
Dongtang
Zhang
a,
Siyu
Wu
a,
Zonghai
Chen
c and
Yugang
Sun
*a
aDepartment of Chemistry, Temple University, 1901 North 13th Street, Philadelphia, Pennsylvania 19122, USA. E-mail: ygsun@temple.edu
bCenter for Nanoscale Materials, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, Illinois 60439, USA
cChemical Sciences and Engineering Division, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, Illinois 60439, USA
First published on 16th April 2018
Abstract
Despite the great promise of molybdenum disulfide (MoS2) as a storage anode for rechargeable lithium-ion batteries, the corresponding rate capacity is limited by the low electrical conductivity and kinetic barriers linked to ion diffusion, which prevents MoS2 from being used in current electrical energy storage devices. A feasible strategy is reported to tackle these challenges by directionally growing MoS2 nanosheets with an expanded interlayer spacing (i.e., 9.4 Å versus 6.15 Å for pristine 2H-MoS2) on multiwalled carbon nanotubes (MWCNTs), which is performed through the microwave-assisted solvothermal reduction of ammonium tetrathiomolybdate [(NH4)2MoS4] in dimethylformamide in the presence of MWCNTs. The intimate interfaces between the MoS2 edges and the MWCNTs as well as the expanded interlayer gaps in MoS2 offer efficient and rapid pathways for ion and electron transport, which is favorable for improving the performance of batteries and enables the highest rate capability to be achieved. Batteries containing the as-synthesized MoS2/MWCNT anodes can constantly deliver a reversible capacity of 390 mA h g−1 at a current rate of up to 57.312 A g−1 (36C), describing the first class of MoS2-based anodes promising for high-rate use.
1. Introduction
Rechargeable lithium-ion batteries (LIBs) have been widely adopted for powering portable electronics, while current technologies remain inadequate for applications such as hybrid or electric vehicles, and electric transportation, in which high rate capability is required. Ideal electrode materials for these emerging applications should possess fast ion and electron transport kinetics in addition to high specific capacity, hence improving the energy storage at high charge/discharge rates. Common strategies explored to enhance ion and electron transport in active electrode materials include using nanostructures, which offer exceptionally short transport lengths and significantly reduced lithium ion diffusion time, and hybridizing them with electrically conductive additives such as carbon.1–3 Besides, for layered intercalation hosts, expanding the interlayer distance could also effectively improve the intrinsic ion diffusivity, owing to the decreased diffusion energy barrier, which has been verified theoretically4–6 and experimentally.5,7–10 However, no significant improvements in rate performance have been observed for the studied layer-structured materials.Recently, transition metal dichalcogenides with layered structures have aroused a lot of interest from the viewpoints of their versatile physics and chemistry as well as natural abundance.11–14 Among these materials, molybdenum disulfide (MoS2) is fundamentally and technologically interesting for a wide variety of applications including electrochemical energy storage,15–17 catalysis,18 chemical sensing,19,20 transistors,21 and photodetectors,22 due to its unique electronic/optoelectronic properties.11,12,15,18,19,21,22 The use of MoS2 as lithium ion storage electrode of LIBs is highly promising as it presents considerable advantages in terms of high rate capability, high capacity retention, small volumetric expansion and low cost.23 MoS2 crystallizes in a graphite-like lamellar structure that is coupled by weak van der Waals forces and readily allows ion intercalation chemistry. Considerable effort has been invested for the development of MoS2 for lithium storage, typically focused on synthesizing highly nanostructured materials to shorten the diffusion path for ions and electrons,8,24–28 and on combining MoS2 with carbon materials to increase its electrical conductivity.29–33 Other strategies for this purpose include introducing dipole molecules (e.g., water) to reduce the polarization of solvated ions, thereby boosting the ion diffusivity,5 or incorporating a suitable polymer such as poly-(ethylene oxide) (PEO) that possesses ion coordination properties to accelerate lithium ion transportation.34 Nevertheless, using MoS2-based materials as candidate electrodes for rapid charging and discharging is still not yet realized.8
In this article, a new class of anode material is reported, consisting of interlayer-expanded MoS2 nanosheets directionally anchored on multiwalled carbon nanotubes (denoted as IE-MoS2/MWCNTs), which enables high specific capacity, excellent capacity retention, satisfactory coulombic efficiency, and especially good rate capability. It exhibits a reversible charge/discharge capacity of 1442 mA h g−1 at 1.592 A g−1 (1C) and retains excellent capacity with a small polarization at a rate as high as 57.3 A g−1 (36C). The successful synthesis benefits from the rational promotion of the heterogeneous nucleation of MoS2 on the controlled oxidized MWCNTs through the microwave-assisted solvothermal reduction of (NH4)2MoS4 in dimethylformamide (DMF). Because of the simplicity, low energy consumption, and ease in scaling up, the method developed for synthesizing IE-MoS2/MWCNTs offers new opportunities for the rational design of electrode materials with enhanced conductivity and rate performances.
2. Experimental section
2.1 Chemical oxidation of MWCNTs
All chemicals were used as received without further purification. The controlled oxidation of MWCNTs was carried out according to the process reported elsewhere.35 In brief, 0.3 g of MWCNT powder was dispersed in a 25 mL aqueous solution formed by mixing sulphuric acid (H2SO4, 96 wt%) and hydrogen peroxide (H2O2, 30 wt%) (VH2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VH2O2 = 7:3) in a 100 mL round-bottom flask equipped with a condenser. The dispersion was constantly stirred for 5 h to completely disperse the MWCNTs. The resulting dispersion was then diluted with an appropriate amount of water, followed by filtration. The collected black solid was washed with water thoroughly until the pH of the washing water became neutral. The washed solid was then placed in a fume hood and dried for 2 days in the ambient environment. The dry solid was re-dispersed in DMF (anhydrous, 99.8%, Sigma-Aldrich) to form a dispersion with a concentration of ∼3 mg mL−1.
:VH2O2 = 7:3) in a 100 mL round-bottom flask equipped with a condenser. The dispersion was constantly stirred for 5 h to completely disperse the MWCNTs. The resulting dispersion was then diluted with an appropriate amount of water, followed by filtration. The collected black solid was washed with water thoroughly until the pH of the washing water became neutral. The washed solid was then placed in a fume hood and dried for 2 days in the ambient environment. The dry solid was re-dispersed in DMF (anhydrous, 99.8%, Sigma-Aldrich) to form a dispersion with a concentration of ∼3 mg mL−1.
2.2 Synthesis of IE-MoS2/MWCNTs
In a typical synthesis, 10 mg of (NH4)2MoS4 (99.97%, Sigma-Aldrich) was added to 6 mL of a DMF dispersion of oxidized MWCNTs, followed by vigorous magnetic stirring for 15 min under ambient conditions. The resulting solution was transferred into a 10 mL microwave reaction vessel, which was sealed and heated to 240 °C at a fast temperature ramp in a Monowave 300 microwave reactor (Anton Paar). The solution temperature was maintained for an appropriate time. The reaction solution was then cooled down to room temperature with pressurized nitrogen flow. The synthesized IE-MoS2/MWCNT product was collected via centrifugation at 6000 r.p.m. (revolutions per minute) for 5 min. After the removal of the colourless supernatant, the precipitate was washed with distilled water and absolute ethanol at least 4 times, followed by drying at 60 °C in an oven for 4 h. Elemental analysis indicated that the MoS2 loading in the IE-MoS2/MWCNT composite was 36 wt%. Since the DMF solution of (NH4)2MoS4 exhibited a brown colour, the colourless supernatant indicated that the synthesis yield reached approximately 100%.2.3 Characterization
The as-synthesized samples were examined by X-ray diffraction (XRD), which was carried out on a Bruker D2 Phaser X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) at 30 kV and a current of 10 mA. TEM and HRTEM images were recorded on a JEOL 2010F(s) transmission electron microscope operated at an acceleration voltage of 200 kV. The energy dispersive X-ray (EDX) spectra were collected with an INCA X-ray microanalysis system equipped on the JEOL 2010F(s) microscope. All the samples were prepared by drop-casting the ethanol suspensions of interesting nanostructures onto carbon-coated copper grids, followed by drying in a fume hood at room temperature. The synchrotron small-angle X-ray scattering (SAXS) measurement was performed at the beam line 12-ID-B of the Advanced Photon Source (APS), Argonne National Laboratory.2.4 Battery assembly and performance evaluation
In a typical process for fabricating working electrodes with the synthesized IE-MoS2/MWCNTs, 850 mg of IE-MoS2/MWCNTs was mixed with N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich), 50 mg of C45 (Timcal), and 100 mg of polyvinylidene (PVdF, Kureha KF-9300) to form a slurry. This slurry was then coated on Cu foil (60 cm × 70 cm) using a doctor blade, followed by drying in a vacuum oven set at 75 °C overnight to evaporate the NMP solvent. The loading of the IE-MoS2/MWCNTs for the electrode was ∼0.2 mg cm−2. Punching the film formed electrode discs with a size of 0.5 cm in diameter, which were then further dried under vacuum at 110 °C to remove any moisture trapped in the electrode. The 2032-type coin cells were assembled in an argon-filled glove box (O2, H2O <5 ppm) using the as-fabricated electrode discs as anodes and lithium foils as the counter electrodes (cathode). The electrodes were separated with a Celgard 2325 separator soaked with an electrolyte (Tomiyama Chemical Industry, Japan) consisting of 1.2 M LiPF6 in ethylene carbonate (EC):ethyl methyl carbonate (EMC) with a weight ratio of 3:7. Anodes made of pure IE-MoS2 nanostructures and MWCNTs were also fabricated and used to assemble coin cells by following the same procedure.
The assembled coin cells were all cycled on a Maccor cycler. They first went through an initial activation step at room temperature, which consisted of three cycles between 3 V and 0.0005 V with C/20 current. Then, the cells were aged within the same voltage range but with 1C rate to evaluate their cycling performance. The specific capacities were calculated based on the mass of the integrated IE-MoS2/MWCNTs including the mass of both MoS2 and MWCNTs. After either activation step or targeted 1C aged cycles, the cells were discharged to 0.9 V and held at that potential for more than 15 hours to wait for the AC impedance measurement. An EG&G 273A potentiostat and a Solartron SI1260 frequency response analyzer controlled by the ZPLOT measurement software were used. The impedance spectra were collected between 100 kHz and 10 mHz, with a 10 mV perturbation around the open-circuit voltage.
3. Results and discussion
3.1 Characterization of the IE-MoS2/MWCNT heterostructures
The IE-MoS2/MWCNT hybrid structures are prepared by microwave-heating a DMF solution of (NH4)2MoS4 in the presence of MWCNTs under solvothermal conditions. Fig. 1 highlights the major steps involved in the synthesis and the structure of the product. It is noteworthy that pure MWCNTs usually present poor wettability with foreign active materials. The MWCNTs are firstly oxidized with a mixture of H2SO4 (96 wt%) and H2O2 (30 wt%) to provide oxygen-containing groups35 that exhibit increased wettability towards the Mo precursor. The enhanced affinity promotes the heterogeneous nucleation of MoS2 upon reduction of MoS42− ions at high temperatures, leading to the formation of MoS2/MWCNT hybrid structures. Due to the increased reducing ability of DMF at elevated temperatures, the oxidized MWCNTs (Ox-MWCNTs) are reduced to MWCNTs by eliminating the oxygen-containing groups. The chemical conversion of the Ox-MWCNTs to MWCNTs stops the continuous nucleation of MoS2 on the Ox-MWCNTs. Therefore, the continuous reduction of MoS42− ions only grow the existing MoS2 nuclei into larger MoS2 nanosheets. The non-wettability between MoS2 and MWCNTs (i.e., hydrophilic MoS2versus hydrophobic MWCNTs) forces the growing MoS2 nanosheets to protrude out of the MWCNT surfaces, enabling a directional assembly of the as-grown MoS2 nanosheets on the outermost walls of the MWCNTs with the formation of intimate interfaces between the MoS2 edges and MWCNT walls. Such a directionally aligned growth of the MoS2 nanosheets on the MWCNTs has also been observed in the synthesis of MoS2/graphene composites.36,37 Similar to the synthesis in the absence of MWCNTs, the resulting MoS2 nanosheets on the MWCNTs also exhibit an expanded interlayer spacing of 9.4 Å (versus 6.15 Å for pristine 2H-MoS2).38 The large interlayer gap is mostly ascribed to the leftover species (e.g., DMF solvent molecules or NH4+ ions) of the synthesis reaction.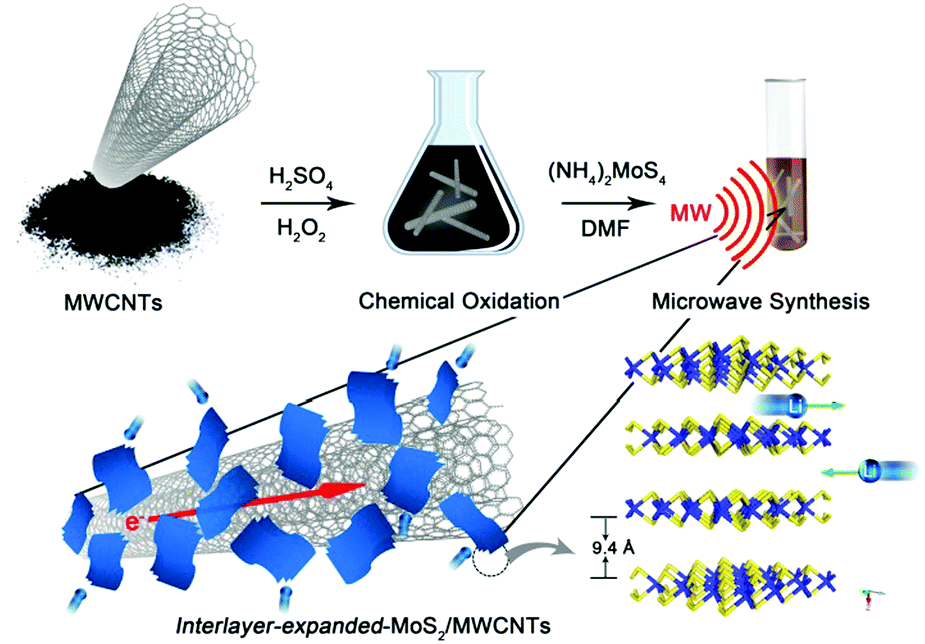 | ||
| Fig. 1 Schematic illustration of the synthesis of the IE-MoS2/MWCNTs through a microwave-assisted solvothermal reduction of (NH4)2MoS4 in DMF in the presence of oxidized MWCNTs. The resulting MoS2 nanosheets exhibit an expanded interlayer spacing of 9.4 Å and directionally anchor on the outermost walls of the MWCNTs, forming interfaces between the MoS2 edges and the MWCNT walls. | ||
Fig. 2 presents a typical sample of IE-MoS2/MWCNTs synthesized from a reaction performed at 240 °C for 100 min. The corresponding transmission electron microscopy (TEM) images (Fig. 2a–c) show the sheet-like MoS2 grown uniformly around each MWCNT. Each MoS2 nanosheet consists of only a few (3–5) S–Mo–S molecular layers and it links to the MWCNT through its edge rather than its basal surface. Most MoS2 nanosheets exhibit sizes less than 20 nm. Control experiments illustrate that using the MWCNTs without oxidation treatment leads to the failure of anchoring MoS2 (Fig. S1, ESI†); while in the absence of the MWCNTs, the exact same synthesis produces sphere-like assemblies of the MoS2 nanosheets, where homogeneous nucleation takes place (Fig. S2, ESI†). Independent from the nucleation process, such microwave chemistry always forms nanostructured MoS2 with an enlarged interlayer spacing of 9.4 Å, which is consistent with the high-resolution TEM (HRTEM) image as shown in Fig. 2c.
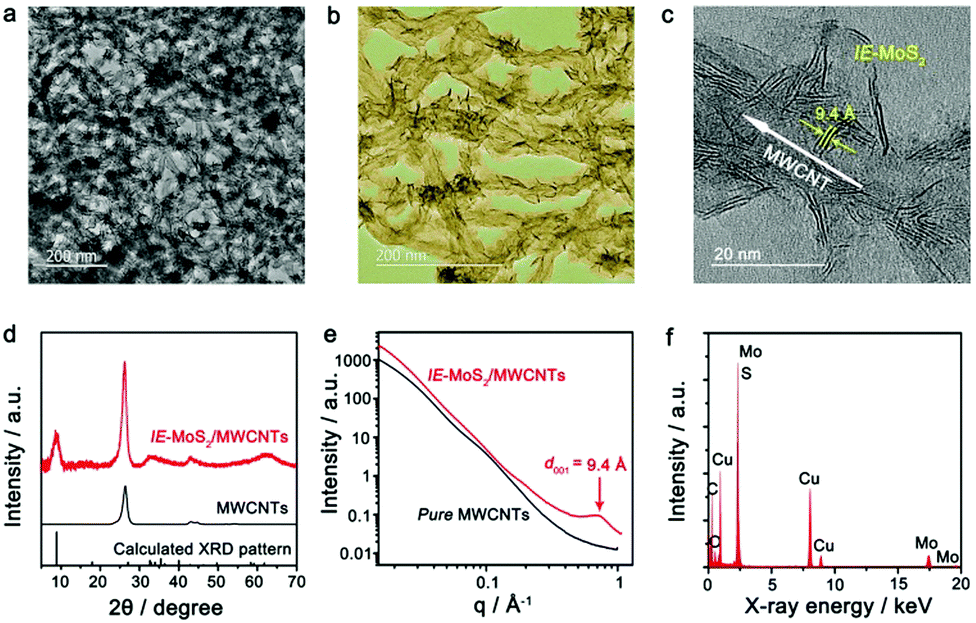 | ||
| Fig. 2 Characterization of the synthesized IE-MoS2/MWCNT hybrid structures. (a and b) Typical TEM images with different magnifications. (c) HRTEM image of a partial MWCNT decorated with IE-MoS2 nanosheets, showing characteristic features including thin thickness of the MoS2 nanosheets, an expanded MoS2 interlayer spacing of 9.4 Å, and interfaces between the MoS2 edges and the MWCNT wall. (d) Comparison of the XRD patterns of the IE-MoS2/MWCNTs (red) and MWCNTs (black), revealing the formation of MoS2 with an expanded interlayer spacing and the intactness of the MWCNTs in the course of the synthesis illustrated in Fig. 1. The sticks represent the theoretical peak positions and their relative intensities corresponding to MoS2 with an expanded interlayer spacing of 9.4 Å. (e) Comparison of synchrotron SAXS patterns of the IE-MoS2/MWCNTs (red) and the corresponding MWCNTs (black). (f) EDX spectrum of the IE-MoS2/MWCNTs. | ||
Fig. 2d shows the X-ray diffraction (XRD) pattern of the synthesized IE-MoS2/MWCNT product, in which the strong Bragg peak at 25.8° corresponds to the (002) reflections of the MWCNTs with a lattice spacing of 3.4 Å.39 The wide-angle reflections in a 2θ range of 30°–70° can be indexed to the hexagonal MoS2 (JCPDS 77-1716). The asymmetric nature of the reflections indicates the turbostratic feature of the grafted MoS2 nanosheets, presumably caused by, for example, the presence of stacking faults among S–Mo–S molecular layers as a result of a–b plane gliding.38,40 The symmetric peak at a low angle of 9.4° originates from the (002) reflections of the layer-structured MoS2 with an interlayer spacing of 9.4 Å, which is consistent with the HRTEM observation. Such an expanded interlayer spacing is also confirmed with the synchrotron small-angle X-ray scattering (SAXS) pattern, which displays a single scattering peak centered at q = 0.67 Å−1 (corresponding to 9.4 Å in d spacing determined from q = 2π/d) (Fig. 2e). In contrast, the corresponding MWCNTs do not show any characteristic SAXS peaks, further confirming the interlayer-expanded feature of the MoS2 nanosheets. Compared to the d spacing of 6.15 Å for pristine 2H-MoS2, about 53% expansion of the S–Mo–S interlayer distance is observed in the synthesized IE-MoS2/MWCNTs, which may facilitate the intercalation/diffusion of lithium ions.4,5 The energy-dispersive X-ray (EDX) spectrum (Fig. 2f) identifies Mo, S, and C as the major elemental components (Cu and part of the C signals emanating from the carbon-coated TEM grid), further demonstrating the formation of IE-MoS2/MWCNT hybrid structures.
3.2 Electrochemical performance of the IE-MoS2/MWCNT heterostructures
Lithium ion storage performance in the as-synthesized IE-MoS2/MWCNTs of Fig. 2 has been evaluated in 2032-type coin cells using lithium metal as the counter electrode. Each cell is initially discharged at a low rate of C/20 (20 h) to activate the electrode material along with storing lithium ions in IE-MoS2 (activation process). Fig. 3a presents cyclic voltammograms (CVs) of an IE-MoS2/MWCNT electrode conducted at a slow scan rate of 0.5 mV s−1. Two cathodic peaks at 0.99 and 0.45 V in the initial cycle can be attributed to lithium intercalation into the layered MoS2, leading to a possible transition from the trigonal prismatic 2H semiconducting phase to the octahedral 1T metallic phase (Fig. S3, ESI†),41,42 followed by a conversion reaction and eventually a deep conversion to a stoichiometrically equivalent Mo- and Li2S-rich composition, which is still a derivative of reduced MoS2 containing Li+ (e.g., Li3MoS2).29,32,42 The peakless current variation between 0.99 and 0.45 V is ascribed to the continuous insertion of lithium ions into the expanded MoS2 interlayer gaps in the course of discharge.43,44 The following anodic process shows two lithium extraction peaks at 1.76 and 2.29 V, which are related to the oxidation of intermediate conversion species (e.g., Li3MoS2 containing possible Mo and Li2S clusters, Li5Mo2S4).29,32,42,45 The subsequent CV profiles of the IE-MoS2/MWCNTs are consistent, showing the disappearance of the two peaks at 0.99 and 0.45 V that are observed in the first cathodic scan. Instead, two pairs of redox peaks show up: the reduction peak at 1.96 V corresponds to the reverse process of the oxidation peak at 2.29 V, highlighting the redox reaction between MoS2 and LiMo2S2 (the shallow insertion intermediate). The reduction peak at 1.23 V corresponds to the reverse process of the oxidation peak at 1.73 V, denoting the reduction/oxidation between the chemical conversion intermediates (e.g., Li3Mo2S4 and Li5Mo2O4) and the shallow insertion intermediate (LiMo2S2). The high consistency in the CV profiles indicates good anode stability and electrochemical reversibility of the synthesized IE-MoS2/MWCNTs. According to the density-functional theory (DFT) simulations, George et al. suggested the stable intermediates in the lithiation process including LiMo2S2 (shallow insertion of Li+), Li3Mo2S4 and Li5Mo2O4 (intermediate insertion of Li+ and chemical conversion), and Li3MoS2 with possible Li2S, Mo atomic chains, and MoLi alloy (deep insertion and chemical conversion).45 The lithium intercalation and conversion reaction increases the interlayer gap, while the integrity of the layered structure still remains. The calculation results indicate that the expanded interlayer spacing of MoS2 in the synthesized IE-MoS2/MWCNT material benefits the lithium intercalation and conversion reaction yet eliminates the structural failure of the layered MoS2 structure.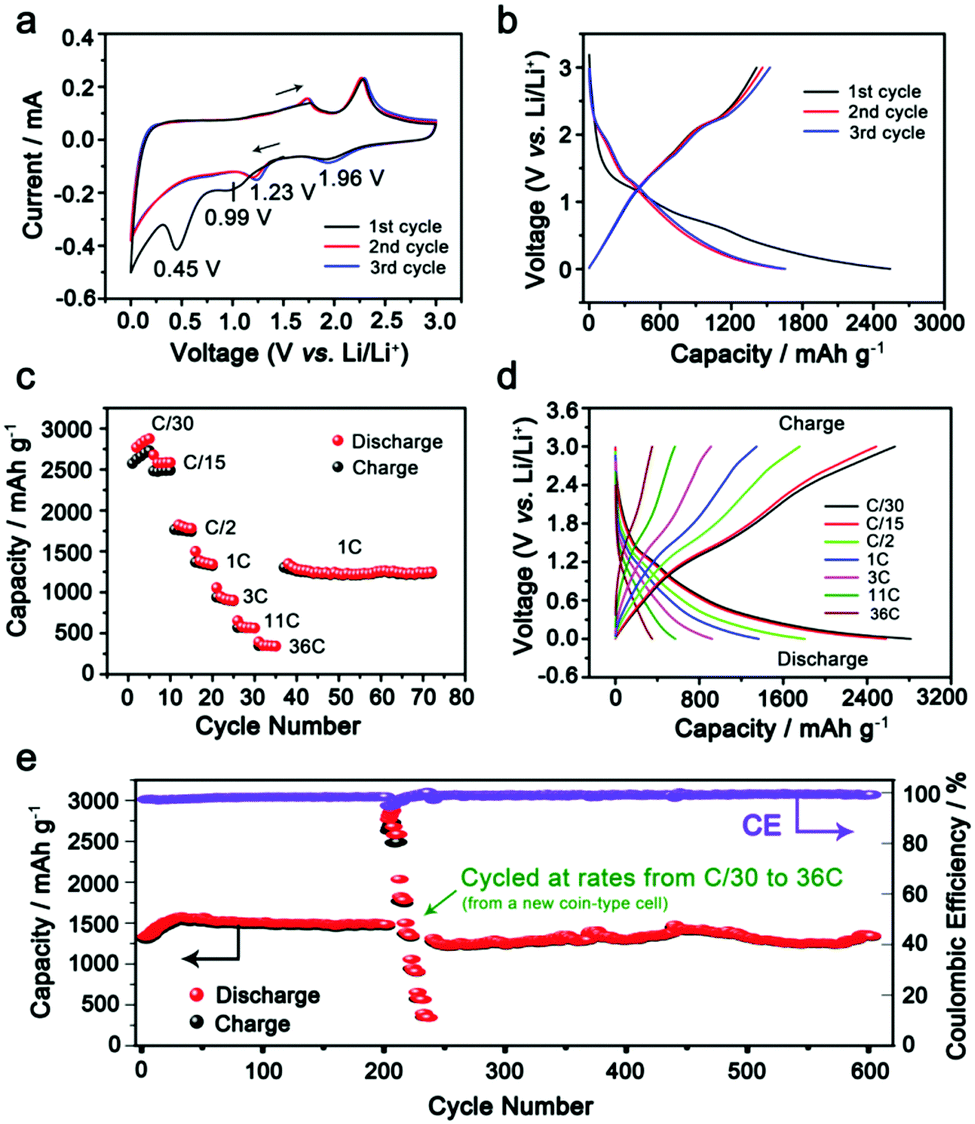 | ||
| Fig. 3 Electrochemical performance of the IE-MoS2/MWCNT anodes in lithium-ion batteries. (a) CV measurement of the IE-MoS2/MWCNTs for the first 3 cycles (i.e., the initial activation step). The sweep rate was 0.5 mV s−1 in a voltage range of 0–3 V versus Li/Li+. (b) Discharge–charge voltage profiles recorded in the first three cycles with the 1C rate. (c) Comparison of discharge/charge capacities of the IE-MoS2/MWCNT anode at various current rates from C/30 to 36C. The measurements were performed after the initial activation step. (d) Representative discharge–charge voltage profiles at various C rates. (e) Discharge/charge capacities and the corresponding coulombic efficiency measured over 600 cycles in the 0.0005–3 V range. | ||
Comparative CV studies on pure IE-MoS2 and MWCNTs show significant electrochemical differences from the IE-MoS2/MWCNTs (Fig. S4 and S5, ESI†), strongly suggesting that the synergistic effect between IE-MoS2 and MWCNTs may enable unique battery performance. Fig. 3b compares the voltage profiles of the IE-MoS2/MWCNTs at the initial cycles. For the first cycle, the IE-MoS2/MWCNT anode exhibits an impressive specific discharge capacity of 2538 mA h g−1 while the specific charge capacity drops to 1414 mA h g−1, which results in a low coulombic efficiency of ∼56%. The coulombic efficiency can rapidly reach 92% in the third cycle with the discharge and charge capacities of 1655 and 1524 mA h g−1, respectively. The difference in the CVs between the first scan and the following scans is consistent with the difference in the discharge curves between the first cycle and the second cycle, indicating that the initial reduction/discharge of the as-synthesized IE-MoS2/MWCNTs involves many side reactions such as reaction with solvent molecules adsorbed on the surfaces and trapped in the interlayer gaps, adsorbed high-oxidation-state species, and possible crystalline defects. The consistency of the profiles of CVs and charge/discharge curves in the 2nd and higher cycles implies that the initial cycle conditions the electrode materials to behave reversibly in electrochemical redox processes relying on the characteristic structure and properties of the IE-MoS2/MWCNTs (e.g., enlarged interlayer gap).
The rate capability of the IE-MoS2/MWCNTs has been assessed after the initial activation process. Fig. 3c exhibits the variation of the discharge capacity from 2686 mA h g−1 to 390 mA h g−1 at a discharge/charge rate varying from C/30 (0.053 A g−1) to 36C (57.312 A g−1). Furthermore, the capacity of 1442 mA h g−1 delivered at 1C (1.592 A g−1) can be recovered and remains stable for numerous cycles after fast-rate operation (Fig. 3c). At a super high current rate of 57.312 A g−1 (i.e., 36C), the electrode is still capable of delivering 390 mA h g−1 capacity in 24.5 s (Fig. 3d). Such high rate capacity value achieved for the IE-MoS2/MWCNTs is remarkable and is far above the documented data for MoS2-based anodes, in which rates higher than 10 A g−1 are barely reported.8Fig. 3e shows the stable capacity retention of the IE-MoS2/MWCNTs anode upon cycling at 1C (1.592 A g−1). An increase of the capacity observed in the initial 35 cycles is presumably caused by the delayed wetting of the electrolyte into the composite electrode. After the conditioning, it demonstrates a capacity of 1442 mA h g−1 with a coulombic efficiency of 98.4% and for a life extending 600 cycles with only 6.2% capacity decay (Fig. 3e). The battery performance of the IE-MoS2/MWCNTs is superior to that of pure IE-MoS2 and MWCNTs (Fig. S6 and S7, ESI†), and also outperforms the reported MoS2-based anode materials.
The electrode after being deeply cycled has been studied to gain insight into the excellent battery performance of the IE-MoS2/MWCNTs. The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image (Fig. 4a, left) reveals that the IE-MoS2/MWCNT hybrid structures still well remain after 200 fatigue cycles but with some extent of aggregation, which is likely due to the mixing with binders in the electrode preparation process. EDX elemental mapping over a typical area of the cycled electrode shows that the spatial distributions of Mo and S are completely the same while they are different from that of C (Fig. 4a, right), implying the high possibility that Mo and S atoms still bond together after cycling. The quantitative analysis of the corresponding EDX spectrum (Fig. 4b) reveals that the atomic ratio of Mo/S is ∼1:2, close to the stoichiometry of MoS2. The high-fidelity retention of spatial overlapping and stoichiometry of the Mo and S elements highlights that the long-time (200 cycles) discharge/charge operation does not completely break the original layered geometry of MoS2 to form segregated nanoparticles. The structural integrity and interlayer distance of the cycled sample can be further studied with TEM and XRD if an appropriate method is developed to prepare a clean sample to avoid the interferences of the electrolyte and the binder.36 The strong signal of the C element is also observed, indicating the good retention ability on electrode constituents. The F and P signals are caused by the residual LiPF6 electrolyte in the vicinity of the studied electrode material. The impedance change of the IE-MoS2/MWCNT electrode has been tracked before cycling and after 35 and 200 cycles. As shown in Fig. 4c, a tremendous impedance decrease can be observed for the cell after 35 cycles, which is attributed to the slow wetting of the electrolyte into the IE-MoS2/MWCNTs, and also to the exclusion of foreign adsorbates and crystalline defects in the initial discharge process. The decreased impedance agrees well with the capacity increase over the first 35 cycles, as observed in Fig. 3e. No significant impedance increase is observed after 200 cycles, indicative of the limited growth of the solid electrolyte interphase during aging,46 which further confirms the good cycling stability of the IE-MoS2/MWCNTs.
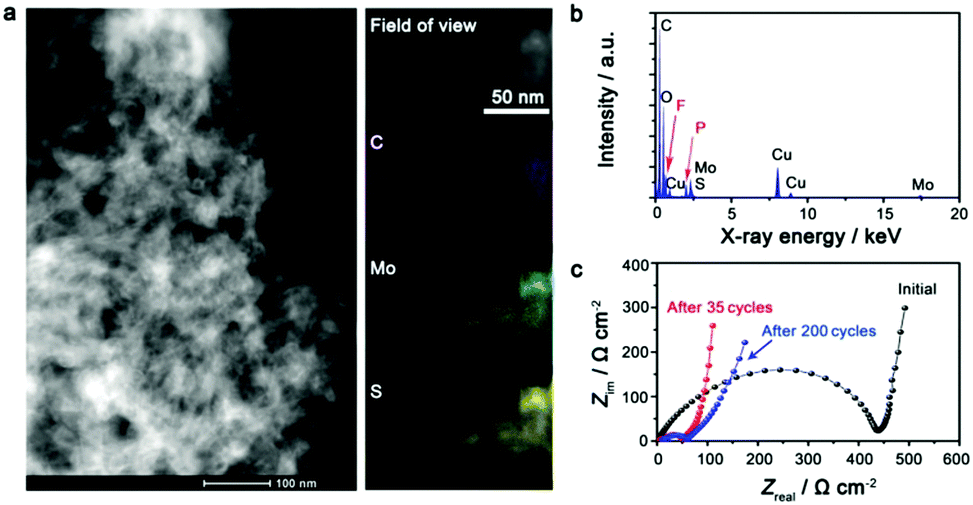 | ||
| Fig. 4 Characterization of the IE-MoS2/MWCNT electrode after discharge/charge cycles. (a) HAADF-STEM image (left) and elemental mapping (right) of the IE-MoS2/MWCNT electrode after 200 cycles. (b) EDX spectrum of the IE-MoS2/MWCNT electrode after 200 cycles. (c) Impedance measurements of the IE-MoS2/MWCNT electrode before (black) and after different numbers of cycles. | ||
3.3 Mechanism for high rate capability
Theoretically, the specific capacity of MoS2 is calculated to be ∼670 mA h g−1 while the value for the MWCNTs is ∼340 mA h g−1.23,30,41 The measured superior capacity of the IE-MoS2/MWCNTs is strongly indicative that a favorable synergistic effect exists in this composite electrode and thus affords capacity gain. Similar improved capacities have also been reported for other nanostructured MoS2-based composites, for example, MoS2 decorated carbon nanofibers,32 MoS2/polyaniline nanowires,42 MoS2/MoC/carbon nanotubes,30 MoS2/graphene29 and others. However, for the existing MoS2-based materials, there is rare published data available for a rate higher than 10 A g−1 (Fig. 5a), and the cycling stabilities are usually demonstrated below 200 cycles. Compared with the reported electrochemical data for the MoS2-based material (Fig. 5a), our IE-MoS2/MWCNTs offer a capacity of 390 mA h g−1 at a very high rate of 57.3 A g−1 (36C), while capacity loss is barely observed in the course of continuous charge/discharge cycles (Fig. 3c), which represents the highest rate capability of the MoS2-based materials.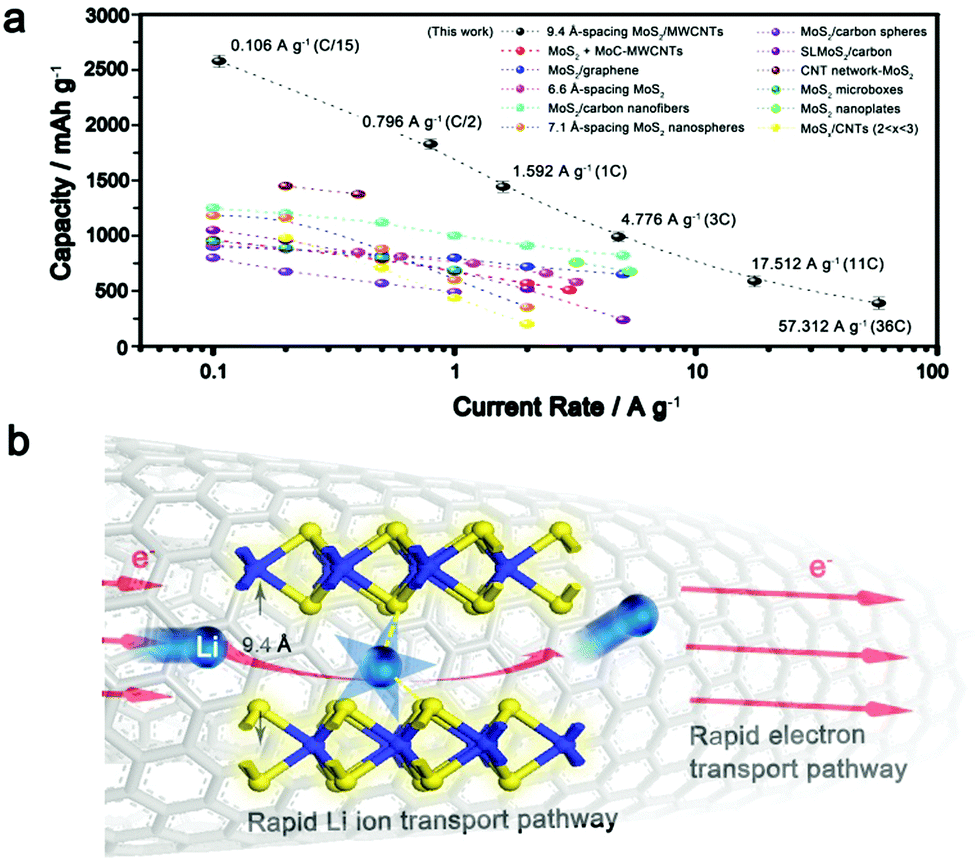 | ||
| Fig. 5 (a) Comparison of rate capability for various MoS2-based electrodes, including MoS2 + MoC-MWCNTs,30 MoS2/graphene,29 6.6 Å-spacing MoS2,10 MoS2/carbon nanofibers,32 7.1 Å-spacing MoS2 nanospheres,57 MoS2/carbon spheres,58 single layer MoS2/carbon,59 CNT network-MoS2,60 MoS2 nicroboxes,27 MoS2 nanoplates,8 and MoSx/CNTs (2 < x < 3).61 (b) Schematic of the plausible mechanism responsible for the enhanced rate performance of the synthesized IE-MoS2/MWCNTs. | ||
The substantially improved battery performance in terms of the high rate capability and capacity can be attributed to the highly conductive MWCNTs, the advantageous interlayer-expanded MoS2 structure, and the synergistic effect between them, which allow for enhancing the ion and electron transport kinetics in the battery. Carbon nanotubes permit nanoscale electrical wiring of active materials to their surfaces due to the striking percolation networks,47,48 creating good composite electrode materials with a high surface area and excellent electric conductivity. The high rate behavior is significantly benefited from the interlayer expansion of MoS2 grafted on the MWCNTs. As for the layered materials, the insertion and transport of lithium ions is mainly determined by the diffusion energy barrier formed between a polarizing cation and a negatively charged host lattice.5,49 For example, bulk MoS2 with an interlayer spacing of 6.2 Å exhibits a calculated diffusion barrier of ∼0.49 eV.4 Recent studies suggest that such slow kinetics can be circumvented by enlarging the interlayer spacing of MoS2,8,24,50 or by reducing dimensionality to the monolayer,4 thus facilitating the insertion and diffusion of lithium ions. Despite these successes, MoS2-based materials performing at high current rates are rarely demonstrated, owing to the lack of effective approaches to further expand the interlayer spacing that permits an optimized lithium–MoS2 interaction (reported values are often <6.9 Å).8,10,24,51–55Fig. 5b highlights how the high electrical conductivity of the MWCNTs and the expanded interlayer spacing of the MoS2 nanosheets cooperate to enable the fast lithium intercalation/diffusion and the following redox reaction involving electron transfer. The significantly expanded interlayer spacing (9.4 Å) facilities the insertion of lithium into the interlayer gaps in the grafted MoS2 nanosheets as well as the following lithium diffusion in the interlayer gaps.56 Once lithium diffuses to the appropriate sites, the redox reaction can occur between lithium and MoS2 and the involving electrons can quickly migrate through the individual S–Mo–S layers to the MoS2 edges interfaced with the MWCNTs, which efficiently guide the electrons to the collecting electrode. Due to the large interlayer gaps that allow all the MoS2 units to (fully) wet with the lithium ions, the molecular layer structure can be maintained even after the IE-MoS2/MWCNTs undergo a deep chemical conversion. In contrast, deep chemical conversion breaks the molecular layers of MoS2 to enable the lithium ions to diffuse to the buried MoS2 units of the MoS2 nanosheets with narrow interlayer gaps. The unique features of the synthesized IE-MoS2/MWCNTs enable outstanding rate capability that outperforms almost all the reported counterpart MoS2-based electrode materials.
4. Conclusion
In summary, we have demonstrated a high-performance anode material composed of MoS2 nanosheets with significantly expanded interlayer spacing, which are directionally assembled around highly percolated MWCNTs to exhibit very short lithium diffusion lengths as well as efficient and rapid pathways for electron and ion transport, boosting discharge/charge kinetics. This newly developed composite material displays remarkable capability at a high current rate even up to 57.3 A g−1 (36C) and is also very stable for over 600 cycles, representing the first MoS2-based material attempting towards high-rate charge and discharge use. These understandings shed light on the rational design of high-performance anode materials with superior rate capability that can be achieved by controlling the nucleation and growth kinetics of colloidal chemistry. The IE-MoS2/MWCNT hybrid structures exhibit major features completely different from the widely studied MoS2/rGO (reduced graphene oxide) composites.62–64 The MoS2/rGO composites usually form layer-by-layer stacking structures with the small-sized MoS2 layers embedded in the stacked rGO sheets, resulting in a difficulty in exposing the MoS2 edges to the electrolyte in batteries. Since the intercalation/deintercalation of ions in MoS2 is dominated by the diffusion of ions through the MoS2 edges into/out of the MoS2 interlayer gaps, embedding the MoS2 in stacked rGO dramatically reduces the diffusion rate of ions (i.e., lower rate capability of the corresponding batteries). In contrast, the IE-MoS2/MWCNT composite (Fig. 2) completely eliminates the disadvantages of the MoS2/rGO composites. The IE-MoS2 nanosheets extend out of the MWCNTs to fully expose the MoS2 edges to the electrolyte, facilitating the intercalation/deintercalation of ions in the MoS2 interlayer gaps and thus improving the rate capability of the corresponding batteries. The unique geometry of the IE-MoS2/MWCNT structure and the expanded interlayer spacing of IE-MoS2 can also benefit the high electrical conductivity in individual S–Mo–S molecular layers, through which the electrons can quickly transfer to the highly conductive MWCNTs, to lower the impedance of the corresponding anodes. It is worthy to note that the high surface area of the nanostructured IE-MoS2/MWCNT electrode materials may induce a non-negligible capacitive behaviour to make a contribution to the electrochemical energy storage capacity of the materials. The actual contribution of storage capacity associated with the lithium intercalation and conversion reaction may be smaller than the measured capacity (Fig. S8, ESI†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Use of the Center for Nanoscale Materials and the Advanced Photon Source (APS) was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS PubMed.
- H. Zhang, X. Yu and P. V. Braun, Nat. Nanotechnol., 2011, 6, 277 CrossRef CAS PubMed.
- Y. Li, D. Wu, Z. Zhou, C. R. Cabrera and Z. Chen, J. Phys. Chem. Lett., 2012, 3, 2221–2227 CrossRef CAS PubMed.
- Y. Liang, H. D. Yoo, Y. Li, J. Shuai, H. A. Calderon, F. C. Robles Hernandez, L. C. Grabow and Y. Yao, Nano Lett., 2015, 15, 2194–2202 CrossRef CAS PubMed.
- S. Yang, D. Li, T. Zhang, Z. Tao and J. Chen, J. Phys. Chem. C, 2012, 116, 1307–1312 CAS.
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, Angew. Chem., 2014, 126, 13008–13012 CrossRef.
- H. Hwang, H. Kim and J. Cho, Nano Lett., 2011, 11, 4826–4830 CrossRef CAS PubMed.
- Y. Li, Y. Liang, F. C. Robles Hernandez, H. Deog Yoo, Q. An and Y. Yao, Nano Energy, 2015, 15, 453–461 CrossRef CAS.
- H. Liu, D. Su, R. Zhou, B. Sun, G. Wang and S. Z. Qiao, Adv. Energy Mater., 2012, 2, 970–975 CrossRef CAS.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699 CrossRef CAS PubMed.
- K. Cao, T. Jin, L. Yang and L. Jiao, Mater. Chem. Front., 2017, 1, 2213–2242 RSC.
- M. Hafeez, L. Gan, A. Saleem Bhatti and T. Zhai, Mater. Chem. Front., 2017, 1, 1917–1932 RSC.
- G. Zhang, H. Liu, J. Qu and J. Li, Energy Environ. Sci., 2016, 9, 1190–1209 CAS.
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang, Y. Wang, P. Liu, Y. Zhang, Y. Wen, L. Gu, G. Ma, Z. Su, Z. Tang, H. Zhao and D. Wang, Nat. Energy, 2016, 1, 16072 CrossRef.
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., Int. Ed., 2013, 52, 6417–6420 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef CAS PubMed.
- Y. Huang, J. Guo, Y. Kang, Y. Ai and C. M. Li, Nanoscale, 2015, 7, 19358–19376 RSC.
- Y. Hu, Y. Huang, C. Tan, X. Zhang, Q. Lu, M. Sindoro, X. Huang, W. Huang, L. Wang and H. Zhang, Mater. Chem. Front., 2017, 1, 24–36 RSC.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147 CrossRef CAS PubMed.
- O. Lopez-Sanchez, D. Lembke, M. Kayci, A. Radenovic and A. Kis, Nat. Nanotechnol., 2013, 8, 497 CrossRef CAS PubMed.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 CAS.
- G. Du, Z. Guo, S. Wang, R. Zeng, Z. Chen and H. Liu, Chem. Commun., 2010, 46, 1106–1108 RSC.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- D. K. Nandi, U. K. Sen, D. Choudhury, S. Mitra and S. K. Sarkar, Electrochim. Acta, 2014, 146, 706–713 CrossRef CAS.
- L. Zhang, H. B. Wu, Y. Yan, X. Wang and X. W. Lou, Energy Environ. Sci., 2014, 7, 3302–3306 CAS.
- J. Wang, H. Tang, H. Wang, R. Yu and D. Wang, Mater. Chem. Front., 2017, 1, 414–430 RSC.
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720–4728 CrossRef CAS PubMed.
- X. Li, J. Zhang, R. Wang, H. Huang, C. Xie, Z. Li, J. Li and C. Niu, Nano Lett., 2015, 15, 5268–5272 CrossRef CAS PubMed.
- J.-Z. Wang, L. Lu, M. Lotya, J. N. Coleman, S.-L. Chou, H.-K. Liu, A. I. Minett and J. Chen, Adv. Energy Mater., 2013, 3, 798–805 CrossRef CAS.
- F. Zhou, S. Xin, H.-W. Liang, L.-T. Song and S.-H. Yu, Angew. Chem., Int. Ed., 2014, 53, 11552–11556 CrossRef CAS PubMed.
- A. Saha, A. Mondal, S. Maiti, S. C. Ghosh, S. Mahanty and A. B. Panda, Mater. Chem. Front., 2017, 1, 1585–1593 RSC.
- J. Xiao, D. Choi, L. Cosimbescu, P. Koech, J. Liu and J. P. Lemmon, Chem. Mater., 2010, 22, 4522–4524 CrossRef CAS.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- G. Jia, D. Chao, N. H. Tiep, Z. Zhang and H. J. Fan, J. Mater. Chem. A, 2018, 14, 136–142 Search PubMed.
- Y. Sun, F. Alimohammadi, D. Zhang and G. Guo, Nano Lett., 2017, 17, 1963–1969 CrossRef CAS PubMed.
- M.-R. Gao, M. K. Y. Chan and Y. Sun, Nat. Commun., 2015, 6, 7493 CrossRef PubMed.
- M. Endo, K. Takeuchi, T. Hiraoka, T. Furuta, T. Kasai, X. Sun, C. H. Kiang and M. S. Dresselhaus, J. Phys. Chem. Solids, 1997, 58, 1707–1712 CrossRef CAS.
- Y. Sun, L. Wang, Y. Liu and Y. Ren, Small, 2015, 11, 300–305 CrossRef CAS PubMed.
- M. A. Py and R. R. Haering, Can. J. Phys., 1983, 61, 76–84 CrossRef CAS.
- L. Yang, S. Wang, J. Mao, J. Deng, Q. Gao, Y. Tang and O. G. Schmidt, Adv. Mater., 2013, 25, 1180–1184 CrossRef CAS PubMed.
- G. X. Wang, S. Bewlay, J. Yao, H. K. Liu and S. X. Dou, Electrochem. Solid-State Lett., 2004, 7, A321–A323 CrossRef CAS.
- J. Xiao, X. Wang, X.-Q. Yang, S. Xun, G. Liu, P. K. Koech, J. Liu and J. P. Lemmon, Adv. Funct. Mater., 2011, 21, 2840–2846 CrossRef CAS.
- C. George, A. J. Morris, M. H. Modarres and M. De Volder, Chem. Mater., 2016, 28, 7304–7310 CrossRef CAS PubMed.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed.
- Y. J. Lee, H. Yi, W.-J. Kim, K. Kang, D. S. Yun, M. S. Strano, G. Ceder and A. M. Belcher, Science, 2009, 324, 1051 CAS.
- J. S. Sakamoto and B. Dunn, J. Electrochem. Soc., 2002, 149, A26–A30 CrossRef CAS.
- G. G. Amatucci, F. Badway, A. Singhal, B. Beaudoin, G. Skandan, T. Bowmer, I. Plitz, N. Pereira, T. Chapman and R. Jaworski, J. Electrochem. Soc., 2001, 148, A940–A950 CrossRef CAS.
- C. Feng, J. Ma, H. Li, R. Zeng, Z. Guo and H. Liu, Mater. Res. Bull., 2009, 44, 1811–1815 CrossRef CAS.
- M. Choi, S. K. Koppala, D. Yoon, J. Hwang, S. M. Kim and J. Kim, J. Power Sources, 2016, 309, 202–211 CrossRef CAS.
- G. Qu, J. Cheng, Z. Wang, B. Wang and S. Ye, J. Mater. Sci., 2016, 51, 4739–4747 CrossRef CAS.
- K. D. Rasamani, F. Alimohammadi and Y. Sun, Mater. Today, 2017, 20, 83–91 CrossRef CAS.
- M. Wang, G. Li, H. Xu, Y. Qian and J. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 1003–1008 CAS.
- Y. Xia, B. Wang, X. Zhao, G. Wang and H. Wang, Electrochim. Acta, 2016, 187, 55–64 CrossRef CAS.
- Y. Ji, Q. Wei and Y. Sun, Ind. Eng. Chem. Res., 2018, 57, 4571–4576 CrossRef CAS.
- S. Zhang, B. V. R. Chowdari, Z. Wen, J. Jin and J. Yang, ACS Nano, 2015, 9, 12464–12472 CrossRef CAS PubMed.
- L. Zhang and X. W. Lou, Chem. – Eur. J., 2014, 20, 5219–5223 CrossRef CAS PubMed.
- C. Zhao, J. Kong, L. Yang, X. Yao, S. L. Phua and X. Lu, Chem. Commun., 2014, 50, 9672–9675 RSC.
- H. Wang, Z. Lu, D. Kong, J. Sun, T. M. Hymel and Y. Cui, ACS Nano, 2014, 8, 4940–4947 CrossRef CAS PubMed.
- Y. Shi, Y. Wang, J. I. Wong, A. Y. S. Tan, C.-L. Hsu, L.-J. Li, Y.-C. Lu and H. Y. Yang, Sci. Rep., 2013, 3, 2169 CrossRef PubMed.
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759–1770 CrossRef CAS PubMed.
- Y. Liu, Y. Zhao, L. Jiao and J. Chen, J. Mater. Chem. A, 2014, 2, 13109–13115 CAS.
- X. Xie, Z. Ao, D. Su, J. Zhang and G. Wang, Adv. Funct. Mater., 2015, 25, 1393–1403 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00117k |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2018 |