
Kinetically controlled assembly of cadmium chalcogenide nanorods and nanorod heterostructures†
Michael J.
Enright
 ,
Harrison
Sarsito
and
Brandi M.
Cossairt
*
,
Harrison
Sarsito
and
Brandi M.
Cossairt
*
University of Washington, Department of Chemistry, Box 351700, Seattle, WA 98195-1700, USA. E-mail: cossairt@chem.washington.edu
First published on 26th March 2018
Abstract
While it is well understood that controlling anisotropic nanostructure growth can be accomplished by establishing kinetic growth conditions, the practical translation of this knowledge to access nanorods with a specific aspect ratio has not been realized. In this study we empirically determine the precursor consumption rates for growing nanorods and use this data to customize the size and shape of anisotropic nanostructures. The purpose of this work is to go beyond simply creating a set of growth conditions to obtain rods, dots, rice, and tetrapods by describing how to synthesize a nanomaterial of desired dimensions and aspect ratio in a pre-meditated fashion. Measured growth rates for model systems of CdSe (3.5 monomers rod−1 s−1 at 250 °C) and CdS nanorods (36 monomers rod−1 s−1 at 340 °C) were used to design elongated nanorods with enhanced aspect ratios and synthesize dot in rod CdS/CdSe and CdSe/CdS heterostructures. These model systems enable us to establish a rubric for the synthesis of customizable nanostructures and serve as a test case for understanding heterostructure assembly in colloidal systems.
 Michael J. Enright | Michael Enright is from Stevens Point, Wisconsin and received his A. B. in Chemistry with a minor in Spanish from Ripon College in 2014. He is currently a PhD candidate in chemistry at the University of Washington under the supervision of Professor Brandi Cossairt. His research focuses on designing synthetic methods to create novel type II semiconductor nanomaterials for solar fuels applications. Michael has received Pacific Northwest National Laboratory and Clean Energy Institute graduate research fellowships along with several departmental honours, including the Lloyd E. and Florence M. West Graduate Merit Fellowship at the University of Washington. |
 Harrison Sarsito | Harrison Sarsito is an undergraduate at the University of Washington and intends to complete his BS degree in Chemical Engineering in June 2019. Harrison is interested in the far-reaching impacts of utilizing nanomaterials in the context of renewable energy production. As a Mary Gates Scholar, he studies the design and synthesis of various photocatalytic nanomaterial systems of different elemental compositions and morphologies under the direction of Professor Brandi Cossairt. He aspires to one day work at the intersection between business and technology in order to play a more active role in the development and implementation of renewable energy technologies. |
 Brandi M. Cossairt | Dr. Brandi Cossairt received her BS in Chemistry from the California Institute of Technology in 2006 as a first-generation college graduate. Brandi went on to receive her PhD in 2010 from the Massachusetts Institute of Technology under the guidance of Professor Christopher Cummins. She then continued her academic training as an NIH NRSA Postdoctoral Fellow at Columbia University in the laboratory of Professor Jonathan Owen. Brandi joined the Department of Chemistry at the University of Washington as an Assistant Professor in 2012. She has received a number of awards for her research including a Packard Fellowship, a Sloan Research Fellowship, and an NSF CAREER Award. Brandi is an associate editor at Inorganic Chemistry and is the co-founder of the Chemistry Women Mentorship Network (ChemWMN). |
Introduction
Since the turn of the 19th century when the concept of interchangeability led to the dawn of modern assembly-line manufacturing, there has been great effort in developing processes that reproducibly yield products of an exact structure and composition from a set of constituent parts. A longstanding goal in the field of nanoscience has been the practical translation of this concept to the bottom-up synthesis of nanomaterials. For nanomaterial synthesis, as in nearly all other fields, it is desirable to have the ability to control the shape and structure of a material and to tune these features to modulate the chemical and physical properties for specific applications.1–3 Anisotropic nanostructures have unique, direction-dependent properties that enable both confinement of electrons, holes, and phonons as well as delocalization of charge carriers in specific and predictable directions.1,2 This directional control has value in applications from energy conversion and storage devices (such as lithium ion batteries, thermoelectric devices, and solar cells)4–11 to electronics (such as conducting platforms, transistors, and electromechanical devices).12–15 Furthermore, anisotropic structures hold great potential for use in optical (i.e. lasers, photodetectors, and OLEDs)16–18 and electrochemical devices (i.e. catalysis and sensors).19,20Metal chalcogenide nanomaterials are a class of semiconductors that possess useful properties for photovoltaic, photodetector, and photocatalytic applications due to their ability to efficiently absorb sunlight to generate excitons.21 Furthermore, the size and shape of metal chalcogenide nanomaterials can be inexpensively controlled using colloidal synthetic techniques.22–24 Specifically, the ability to exploit structural differences between wurtzite and zinc blende crystal structures facilitates colloidal synthesis of nanocrystals with unique crystal facets and distinct shapes. Axial growth on wurtzite nanocrystals occurs on the (0001) axis to give high aspect ratio nanorods. Existing mechanistic studies show the importance of monomer concentration on rod growth,25 as well as support a magic size cluster mediated pathway.26 The impact of ligands on directional nanocrystal growth has also been examined, showing a clear impact on growth rates and specific facet coordination on asymmetric wurtzite seeds.27
While it is well understood that controlling anisotropic structure formation can be accomplished by establishing kinetic growth conditions, the practical extension of this knowledge to access nanorods with a specific aspect ratio has not been realized. The approach adopted in most synthetic reports is to devise a set of conditions that give nanorods. However, for nanomaterial application development, the logic of this approach must be flipped so that the nanorod structure is considered first and then synthetic conditions are devised for obtaining the target structure. Here we seek to build on existing crystal growth precedent to establish methods for accessing nanorods of specific lengths and widths. The purpose of this work is to go beyond simply creating a set of growth conditions to obtain rods, dots, rice, and tetrapods,23 and to demonstrate how to synthesize a nanomaterial of desired aspect ratio in a premeditated fashion. Specifically, we measure monomer addition rates on seeded and unseeded CdSe nanorods. However, simply using these growth rates as monomer resupply rates during nanorod growth is insufficient to maintain kinetic, 1-D growth. Instead, we show that maintaining high precursor concentrations is more important for extending 1-D growth and we quantify the Cd/Se to ligand ratios required for preserving a 1-D growth environment. Understanding these parameters in any system enables premeditated design of nanorods of specific aspect ratios and reduces the amount of waste from unused precursors. This work also describes how to control nanocrystal growth on a pre-existing seed, enabling greater customizability of anisotropic nanostructures. We envision this model system serving as a rubric for the development of customizable anisotropic nanocrystals, and as a platform for understanding heterostructure assembly in colloidal systems.
Results and discussion
Extensive work has been carried out to synthesize high aspect ratio CdSe nanorods for a variety of diverse applications.28 Most of these procedures are based on the same recipe where a long chain cadmium alkyl phosphonate is heated to high temperature (above 300 °C) in trioctylphosphine oxide (TOPO) solvent. Then, a solution of selenium precursor, most commonly trioctylphosphine selenide (TOP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se), is injected rapidly to facilitate nucleation of wurtzite CdSe crystals before a growth temperature (around 250 °C) is established to grow the nanorods. For many investigations (i.e. functionalization with catalytically active nanoparticle tips,29–32 exciton generation and mobility studies,33,34 and device assembly35,36) simply obtaining a reasonably monodisperse sample of nanorods with a high aspect ratio is sufficient. Mechanistic studies of anisotropic assembly and shape control have illuminated the specific conditions required to enable rod growth as well as outline the general stages of rod growth construction.26,27,37 Despite these contributions and the apparent ease of obtaining non-isotropic cadmium chalcogenide structures, we still do not yet have sufficient synthetic control to enable the a priori setting of reaction conditions to obtain nanorods of a specific aspect ratio and length. In this report, we outline a rubric for how to determine rod growth rates and use that information to design rods or heterostructures of desired dimensions.
Se), is injected rapidly to facilitate nucleation of wurtzite CdSe crystals before a growth temperature (around 250 °C) is established to grow the nanorods. For many investigations (i.e. functionalization with catalytically active nanoparticle tips,29–32 exciton generation and mobility studies,33,34 and device assembly35,36) simply obtaining a reasonably monodisperse sample of nanorods with a high aspect ratio is sufficient. Mechanistic studies of anisotropic assembly and shape control have illuminated the specific conditions required to enable rod growth as well as outline the general stages of rod growth construction.26,27,37 Despite these contributions and the apparent ease of obtaining non-isotropic cadmium chalcogenide structures, we still do not yet have sufficient synthetic control to enable the a priori setting of reaction conditions to obtain nanorods of a specific aspect ratio and length. In this report, we outline a rubric for how to determine rod growth rates and use that information to design rods or heterostructures of desired dimensions.
The generally accepted mechanism for syntheses that yield nanorods proceeds through three phases.37 First, when reagent concentrations are high, growth along the c-axis of a wurtzite nanocrystal is promoted and known as the 1-D growth regime. As precursors are consumed by the growing nanorod, the solubilized monomer concentration decreases, and growth is facilitated on all axes, known as the 3-D growth regime. Understanding when to expect axial growth to slow down relative to radial growth is vital for targeted structure assembly. The synthesis time it takes to reach the transition point between the two regimes is highly sensitive to each system. Evaluating precursor amounts and concentrations at these observable transition points can help accurately determine when to expect synthetic conditions to promote 1-D or 3-D nanocrystal growth. The third regime is rod-to-sphere ripening, which occurs when the concentration of precursors falls below a second threshold and no longer facilitates growth of new monomer on the nanocrystal. During this phase, monomers from the rod ends migrate to the sides of the rod to give spheres, the more thermodynamically favoured product over the kinetically derived rods.
To better understand how to design nanorods of desired dimensions, a baseline evaluation of unseeded nanorod growth rate is a vital first step. As can be seen in the absorbance spectrum in Fig. 1A and as detailed by Jiang and Kelley, the mechanism of nanorod growth proceeds via a magic size cluster (MSC) mediated pathway.26 The sharp feature at 348 nm is indicative of the presence of the MSC throughout the initial 30 min of nanorod growth. Aspect ratio evaluation of the nanomaterials over this timeframe shows nanorods, once seeded, grow nearly exclusively along the wurtzite c-axis, for the first 16–18 min. Fig. 1 shows that at approximately 18–20 min, the aspect ratio, as well as rate of length increase begin to slow as the growing nanorods begin to transition from the 1-D growth regime into the 3-D growth regime. Despite the tapering of length growth rate, the rate of nanocrystal volume growth remains relatively unchanged. At this transition point, there is still an abundance of precursor present to exceed the critical saturation point to make MSCs, as well as sufficient precursor present to enable nanocrystal growth at an unimpeded rate. Despite this, the available concentration of monomer has decreased enough to no longer support 1-D growth kinetics.
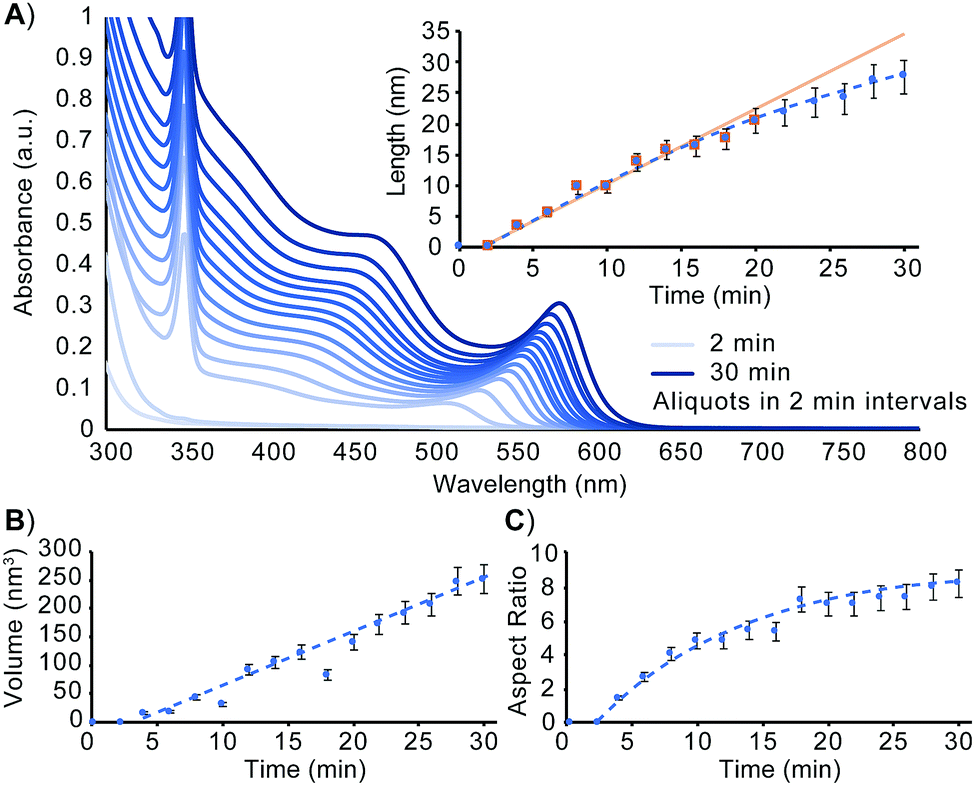 | ||
| Fig. 1 Evaluation of unseeded CdSe nanorod growth. (A) UV-Vis at 2 min intervals over the first 30 min of nanorod growth. Nanorod dimensions measured using TEM analysis as a function of time are plotted for length (A, inset), volume (B), and aspect ratio (C). The equation for the line of best fit for the volume vs. time relationship is y = 9.7919x − 44.591. Associated TEM images are included in Fig. S1 (ESI†). | ||
Since the 1-D growth regime only predominates over the first 20 minutes of CdSe nanorod growth, it is necessary to devise methods to prolong the duration of length growth to continue to access nanorods of increasing length. One way the duration of the 1-D growth regime is increased is to halt CdSe nanorod growth within the first 20 min and use those nanorods as seeds to restart rod growth in a fresh bath of precursor. Restarting the reaction with fresh precursor effectively reinstates the original 1-D growth regime conditions and rod growth begins again upon the nanorod seeds. The CdSe rod growth reaction can be halted and restarted by removing or providing heat to the system. Nucleation of wurtzite seeds requires high temperatures (above 300 °C), which is why rod growth begins with rapid, hot-injection of the selenium precursor. Growth of elongated wurtzite rods upon a wurtzite seed (dots or rods), however, is readily facilitated at 250 °C when sufficient monomer is present. As long as there is a source of seeds and a supply of monomers, or available precursor to give monomers, rod growth can be restarted and maintained at 250 °C. Independent nucleation of new nanomaterials is not observed when the reaction is restarted from seeds below the nucleation temperature. Fig. S2 (ESI†) illustrates that nanorods can be used as seeds for extended nanorod growth in fresh precursor and that successive additions of new precursor enable continued nanorod growth to 100 nm.
Restarting nanorod growth with a fresh supply of precursor gives longer nanorods with greater aspect ratios over the same total growth time. Fig. 2 displays the set of nanorod seeds (column 1) used as scaffolds for continued rod growth. These rods are extracted from a solution of growing nanorods. An identical volume of each sub-sample of nanorods was extracted at each time point (10 min, 20 min, 30 min, and 60 min) and each sample of rods was purified in an identical manner to remove unreacted precursor and unbound ligand. A combination of quantification of CdSe composition using Inductively Coupled Plasma-Optical Emission Spectroscopy (ICP-OES) and TEM evaluation to determine rod volume was used to verify that the number of rod seeds to be added to fresh precursor remains the same across all growth restart procedures. Rod growth was restarted upon the purified, intermediate seeds through rapid injection of selenium precursor to a solution of seeds and cadmium tetradecylphosphonate (Cd-TDPA) at 250 °C. Fig. 2 and 3 show that restarting rod growth upon nanorods originally grown for 10 min and 20 min facilitates greater rod elongation than the expectation for a growing nanorod over the same total reaction time. A nanorod that experiences 10 min of growth, purification, and an additional 10 minutes of growth in fresh precursor is both longer and exhibits a greater aspect ratio than a nanorod that grows for 20 min continuously. Every time rod growth conditions are reinitiated; width growth is curtailed, and aspect ratio increases. A comparison of nanorods that have experienced 30 min of total growth time shows that nanorods that undergo a growth–purify–restart procedure with purification at 10 min or 20 min gives higher aspect ratio nanorods than 30 min of continuous growth. Curiously, a sample that first grows for 10 min with an additional 20 min of growth has a smaller aspect ratio than its converse (20 min growth initially followed by 10 additional minutes). One explanation for this phenomenon is that there is a 4–5 min induction time between precursor injection and the start of nanocrystal growth when starting a reaction without seeds. The restarted reactions have seeds and do not require this additional pre-growth time. Since the greatest enhancements to aspect ratio occur between 5–20 min (15 min of growth time) the 10 min + 20 min sample experiences 5 min of 1-D growth before purification but only 15 min of 1-D growth when restarted. The 20 min + 10 min sample experiences 15 min of 1-D growth pre-purification and 10 min after restart. Thus, the 10 min + 20 min sample experiences 5 fewer minutes of 1-D growth and has a smaller aspect ratio. Major caveats to rod elongation via the growth–purify–restart method is the increased level of waste and significant time increase with each subsequent restart.
 | ||
| Fig. 2 TEM images showing unseeded and seeded nanorod growth as a function of time. Nanorod dimensions (length by width) are summarized explicitly in the tables at the upper right. | ||
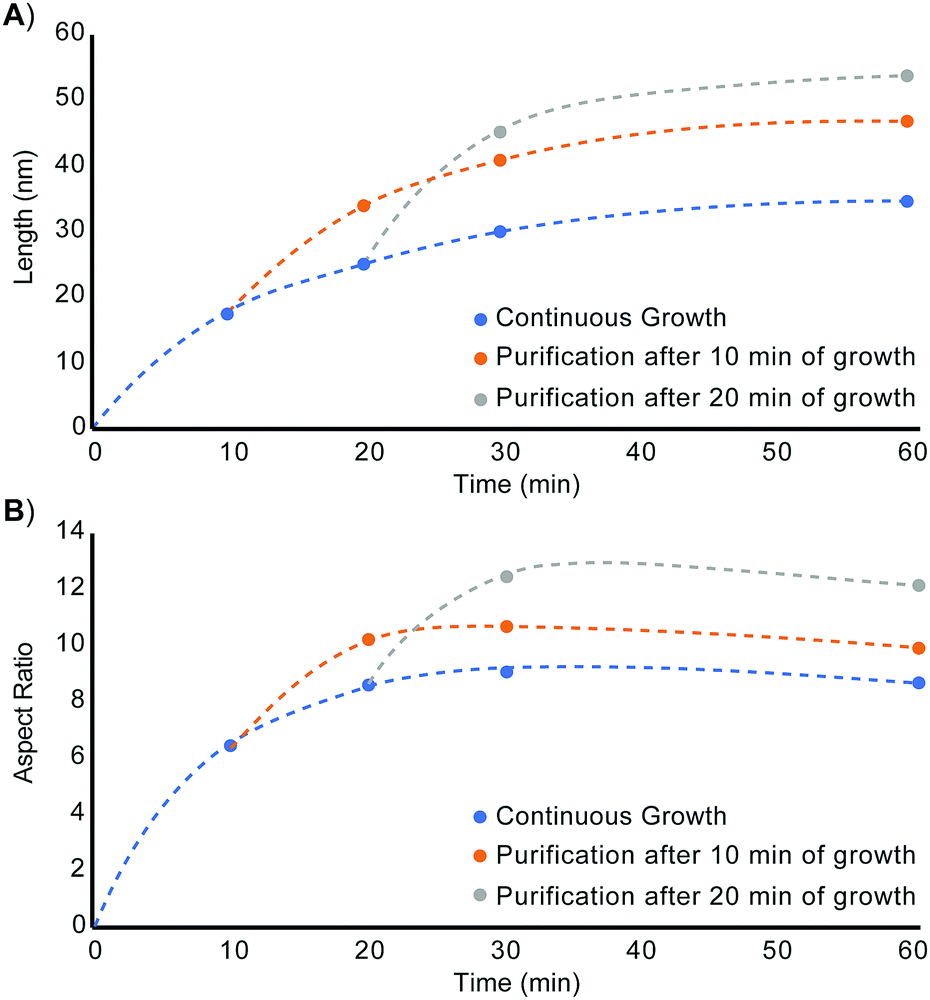 | ||
| Fig. 3 (A) Length and (B) aspect ratio evolution of unseeded CdSe nanorods (blue), nanorods seeded from nanorods grown for 10 minutes (orange), and nanorods seeded from nanorods grown for 20 minutes (gray). Width and volume vs. time plots are included in Fig. S3 (ESI†). | ||
In order to more readily access nanorods of prescribed dimensions without extensive intermediary purification, a more comprehensive understanding of rod growth conditions beyond knowing when a synthesis transitions between 1-D and 3-D growth regimes is needed. Revisiting the data presented in Fig. 1, an evaluation of unseeded nanorod growth shows that nanorods increase in volume at a rate of 2.9 monomers rod−1 s−1 (see ESI†). While tracking length increase and elucidating monomer addition rates can assist in understanding how to better target a specific nanostructure, it is an incomplete description of how to establish synthetic conditions for prolonging rod growth over an extended period of time. Quantifying the monomer consumption rate for a known concentration of seeds, however, is sufficient for determining the exact amount of reagent consumed by the growing nanorod ensemble. This assembly rate can be subsequently applied to future systems in which precursor is resupplied to maintain 1-D growth regime conditions.
The number of nanorods is estimated using the CdSe extinction coefficient that has been measured for CdSe quantum dots.38 Notably, this estimation appeared consistent with nanorod quantification using a combination of ICP-OES and TEM. In an effort to best emulate the unseeded rod growth synthesis, three concentrations of wurtzite CdSe nanoparticle seeds were used as scaffolds for CdSe nanorod growth: 2.73 × 10−7 mol, 2.73 × 10−8 mol, and 1.00 × 10−8 mol seed samples. As can be seen in Fig. S4–S7 (ESI†), none of the three seed concentrations yielded an exact match to the growth rate of unseeded nanorods, however, the 2.73 × 10−8 mol seed synthesis tracked the closest. The 2.73 × 10−7 mol appears to have too many seeds because the length and volume of the nanocrystals appears to reach a limit of length and volume prior to 15 min of growth and this nanorod size is both shorter and has less volume than that observed for unseeded nanorods grown for 30 min. The higher seed count leads to less precursor available to grow upon each nanorod. It also gives a lower effective precursor concentration leading to a shorter period of time in the 1-D growth regime and an earlier entrance into the 3-D growth regime. While the 1.00 × 10−8 mol seed sample appears to track well to the unseeded case by volume, the rate of length growth is slower. This appears to point to a lower limit for nucleus concentration given the set of precursor conditions. The low seed concentration may lead to nucleation of new CdSe. The 2.73 × 10−8 mol seeded growth rates tracked closely to the unseeded growth rates by both length and width evaluations and is used as the starting seed concentration for precursor replenishment studies. Evaluation of the volume growth rates of all of the seeded samples over the early time points (before the rate of volume increase tapers off) shows that all three seeded samples grow at a rate of 3.5 monomers rod−1 s−1.
Similar CdSe growth rates are observed when using wurtzite CdS seeds as a scaffold for CdSe nanorod growth. As can be seen in Fig. S8–S10 (ESI†), CdSe growth on two different concentrations of CdS seeds demonstrated a nearly identical volume, length, and growth rate between 5–20 min of the reaction when compared to unseeded CdSe growth. This is important for heterostructure development since changing the seed material appears to have minimal impact on the rate of the growing nanorod. It should be noted that there is an initial etching step when using CdS seeds. The width of the material when using 2.73 × 10−7 mol seeds initially decreases before retaining its original 4 nm size. When a greater concentration of seeds is provided to the system (8.19 × 10−7 mol) the etching step is less readily observable. In the high seed concentration heterostructure, the growing nanorods exit the 1-D growth regime more quickly than in any other lower seed concentration conditions. Thus, the conditions for the high seed concentration rod growth likely enters the 3-D growth regime at an earlier time point, thereby replacing the etched surface with new monomer more quickly.
Conversely, growth rates for CdS nanorod growth on CdSe seeds can be determined using the same type of TEM evaluation. CdS nanorod growth is typically carried out at greater temperatures (340 °C) and higher chalcogenide precursor concentrations when compared to CdSe nanorod growth.39 This is due to the decreased reactivity of TOPS versus TOPSe, which arises from the stronger binding of S to P, resulting in less available S2− on a per molecule basis.40 CdS monomers assemble on growing CdS rods at a rate of 36 monomers rod−1 s−1. While this measured rate is about 10 times more rapid than CdSe at 250 °C, it does not serve as a direct comparison to CdSe nanorod growth due to disparate temperature and concentration conditions. Despite these differences, seeded rod growth of CdS and CdSe nanorods both exhibit an abrupt curtailment to the length extension rate of the growing material. After 15 min, 1-D growth abruptly slows, and volume increases are predominately from increases in nanorod width. Figures detailing the rates of length, width, volume, and aspect ratio of the growing nanorod sample are provided in the ESI† (Fig. S11 and S12).
Knowing the exact number of seeded nanorods in the system enables accurate determination of precursor consumption and precursor to rod incorporation yields. Over the first 30 min of the reaction, the time where the highest aspect ratio nanorods are obtained, less than 10% of the precursors have been incorporated into nanorods. Evaluation of the aspect ratios for both seeded and unseeded CdSe nanorods shows that enhancements to aspect ratio decline for both seeded and unseeded rods before 20 min of reaction. At 20 min, only 6.1% and 4.3% of the available cadmium supply has been incorporated into nanorods for the seeded and unseeded cases, respectively. These low percentage yields and narrow window for reaction conditions that facilitate nanorod growth are two major challenges for anisotropic nanocrystal and heterostructure development.
Since favourable rod growth conditions only exist in a small window with abundant precursor, spiking the system with excess reagent during the reaction should replenish the reagent consumed during the early time points of the synthesis and retain conditions for the 1-D growth regime. As can be seen in the unseeded and seeded growth cases, there is never more than a 5 min induction time to build up sufficient monomer reserves before rod growth begins. We propose therefore that replenishment at 7.5 min is late enough that it should not disrupt initial monomer formation and rod nucleation. Since rods remain in the 1-D growth regime beyond the first 15 min, there is also sufficient time to generate new monomers from the fresh precursor to promote continued length growth. As can be seen in Fig. 4, doubling the amount of fresh precursor at 7.5 min does in fact prolong the amount of time spent in the 1-D growth regime and yields longer and thinner nanorods compared to their counterparts from non-replenishment syntheses across the same time points. Doubling the starting precursor extends the duration of 1-D growth to about 35 min, while adding 50% more precursor leads to exiting the 1-D growth regime before 25 min. The width, volume, and aspect ratio profiles over time for addition of fresh precursor at a single event are depicted in Fig. S13 (ESI†) with accompanying TEM images in Fig. S14 and S15 (ESI†) for precursor doubling and 50% more precursor respectively.
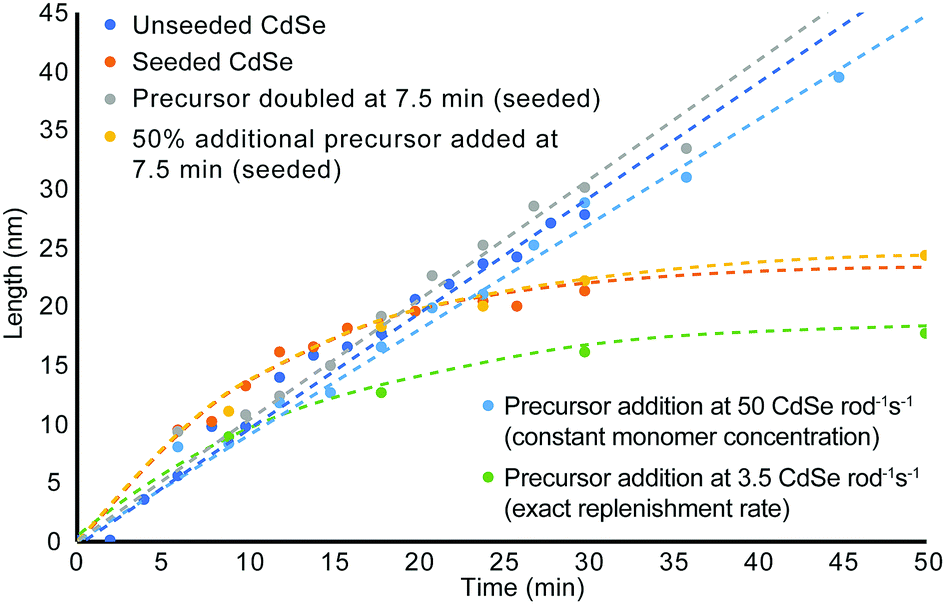 | ||
| Fig. 4 Comparison of length evolution across reaction conditions. For all precursor addition reactions, nanorod growth was seeded by wurtzite CdSe seeds and the precursor addition was initiated at 7.5 minutes. | ||
Real-time monomer concentration is challenging to quantify directly, so instead we can project the concentrations of cadmium and selenium to provide insights into precursor availability. Looking at the ratio of cadmium to its native ligand, TDPA, and selenium to trioctylphosphine, also assists in deconvoluting the availability of monomer. Fig. S16 (ESI†) shows the simulated molarities of cadmium and selenium as well as the ratios of cadmium and selenium to their native ligand based on our calculated precursor consumption rates. These simulations quantify precursor availability throughout the experiment and project what the expected precursor molarities would be assuming constant nanorod growth rate over the first 30 min of the reaction. The simulation uses the measured growth rates by volume observed over the 1-D growth regime. The constant monomer consumption measured during 1-D growth are not maintained throughout the 3-D growth regime. However, in all aforementioned unseeded, seeded, and precursor replenishment conditions, the constant growth rates by volume extend through the entire 1-D growth regime and into the early part of 3-D growth.
When the ratio of cadmium or selenium to ligand, TDPA or TOP respectively, is high, the equilibrium between bound and free precursor that can form monomers is also high. As this ratio decreases (as is the case when cadmium and selenium is consumed by the growing nanorods) the excess ligand binds available monomer precursors and reduces the relative concentration of monomer in solution available for the growing nanorods. Thus, in order to facilitate a 1-D growth environment for sustainable elongation of CdSe nanorods, a minimum ratio of cadmium to TDPA and selenium to TOP must be retained. For this system, the minimum ratio for cadmium to TDPA is approximately 0.475 (or 2.1 TDPA per cadmium) and 0.195 selenium to TOP (or 5.1 TOP per selenium). As long as these thresholds are not exceeded, conditions that support rod growth can be maintained. The known chemical equilibrium constants between trialkyl phosphine selenides lies towards the bound phosphine selenide.40,41 As selenium is consumed by the growing nanorods, the shift in equilibrium back towards free selenide (the reactive species in monomer formation) is insufficient to resupply selenium at a rate required for nanorod growth. To maintain conditions for length elongation, the resupplied precursor must be provided at a concentration capable of shifting the equilibrium back to a concentration of available selenium high enough to continue rod growth (eqn (1)).
| Precursor (i.e. TOPSe) ⇆ monomer + free ligand → rods | (1) |
From these quantitative observations, we derive the hypothesis that rod growth can be sustained as long as the minimum ligand to cadmium and selenium ratios are not exceeded. To test this theory, fresh precursor was resupplied at a rate sufficient to stay below the Cd/Se to ligand threshold to enable these thresholds to be un-exceeded for an extended period of time without contributing large volumes of excess waste. As can be seen in Fig. 3 and Fig. S17, S18 (ESI†), replenishing the cadmium and selenium precursor at a rate of 0.083 mmol cadmium and selenium per min results in favourable rod growth conditions that can be maintained beyond 45 min to give longer, higher aspect ratio nanorods. While the rod growth rate proceeds at 3.5 monomers rod−1 s−1, the necessary resupply rate for extended 1-D growth is 14.4× greater and requires precursor to be resupplied at a rate of 50 Cd or Se rod−1 s−1.
To demonstrate that precursor replenishment is not simply a function of replacing the amount of consumed precursor, cadmium and selenium are replaced by syringe pump at the exact consumption rate. Fig. 3 and Fig. S17, S19 (ESI†) shows this instead leads to exiting the 1-D growth regime prematurely and gives nanorods of reduced aspect ratio. While this is greatly inhibitory to growing longer, high aspect ratio nanorods, it is a feature that provides greater customizability to the shape of the rod. If rods with reduced aspect ratios or larger diameters than the starting seed are desired, the synthesis can be easily modified by adding additional TOP or TDPA (an amount to exceed the Cd/Se to ligand threshold) to the system. Adding TOP or TDPA led to a premature exit from the 1-D growth regime and early aspect ratio curtailment within minutes of supplying new ligand (Fig. S20–S22, ESI†).
Providing additional TOP shifts the equilibrium between bound Cd-TDPA and TOPSe and monomer back towards the bound precursor, thereby reducing the monomer concentration. This leads to earlier 3-D growth and a slower rate of monomer addition to the nanorod, as shown in Fig. S20 (ESI†). While additional TDPA also leads to an early exit from the 1-D growth regime, the extra acid facilitates an environment in which the growth rate increases but the rate of length growth is unperturbed. The addition of phosphonic acid likely leads to a systematic change in the equilibrium between ligand-bound precursor, monomer, and free ligand as well as perturbing the off-path equilibrium between monomer and magic size clusters. As shown previously, additional phosphonic acid perturbs CdSe magic size clusters and leads to more rapid nanocrystal growth, as observed by a more rapid red shift of the excitonic peak.26,42
This pair of added ligand studies illustrate two distinct methods to obtain lower aspect ratio nanorods. While both TOP and TDPA addition led to a premature exit from the 1-D growth regime, additional TOP dramatically slowed nanorod growth by reducing the concentration of available monomer. TDPA addition, however, accelerated the nanorod growth rate and promoted more rapid growth about the circumference of the nanorods while axial growth continued. From a practical perspective, addition of TOP can be used for synthetic approaches where shorter and wider rods are desired. If the target rod product is wider but of a length accessible along a typical rod growth profile (Fig. 1), addition of TOP minutes before the target length is achieved will enable length to be set while circumferential growth proceeds towards the favoured structure. To access wider rods with lengths only obtainable beyond the first 15 min of rod growth, addition of TDPA will help to enhance circumferential growth without significantly inhibiting length extension.
The exact replenishment rates reported here will vary from system to system depending on the monomer to rod conversion rate and the concentrations of the starting and resupplied precursors. However, this quantitative control over nanorod growth offers a 3-step rubric that goes beyond customizability of shape and structure of CdSe, CdS, and CdS/CdSe heterostructures. First, determine the growth rate of the nanocrystal. In these model systems we demonstrate CdSe nanorods assemble at a rate of 3.5 monomers rod−1 s−1 (at 250 °C) while CdS grows at 36 monomers rod−1 s−1 (at 340 °C). This growth rate is determined by evaluating changes in total volume of the growing nanorods. As can be seen throughout this set of studies, changes in rates of monomer addition to the volume of the nanorod do not directly correlate with 1-D to 3-D growth regime transitions. Some conditions, such as seeded growth with a large quantity of seeds, lead to plateauing volume growth rates after exiting the 1-D growth regime, while unseeded nanorod growth showed very little change in volume increases across this transition point. The second step is to determine the 1-D to 3-D transition time point by evaluating length growth rates or aspect ratio changes. When sufficient precursor is provided to create rod growth conditions, length growth initially proceeds at a linear rate. Departure from 1-D growth conditions are indicated by a plateau or taper away from the initial linear length growth rate. Both of these steps can be accomplished with the same sample. Unseeded growth, for example, transitions into the 3-D growth regime after 20 min. This transition is unobservable by exclusively tracking rod volume but is readily apparent from length and aspect ratio evaluations. The final step to obtain longer rods or increase aspect ratio, is to resupply precursors at high concentrations to retain high Cd/Se to ligand ratios and push the equilibrium between precursor and available monomer towards monomer formation. For radial increases and reduced aspect ratios, evaluate length growth rates to determine when rods have nearly attained the targeted length, rapidly add free ligand (i.e. TOP) and solvent to force premature departure from the 1-D growth regime. Note that length growth, while curtailed, does not completely stop and growth along the long axis still needs to be considered.
Using this rubric, this work demonstrates three methods of aspect ratio enhancement. The growth–purify–restart method (illustrated in Fig. 2) offers excellent control to reliably target a particular size and can be used repeatedly to give rods of significantly increased length with minimized radial growth. Abundant precursor replenishment in a single step can also prolong 1-D growth conditions by supplying concentrations of precursor that are sufficient to continue to force the equilibrium between ligand bound precursor and available monomer back towards the monomer. The third method, which arises from our improved mechanistic understanding of nanorod growth, is to directly replenish consumed monomer at the rate of its consumption. This method yields less precursor waste and offers the greatest control over the exact dimensions of the nanomaterial. For retention of length growth in this CdSe system, precursor must be provided at a rate of 50 Cd or Se rod−1 s−1. While the physical technique for resupplying precursor is similar to dropwise addition and successive ionic layer adsorption and reaction synthetic methods, these methods are typically employed to limit the amount of precursor available in the system to prevent new, independent, nanomaterial nucleation. Quantitative resupply is more calculated and is employed to stay above a minimum threshold rather than stay below a critical concentration limit. This work provides further insights into how to construct desirable anisotropic heterostructures.43–46
Experimental
General methods
All manipulations were carried out using standard Schlenk or glovebox techniques under dry nitrogen. Cadmium oxide (CdO, >99.99%), trioctylphosphine (TOP, 97%), tributylphosphine (TBP, 95%), propylphosphonic acid (95%), selenium (99.99%), and sulfur (99.5%) were all purchased from Sigma Aldrich and used as received without further purification. Tetradecylphosphonic acid (TDPA, 99%) and octadecylphosphonic acid (ODPA, 99%) were purchased from PCI Synthesis and used as received. Anhydrous methanol, toluene, and pentane were purchased from various sources. Trioctylphosphine oxide (TOPO, 90%) was purchased from Sigma Aldrich and purified by repeated recrystallization until impurities were no longer present by evaluation using 1H and 31P NMR spectroscopy.47 Solutions of trioctylphosphine-selenide (TOP–Se) and trioctylphosphine-sulfide (TOP–S) were pre-prepared by dissolving metallic selenium or sulfur powder into TOP in a glovebox. Concentrations and exact solution preparation procedures are reported individually for the synthesis of each nanomaterial.Synthesis of CdSe and CdS seeds
The syntheses of wurtzite CdSe and CdS seeds were adopted from the procedures reported by Manna, et al.48 For CdSe, CdO (0.060 g, 0.47 mmol), ODPA (0.280 g, 0.837 mmol) and TOPO (3.00 g, 7.76 mmol) were loaded into a 50 mL three neck flask. This mixture was flushed with nitrogen and degassed at room temperature for 30 min before heating to 150 °C. The mixture was held under vacuum for 1 h. Then, the solution was heated to 300 °C under flowing nitrogen. Upon reaching 300 °C, TOP (1.50 g, 4.05 mmol) was injected by syringe and the temperature was increased to 370 °C. The temperature of the cadmium precursor was allowed to stabilize at 370 °C before addition of the selenium precursor. Once the cadmium precursor turned clear (this typically happens by the time the temperature stabilizes), TOP–Se was rapidly injected by syringe addition. The TOP–Se precursor was prepared by dissolving 0.058 g of selenium powder (0.74 mmol) in 0.360 g of TOP (0.971 mmol). The reaction was allowed to proceed for 10 s before the heat was removed. When the reaction mixture cooled to about 100 °C, 10 mL of toluene was added to the sample to better facilitate purification. The quantum dots were purified under a nitrogen atmosphere by repeated dissolution in toluene and centrifuge assisted precipitation with methanol. Purified CdSe seeds were suspended in anhydrous toluene or pentane and centrifuged after 8–24 h to remove excess ligand before filtration with a 0.45-micron PDFE syringe filter. The quantum dot size and concentration were quantified by UV-Vis spectroscopy.38,49 The size of the wurtzite seed can be adjusted by adjusting the reaction time prior to quenching. Immediate removal of the heating mantle yields ∼2.5 nm seeds, 10 s reaction time yields ∼3.6 nm particles, and 30 s reaction time yields ∼5.2 nm nanocrystals although exact reaction conditions will vary slightly across set-ups.Synthesis of CdS nanorods
The synthesis of CdS nanorods from wurtzite CdSe seeds is based on the procedure reported by Alivisatos, et al.39 In a 50 mL three-neck flask, CdO (0.230 g, 1.79 mmol), ODPA (1.08 g, 3.22 mmol), propylphosphonic acid (0.075 g, 0.604 mmol) and TOPO (3.35 g, 8.66 mmol) were loaded and degassed for 30 min at room temperature. The sample was then heated to 120 °C and held under vacuum for 1 h. After degassing, the solution was heated to 320 °C under nitrogen until the solution was clear, indicating the formation of cadmium octadecylphosphonate. After the cadmium precursor was formed, the solution was cooled to 120 °C and exposed to a vacuum for 2 h to remove water that is generated during precursor formation. After 2 h, the solution was placed back under nitrogen and heated to 340 °C and TOP (1.00 g, 2.70 mmol) was added by syringe addition. When the temperature re-stabilized at 340 °C, TOP–S was added by rapid syringe injection. The TOP–S precursor was made by dissolving sulfur (0.0518 g, 1.62 mmol) in TOP (0.598 g, 1.62 mmol) and was stirred overnight. Gentle heating (60 °C) and sonication may be necessary to fully dissolve the sulfur. 20 s after the injection of TOP–S, the wurtzite CdSe seeds dissolved in 1.00 g TOP (2.70 mmol) were rapidly injected into the reaction mixture. Approximately 5.5 × 10−7 mol of CdSe quantum dots were added in this reaction. This reaction was quenched by removing heat after 10 min of growth to obtain nanorods with dimensions of 16.9 ± 1.0 nm by 4.5 ± 0.3 nm. Reaction times can be varied to obtain structures of desired length/width. When the CdS nanorods cooled to about 100 °C, 10 mL of toluene was added to solubilize the nanocrystals. Several repeated purification steps of suspension in toluene, precipitation with methanol, and centrifugation were carried out in a nitrogen atmosphere to remove unreacted reactants, solvent, and excess ligand. Purified nanorods were suspended in anhydrous toluene or pentane and centrifuged after 8–24 h to remove excess ligand before filtration with a 0.45-micron PDFE syringe filter.Seed concentration, reaction time, and TOP–S volume all impact the resulting structure. Increasing seed concentration yields shorter rods (less CdS per rod) but longer reaction times yields longer rods (longer growth time). Increasing the volume of TOP–S added to the reaction also facilitates longer, thinner rods over 10 min while the rod length is dictated by absolute precursor concentration.
Synthesis of CdSe nanorods
CdSe nanorod growth experiments were modified off of procedures reported by Kim and Korgel.50 CdO (0.241 g, 1.87 mmol), TDPA (1.04 g, 3.74 mmol), and TOPO (0.75 g, 1.94 mmol) were mixed together in a 50 mL three-neck flask and degassed under vacuum for 1 h. Then, the mixture was heated to 300 °C under nitrogen to form cadmium tetradecylphosphonate. Note: due to the small volume of TOPO, CdO will sometimes cake to the side of the flask. Ensure all of the reagents are well mixed to evolve the clear cadmium precursor. This solution was then cooled to room temperature and was aged for 24 h under nitrogen. The aging step is important for reproducible rod growth.50 After 24 h of aging, and additional 2.325 g TOPO (6.01 mmol) was added to the flask and the mixture of white solids was degassed at room temperature for 1 h. Next the reaction mixture was heated to 320 °C under nitrogen for rod growth. Once the temperature stabilized at 320 °C, the selenium precursor was added rapidly by syringe injection. The temperature was immediately dropped to about 250 °C for the duration of the rod growth experiment. The selenium precursor was prepared by dissolving selenium powder (0.126 g, 1.61 mmol) in TOP (3.482 mL, 7.81 mmol), TBP (0.468 mL, 1.90 mmol), and toluene (0.694 mL, 6.53 mmol) in a glovebox. After about 30 min of CdSe rod growth, increases to aspect ratio cease and the reaction can be stopped unless low aspect ratio nanorods (less than 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) are desired. The reaction is terminated by removal of the heat source and 10 mL of toluene is added to solubilize the reaction mixture for purification. Methanol or ethanol are used as the anti-solvent. Repeated resuspension in toluene followed by methanol addition and centrifugation purifies the nanorods. Purified nanorods are suspended in anhydrous toluene and centrifuged after 8–24 h to remove excess ligand before filtration with a 0.45-micron PDFE syringe filter.
:1) are desired. The reaction is terminated by removal of the heat source and 10 mL of toluene is added to solubilize the reaction mixture for purification. Methanol or ethanol are used as the anti-solvent. Repeated resuspension in toluene followed by methanol addition and centrifugation purifies the nanorods. Purified nanorods are suspended in anhydrous toluene and centrifuged after 8–24 h to remove excess ligand before filtration with a 0.45-micron PDFE syringe filter.
For seeded CdSe rod growth, the desired quantity of seeds in pentane or toluene is added to the reaction flask 1 h after adding the additional 2.325 g TOPO. The solvent is evaporated at 50 °C for pentane and 100 °C for toluene. Additionally, instead of heating to 320 °C for the selenium injection, heat to the rod growth temperature of 250 °C to avoid independent nucleation of CdSe and to facilitate CdSe growth directly on the seeds.
Sample characterization
For evaluation of structures presented in this report, UV-Vis spectra were recorded using an Agilent Cary 5000 spectrophotometer. TEM images were obtained on an FEI Technai G2 F20 microscope. Analysis of TEM images was performed via manual counting using the ImageJ software package. Elemental quantification was carried out using a Perkin Elmer Optima 8300 Inductively Coupled Plasma-Optical Emission Spectrophotometer.ICP-OES was used to quantify the amount of cadmium in a sample. Calculating the rod volume by TEM approximates the per rod cadmium content. Concentrations of rods/sample can be calculated by dividing the cadmium/sample measured using ICP-OES by cadmium/rod calculated from TEM. The purification procedure used to remove excess ligand, solvent, and unreacted precursor and monomer from the nanorod sample is rigorous and leads to loss of an unquantifiable number of rods. Thus, this concentration estimation for the number of growing rods in an unseeded CdSe nanorod synthesis is likely an overestimation of the number of nanorods that seed and grow during the reaction.
Conclusions
In summary, this report quantifies the growth rates and establishes the conditions needed to support the anisotropic extension of CdSe, CdS, and CdS/CdSe nanorods. These measured parameters are derived from widely used synthetic methods for cadmium chalcogenide nanorods and serve as a model system for controlling the dimensions and aspect ratios of other materials and systems that follow similar assembly mechanisms. The methods used for extending 1-D growth conditions serve as a rubric for customizability of nanorod dimensions. In particular, this work outlines the steps for customizability of anisotropic colloidal nanocrystals. Despite extensive investigations of CdS and CdSe nucleation and growth over the past 20 years, this is one of the first reports to bridge the gap between scientific knowledge and practical implementation of this knowledge to obtain nanostructures of specific dimensions. This report shows how to approach studying and subsequently modulating reaction conditions to obtain kinetic nanocrystal products.While there exist many methods for aspect ratio enhancement, this report highlights three ways to achieve nanostructures of a set of exact dimensions: growth–purify–restart, abundant precursor replenishment, and direct replenishment/addition of precursors or additives. This report highlights the ability to grow nanorods of exact dimensions with spherical and anisotropic nanocrystals as seeds for enhanced control over the parameters of the final product. The rubric outlined here for CdS and CdSe shows that much of the information required to determine how to develop a nanostructure for any colloidal system can be learned by first quantitatively mapping the progress of the developing nanocrystals. Once this baseline has been established, a determination of the most appropriate way was (i.e. based on workability of precursors, cost, and available materials, etc.) to retain or disrupt kinetic growth conditions can be made. This rubric also extends to heterostructures and provides an outline for how to generate rod–rod heterostructures with exact control of the length of each component.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the University of Washington Clean Energy Institute, the Alfred P. Sloan Foundation, and the David and Lucile Packard Foundation. Part of this work was conducted at the Washington Nanofabrication Facility/Molecular Analysis Facility, a National Nanotechnology Coordinated Infrastructure (NNCI) site at the University of Washington, which is supported in part by funds from the National Science Foundation (Grants 1542101, 1337840, and 0335765), the National Institutes of Health, the Molecular Engineering & Sciences Institute, the Clean Energy Institute, the Washington Research Foundation, the M. J. Murdock Charitable Trust, Altatech, ClassOne Technology, GCE Market, Google, and SPTS.Notes and references
- W. Shi, R. W. Hughes, S. J. Denholme and D. H. Gregory, CrystEngComm, 2010, 12, 641–659 RSC.
- P. R. Sajanlal, T. S. Sreeprasad, A. K. Samal and T. Pradeep, Nano Rev., 2011, 2, 5883 CrossRef PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- H. Li, Z. Wang, L. Chen and X. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- A. I. Hochbaum and P. Yang, Chem. Rev., 2010, 110, 527–546 CrossRef CAS PubMed.
- T. C. Harman, P. J. Taylor, M. P. Walsh and B. E. LaForge, Science, 2002, 297, 2229–2232 CrossRef CAS PubMed.
- K. F. Hsu, S. Loo, F. Guo, W. Chen, J. S. Dyck, C. Uher, T. Hogan, E. K. Polychroniadis and M. G. Kanatzidis, Science, 2004, 303, 818–821 CrossRef CAS PubMed.
- R. Venkatasubramanian, E. Siivola, T. Colpitts and B. O'Quinn, Nature, 2001, 413, 597–602 CrossRef CAS PubMed.
- X. Wang, J. Song and Z. L. Wang, J. Mater. Chem., 2007, 17, 711–720 RSC.
- J. Zhu, B. S. Shim, M. Di Prima and N. A. Kotov, J. Am. Chem. Soc., 2011, 133, 7450–7460 CrossRef CAS PubMed.
- S. De, P. E. Lyons, S. Sorel, E. M. Doherty, P. J. King, W. J. Blau, P. N. Nirmalraj, J. J. Boland, V. Scardaci, J. Joimel and J. N. Coleman, ACS Nano, 2009, 3, 714–720 CrossRef CAS PubMed.
- J.-Y. Lee, S. T. Connor, Y. Cui and P. Peumans, Nano Lett., 2008, 8, 689–692 CrossRef CAS PubMed.
- D. Azulai, T. Belenkova, H. Gilon, Z. Barkay and G. Markovich, Nano Lett., 2009, 9, 4246–4249 CrossRef CAS PubMed.
- C.-H. Cho, C. O. Aspetti, M. E. Turk, J. M. Kikkawa, S.-W. Nam and R. Agarwal, Nat. Mater., 2011, 10, 669–675 CrossRef CAS PubMed.
- J. A. Schuller, E. S. Barnard, W. Cai, Y. C. Jun, J. S. White and M. L. Brongersma, Nat. Mater., 2010, 9, 193–204 CrossRef CAS PubMed.
- S. A. McDonald, G. Konstantatos, S. Zhang, P. W. Cyr, E. J. D. Klem, L. Levina and E. H. Sargent, Nat. Mater., 2005, 4, 138–142 CrossRef CAS PubMed.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- D. R. Kauffman, D. C. Sorescu, D. P. Schofield, B. L. Allen, K. D. Jordan and A. Star, Nano Lett., 2010, 10, 958–963 CrossRef CAS PubMed.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- X. Peng, L. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisatos, Nature, 2000, 404, 59–61 CrossRef CAS PubMed.
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2002, 124, 3343–3353 CrossRef CAS PubMed.
- X. Peng, Adv. Mater., 2003, 15, 459–463 CrossRef CAS.
- L. Manna, E. C. Scher and A. P. Alivisatos, J. Am. Chem. Soc., 2000, 122, 12700–12706 CrossRef CAS.
- Z.-J. Jiang and D. F. Kelley, ACS Nano, 2010, 4, 1561–1572 CrossRef CAS PubMed.
- D. Kim, Y. K. Lee, D. Lee, W. D. Kim, W. K. Bae and D. C. Lee, ACS Nano, 2017, 11, 12461–12472 CrossRef CAS PubMed.
- L. Zhao, L. Hu and X. Fang, Adv. Funct. Mater., 2012, 22, 1551–1566 CrossRef CAS.
- T. Mokari, E. Rothenberg, I. Popov, R. Costi and U. Banin, Science, 2004, 304, 1787–1790 CrossRef CAS PubMed.
- Y. Nakibli and L. Amirav, Chem. Mater., 2016, 28, 4524–4527 CrossRef CAS.
- S. M. Kim, S. J. Lee, S. H. Kim, S. Kwon, K. J. Yee, H. Song, G. A. Somorjai and J. Y. Park, Nano Lett., 2013, 13, 1352–1358 CrossRef CAS PubMed.
- L. Amirav and A. P. Alivisatos, J. Phys. Chem. Lett., 2010, 1, 1051–1054 CrossRef CAS.
- L. T. Kunneman, M. Zanella, L. Manna, L. D. A. Siebbeles and J. M. Schins, J. Phys. Chem. C, 2013, 117, 3146–3151 CAS.
- H. Zhu and T. Lian, J. Am. Chem. Soc., 2012, 134, 11289–11297 CrossRef CAS PubMed.
- A. Salant, M. Shalom, Z. Tachan, S. Buhbut, A. Zaban and U. Banin, Nano Lett., 2012, 12, 2095–2100 CrossRef CAS PubMed.
- S. Lee, J. C. Flanagan, J. Kang, J. Kim, M. Shim and B. Park, Sci. Rep., 2015, 5, 17472 CrossRef CAS PubMed.
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2001, 123, 1389–1395 CrossRef CAS.
- J. Jasieniak, L. Smith, J. van Embden, P. Mulvaney and M. Califano, J. Phys. Chem. C, 2009, 113, 19468–19474 CAS.
- D. V. Talapin, J. H. Nelson, E. V. Shevchenko, S. Aloni, B. Sadtler and A. P. Alivisatos, Nano Lett., 2007, 7, 2951–2959 CrossRef CAS PubMed.
- R. García-Rodríguez, M. P. Hendricks, B. M. Cossairt, H. Liu and J. S. Owen, Chem. Mater., 2013, 25, 1233–1249 CrossRef.
- R. D. Baechler, M. Stack, K. Stevenson and V. Vanvalkenburgh, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 48, 49–52 CrossRef CAS.
- H. Liu, J. S. Owen and A. P. Alivisatos, J. Am. Chem. Soc., 2007, 129, 305–312 CrossRef CAS PubMed.
- F. Shieh, A. E. Saunders and B. A. Korgel, J. Phys. Chem. B, 2005, 109, 8538–8542 CrossRef CAS PubMed.
- M. J. Enright, H. Sarsito and B. M. Cossairt, Chem. Mater., 2017, 29, 666–672 CrossRef CAS.
- J. E. Halpert, V. J. Porter, J. P. Zimmer and M. G. Bawendi, J. Am. Chem. Soc., 2006, 128, 12590–12591 CrossRef CAS PubMed.
- S. Kumar, M. Jones, S. S. Lo and G. D. Scholes, Small, 2007, 3, 1633–1639 CrossRef CAS PubMed.
- F. Wang, R. Tang and W. E. Buhro, Nano Lett., 2008, 8, 3521–3524 CrossRef CAS PubMed.
- L. Carbone, C. Nobile, M. De Giorgi, F. D. Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- B. Koo and B. A. Korgel, Nano Lett., 2008, 8, 2490–2496 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00056e |
| This journal is © the Partner Organisations 2018 |