
Alternating donor–acceptor indigo-cyclopentadithiophene copolymers: competition between excited state conformational relaxation, energy transfer and excited state proton transfer†
João
Pina
 a,
Mohamed
Alnady
a,
Anika
Eckert
b,
Ullrich
Scherf
b and
J. Sérgio
Seixas de Melo
*a
a,
Mohamed
Alnady
a,
Anika
Eckert
b,
Ullrich
Scherf
b and
J. Sérgio
Seixas de Melo
*a
aCQC, Department of Chemistry, University of Coimbra, P3004-535 Coimbra, Portugal. E-mail: sseixas@ci.uc.pt
bBergische Universitat, Wuppertal, Macromolecular Chemistry Group (buwmakro) and Institute for Polymer Technology, Gauss-Str. 20, D-42097, Wuppertal, Germany
First published on 30th November 2017
Abstract
A comprehensive characterization of the electronic excited states of an alternating donor–acceptor indigo-[didodecyl-cyclopentadithiophene] copolymer (IndC12CPDT) together with a model compound (with one indigo unit and two C12CPDT units) was undertaken. The two chromophoric units of the copolymer, indigo and C12CPDT, were independently investigated. The study encompasses qualitative absorption, fluorescence, phosphorescence emission and transient singlet–singlet and triplet–singlet difference spectra, together with quantitative measurements of quantum yields and lifetimes. The excited state dynamics of the trimeric model compound and copolymer was investigated in solution and in the solid state. Namely the competition of conformation relaxation processes with energy transfer/migration between the didodecyl-cyclopentadithiophene moiety (donor) and indigo (acceptor) units, together with the occurrence of fast excited state proton transfer (ESPT, from the N–H to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of indigo units), was observed. Our results demonstrate that ESPT and intrachain energy transfer are competitive processes in the IndC12CPDT copolymer excited state both in solution and in the solid state while conformational relaxation was found to be absent.
O group of indigo units), was observed. Our results demonstrate that ESPT and intrachain energy transfer are competitive processes in the IndC12CPDT copolymer excited state both in solution and in the solid state while conformational relaxation was found to be absent.
Introduction
In the organic solar cells field thiophene-based polymers cyclopentadithiophene (CPDT) polymers have received particular attention because of their high conductivities and narrow band gaps.1 Cyclopentadithiophene-based copolymers have shown promising power conversion efficiencies in bulk heterojunction organic photovoltaic devices of up to 3.8%.2 CPDT-based copolymers showed much higher PCEs, e.g. for PCPDTBT a PCE of 5.5% was reported 10 years ago3 and more recently a value of 10.6% was reported for CPDT-based copolymers when incorporated into tandem solar cells. This value approaches the maximum value obtained to date for organic ternary solar cells of 12%.4 The absorption behavior of indigo as a small molecule makes it attractive for application in organic solar cells. However, the poor solubility of indigo itself makes the design of solar cells challenging. Hence, coupling indigo as an acceptor compound with solubilizing donor groups to synthesize indigo-containing polymers is a strategy to obtain soluble indigo copolymers.Much research has been focussed on alternating donor–acceptor copolymers containing isoindigo and diketopyrrolopyrrole (DPP) chromophores for applications in photovoltaic devices and organic field-effect transistors, while the corresponding copolymers containing indigo units have been poorly investigated until now.5,6 Indigo seems to be a unique case as an acceptor building block since the longest wavelength absorption band is weakly influenced during the incorporation of indigo units into alternating copolymers as demonstrated for the previously investigated fluorene–indigo system.6 In the present study we found that it was interesting to switch to copolymers with a stronger donor–acceptor character by replacing the weak donor unit fluorene by a stronger donor unit CPDT. The hereby, synthesized and investigated, resulting low bandgap copolymers are likely to be attractive for applications in organic electronic devices. However, the motivation of the present work consists of a fundamental study of the electronic and optical properties of indigo-containing donor–acceptor copolymers.
The remarkable (photo)stability of indigo is imparted to the fast intramolecular excited state proton transfer involving the proton from the amine group and oxygen from the carbonyl group, which was found to involve a single proton transfer.7,8 In the ground state the planar cis-conformation of indigo is due to the hydrogen bonding involving the two N–H and two CO groups. These ground- and excited-state properties of indigo preclude the photoinduced trans–cis isomerization that is observed for other derivatives such as thioindigo.
Although a polymeric indigo has been known since 1990 – first prepared by Tanaka et al. consisting of indigo units connected by methylene bridges9 – the literature regarding this type of polymer is scarce in comparison with its isomeric isoindigo. This is likely due to the weaker hydrogen bonds that are established with isoindigo in comparison with indigo. The Tanaka polymeric indigo was found to be insoluble due to the lack of solubilizing units. More recently, Voss et al. and Seixas de Melo et al. investigated other indigo-based polymers, with indigo units linked by methylene bridges and in some of them with N-acetyl instead of N–H units (these aimed to avoid the strong hydrogen bonds between amine and carbonyl groups, by using indoxyl and indolone co-monomers for the coupling with indigo), which were found to be soluble in various media.10,11
Other alternative synthetic methodologies to make indigo-based soluble polymers were carried out more recently by Głowacki et al.12 (2014), and Guo et al.13 (2015), in which the indigo-containing polymers are protected by tert-butyloxycarbonyl groups (boc protecting group) which can be further cleaved under thermal or acidic conditions. More recently, Bronstein et al. (2016)14 synthesized another (rigid) indigo derivative linked to thiophene units – the indolo-naphthyridine-6,13-dione thiophene – which showed, due to its rigid structure, good appetence for organic electronic applications.
This work aims to contribute to the first detailed study of the photophysical properties of an indigo-[didodecyl-CPDT] copolymer together with the oligomeric model compound, in which indigo is sandwiched by two donor units, which will allow conclusions to be drawn regarding the competition between all the excited state decay processes potentially involved in these polymers: energy transfer, conformational relaxation and excited state proton transfer.
Experimental section
All the solvents were of spectroscopic grade and were used as received. Absorption and fluorescence spectra were recorded on Cary 5000 UV-Vis-NIR and Horiba-Jobin-Ivon Fluorog 3-22 spectrometers respectively. Phosphorescence measurements were made in methylcyclohexane glasses at 77 K and the Horiba-Jobin-Ivon Fluorog 3-22 spectrometer was used in phosphorescence mode (using a pulsed excitation source).The fluorescence quantum yields were measured using α,α′-bithiophene in dioxane solution (ϕF = 0.017)15 and cresyl violet perchlorate (ϕF = 0.54)16,17 in methanol solution as standards, while the phosphorescence quantum yields were determined using benzophenone (ϕPh = 0.84)18 in ethanol solution as a standard. The errors associated with these determinations are <5%.
Fluorescence decays were measured using a home-built picosecond time correlated single photon counting, TCSPC, apparatus (3 ps time resolution) described elsewhere.19 The fluorescence decays and the instrumental response function (IRF) were collected using a time scale of 1024 (or 4096) channels, until 5 × 103 counts at maximum were reached. Deconvolution of the fluorescence decay curves was performed using the modulating function method, as implemented by G. Striker in the SAND program, as previously reported in the literature.20
The experimental setup used to obtain the triplet–triplet absorption spectra has been described elsewhere.21
The experimental setup for ultrafast spectroscopic and kinetic measurements consists of a broadband (350–1600 nm) HELIOS pump–probe femtosecond transient absorption spectrometer from Ultrafast Systems, equipped with an amplified femtosecond Spectra-Physics Solstice-100F laser (800 nm central wavelength displaying a pulse width of 128 fs at 1 kHz repetition rate) coupled with a Spectra-Physics TOPAS Prime F optical parametric amplifier (195–22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nm) for pulse pump generation. Probe light in the Vis and NIR range was generated by passing a small portion of the 800 nm light from the Solstice-100F laser through a computerized optical delay (with a time window up to 8 ns) and focusing it on a sapphire plate to generate a white-light continuum in the 440–800 nm and 800–1600 nm range, respectively. All the measurements were obtained in a 2 mm quartz cuvette with optical densities lower than 0.3 at the pump excitation wavelength. The instrumental response function of the system was assumed to be equal to that of the pump–probe cross correlation determined from the measurement of the instantaneous stimulated Raman signal from the pure solvent (in a 2 mm cuvette). Typical values for the IRF of the system were found to be better than 250 fs. To avoid photodegradation the solutions were stirred during the experiments or in movement using a motorized translating sample holder. Transient absorption data were analyzed using the Surface Xplorer PRO program from Ultrafast Systems and the global analysis of the data (from which the lifetimes of the observed transients were obtained) was performed using Glotaran software.22
000 nm) for pulse pump generation. Probe light in the Vis and NIR range was generated by passing a small portion of the 800 nm light from the Solstice-100F laser through a computerized optical delay (with a time window up to 8 ns) and focusing it on a sapphire plate to generate a white-light continuum in the 440–800 nm and 800–1600 nm range, respectively. All the measurements were obtained in a 2 mm quartz cuvette with optical densities lower than 0.3 at the pump excitation wavelength. The instrumental response function of the system was assumed to be equal to that of the pump–probe cross correlation determined from the measurement of the instantaneous stimulated Raman signal from the pure solvent (in a 2 mm cuvette). Typical values for the IRF of the system were found to be better than 250 fs. To avoid photodegradation the solutions were stirred during the experiments or in movement using a motorized translating sample holder. Transient absorption data were analyzed using the Surface Xplorer PRO program from Ultrafast Systems and the global analysis of the data (from which the lifetimes of the observed transients were obtained) was performed using Glotaran software.22
Time-resolved fluorescence emission and transient absorption studies as a function of temperature were performed using an Oxford Instruments Optistat DN2 cryostat or a Quantum Northwest Flash 300 cuvette holder and temperature controller TC125.
Thin films from the compounds were obtained with a Desk-Top Precision Spin Coating System, model P6700 series from Speedline Technologies. The solid-state thin film from the samples was obtained by deposition of a few drops of solution onto a circular sapphire substrate (10 mm diameter) followed by spin coating (2500 rpm) in a nitrogen-saturated atmosphere (2 psi). Solutions for spin-coating were prepared by dissolving 1.8 mg of the samples in 200 μL of chloroform solution with stirring at 40 °C for 30 min.
Synthesis and characterization
:40 mixture of monostannylated (4,4-didodecyl-4H-cyclopenta[1,2-b:5,4-b′]dithiophene-2-yl)trimethylstannane and non-substituted 4,4-didodecyl-4H-cyclopenta[1,2-b:5,4-b′]dithiophene (containing 0.887 mmol stannyl equivalents) were dissolved in 15 mL tetrahydrofuran. The microwave-assisted reaction was done at 125 °C for 15 minutes followed by stirring for 14 h at 90 °C (see Scheme 1). The blue solution was diluted with chloroform and washed with 2 M hydrochloric acid. After multiple silica column chromatographies with hexane/dichloromethane 187 mg (39%) of the trimer were obtained.
 | ||
| Scheme 1 Synthesis of the trimeric model compound, MC. | ||
1H-NMR (600 MHz, CDCl3): δ [ppm] = 9.36 (bs, 2H), 7.64 (d, 2H), 7.30 (s, 2H), 7.21–7.17 (m, 4H), 7.07 (d, 2H), 6.94 (d, 2H), 1.95–1.78 (m, 8H), 1.40–1.11 (m, 72H), 1.08–0.93 (m, 8H), 0.86 (t, 12H).
13C-NMR (150 MHz, CDCl3): δ [ppm] = 186.9, 159.1, 158.9, 152.5, 143.1, 142.6, 138.9, 136.3, 126.2, 124.9, 122.6, 121.6, 119.8, 118.3, 117.6, 107.5, 53.6, 37.7, 31.9, 30.0, 29.7, 29.6, 29.4, 29.3, 24.6, 22.7, 22.6, 14.1.
MS (MALDI-TOF): [C82H114N2O2S4] m/z = 1288.4.
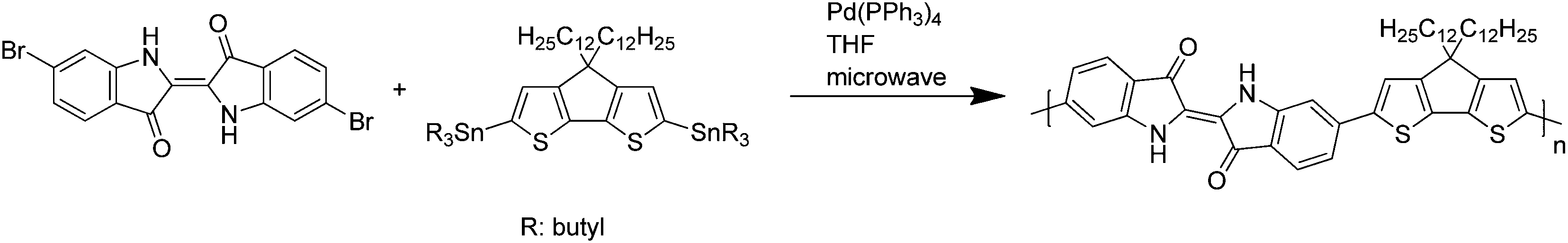 | ||
| Scheme 2 Synthesis of the INDC12CPDT copolymer. | ||
GPC: (DCM fraction, solvent: trichlorobenzene): Mn [g mol−1] = 7400; Mw [g mol−1] = 49400; PDI = 6.68.
1H-NMR (600 MHz, C2D2Cl4): δ [ppm] = 8.91, 7.67, 7.33, 7.21, 6.93, 3.03–0.55.
Recording 13C-NMR spectra was impossible due to the limited solubility.
Results and discussion
Synthesis and characterization of the copolymer
For the synthesis of the IndC12CPDT copolymer a Stille-type aryl–aryl coupling reaction was applied. Equimolar amounts of 6,6′-dibromoindigo, (4,4-didodecylcyclopenta[1,2-b:5,4-b′]dithiophene-2,6-diyl)bis(tributylstannane) and 17.6 mol% of (4,4-didodecyl-4H-cyclopenta[1,2-b:5,4-b′]dithiophene-2-yl)trimethylstannane as an end capper as well as tetra(triphenylphosphine)palladium(0) as a catalyst were dissolved in tetrahydrofuran. The coupling took place under microwave heating for 15 minutes at 125 °C followed by stirring at 90 °C for a further 72 h. After an aqueous work-up the concentrated polymer solution was precipitated into methanol. Subsequent solvent extraction gave a DCM fraction of the copolymer in 48% yield with a molecular weight of 7400 g mol−1. The synthesis of the trimeric model compound (MC) was accomplished by a Stille-type coupling with tetra(triphenylphosphine)palladium(0) as a catalyst and tetrahydrofuran as a solvent for 15 minutes microwave heating at 125 °C, followed by stirring at 90 °C for a further 14 hours. The crude blue trimer was purified by multiple column chromatography. The synthesis of C12CPDT followed procedures described in the literature and will be published in an independent work.23–25The structures and acronyms of the investigated compounds are depicted in Scheme 3, consisting of an alternating indigo-[didodecyl-cyclopentadithiophene] copolymer, IndC12CPDT, together with the trimeric model compound, MC, and the two individual chromophores: C12CPDT and indigo.
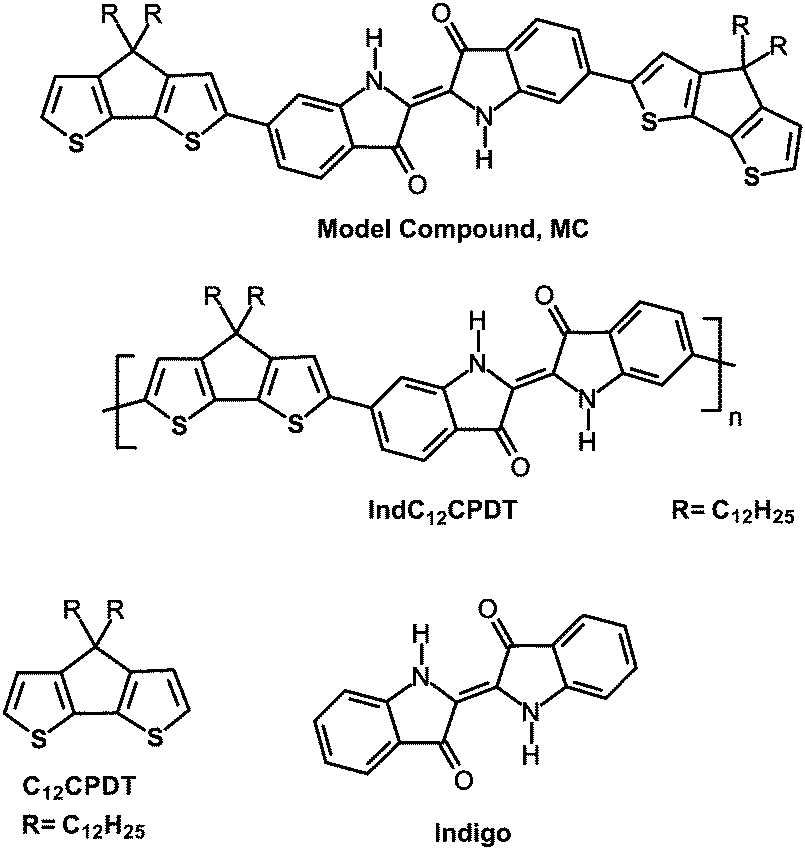 | ||
| Scheme 3 Structures of the alternating indigo-[didodecyl-cyclopentadithiophene] copolymer together with the trimeric (model) compound, MC, of C12CPDT and indigo. | ||
Spectroscopic properties
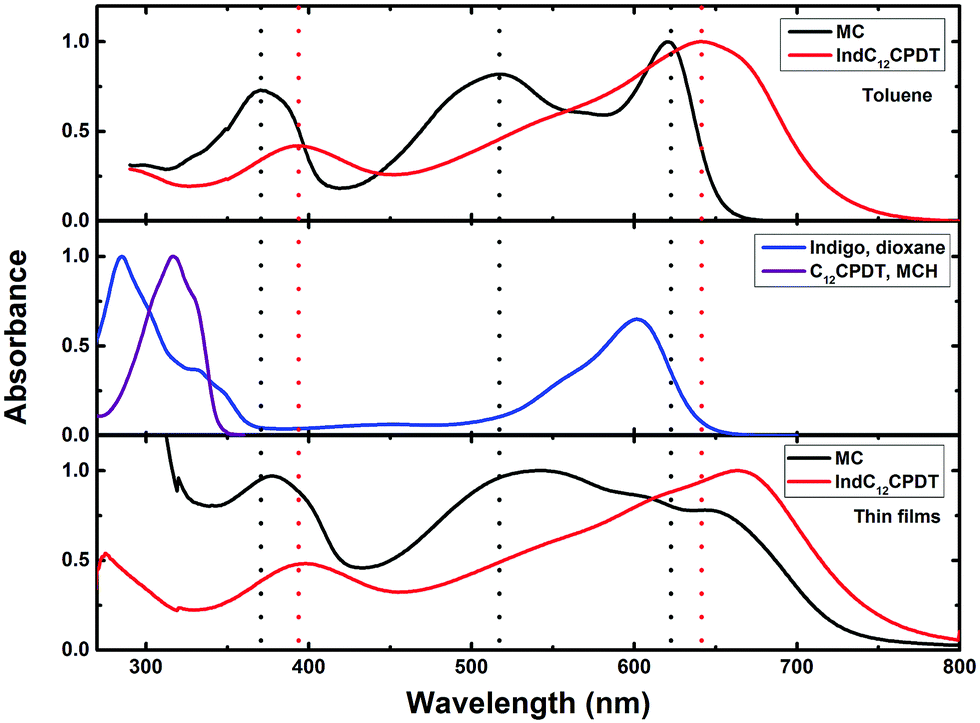 | ||
| Fig. 1 Room temperature normalized absorption spectra for the model compound (MC) and indigo-[didodecyl-cyclopentadithiophene] copolymer (IndC12CPDT) in toluene solution and in the solid state (thin films), together with the spectra of indigo in dioxane solution and cyclopentadithiophene in methylcyclohexane (MCH) solution. The vertical dashed lines are just meant to be guides to the eye. | ||
The absorption spectra of the oligomeric and polymeric IndC12CPDT derivative (consisting of one indigo unit linked to two didodecyl-CPDT units) show the characteristic absorption features of its chromophoric constituents (although red-shifted) together with an additional broad band, in-between the two above-mentioned (indigo and C12CPDT). In the case of the model compound (MC) a clear additional absorption band, with a maximum at ∼517 nm, also present in IndC12CPDT, but in this compound buried underneath the intense long wavelength absorption, is observed (Fig. 1 and Table 1).
| Compd | λ Absmax (nm) 293 K | λ Fluomax (nm) 293 K | λ Phosphmax (nm) 77 K |
|
ϕ F 293 K | τ F (ps) 293 K | ϕ Ph (77 K) | τ Ph (ms) 77 K | τ T (μs) 293 K |
|---|---|---|---|---|---|---|---|---|---|
| a Data obtained with λexc = 552 nm. b Data in DMF taken from ref. 27 in which it constitutes the major decay component. c Obtained by Pulse Radiolysis in benzene solution in the presence of biphenyl or naphthalene as triplet sensitizers.27 d In the case of the fluorescence decay times presented, these are the major decay components. Other decay times are present and associated with the ESPT and ET processes; see the text for more details. e The detailed data on C12CPDT will be the subject of an independent work.26 | |||||||||
| C12CPDTe | 316 | 363 | 610 | 370 | 0.05 | 166 | 0.004 | 5.9 | 64 |
| Indigo | 285, 601 | 645 | — | 540c | 0.0023b | 140b | — | — | 30 |
| MC | 372, 517, 620 | 650 | — | 720 | 0.004a | 50 | — | — | 2.9 |
| (378, 540, 645) | — | ||||||||
| IndC12CPDT | 393, 642 | 656 | — | 740 | 0.002a | 70 | — | — | 6.5 |
| (397, 663) | — | ||||||||
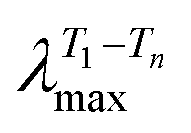
Upon going from the oligomeric model compound to the copolymer a significant red shift of the absorption spectra (∼21–33 nm depending on the band considered) is observed. In addition, for IndC12CPDT, the significant red shift of the in-between (517 nm) absorption band when compared to the model compound originates a high superposition, appearing as a shoulder, with the low-energy absorption band (Fig. 1). Additionally, the absorption features of the 517 nm absorption band in the investigated compounds are in agreement with what was previously observed for indigo-fluorene copolymers and thus can be assigned to a charge transfer transition.6
When comparison is now made with indigo and (i) the MC and (ii) the copolymer (IndC12CPDT), a significant red-shift of 19 nm and 41 nm, respectively, is found for the low-energy absorption. This clearly suggests that, in the copolymer (IndC12CPDT) due to the high superposition of the in-between with the lowest absorption band (when compared to the MC), the nature of the CT transition is more pronounced in the copolymer than in the MC.
Upon going to the solid state (thin films) the absorption bands for the MC and IndC12CPDT are found to be red shifted with a good match, relative to the solution spectra (Fig. 1 and Table 1). This is clearly more evident in the case of IndC12CPDT with an almost identical shape of the solution and the solid state spectra (although with a different maxima).
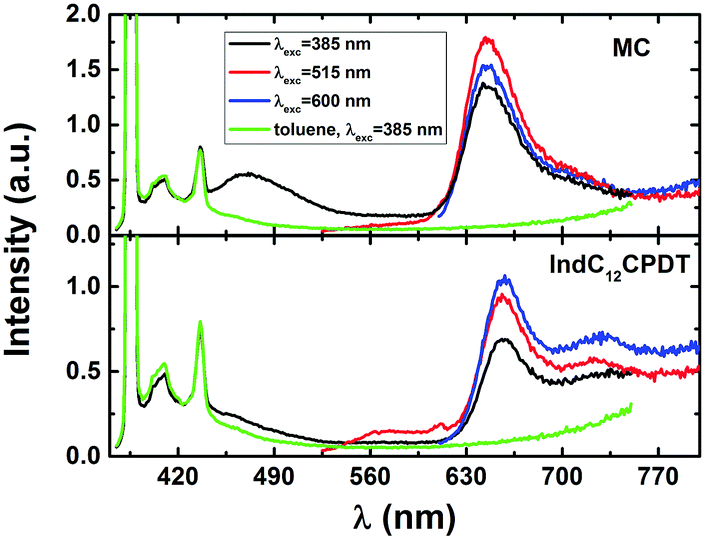 | ||
| Fig. 2 Fluorescence emission spectra for the copolymer (bottom panel) and model compound (top panel) collected at different excitation wavelengths in toluene solution at 293 K. For comparison purposes the spectra of the solvent toluene with excitation at 385 nm were also included. | ||
In order to explore and support the possible occurrence of intramolecular energy transfer from the C12CPDT moiety to the indigo unit in the IndC12CPDT copolymer, the fluorescence excitation spectra were collected on the onset and tail of the low-energy emission band, see Fig. 2 and Fig. S1 in ESI.† Although (i) the emission spectra of the investigated copolymer when excitation is performed in the C12CPDT-related absorption band (λexc = 385 nm, see Fig. 2) display the characteristic emission of the indigo moiety and (ii) the fluorescence excitation spectra collected in the indigo-related emission band match the characteristic absorption bands of the copolymer, it is not possible to conclude unequivocally, based only on the steady-state fluorescence study, on the occurrence of intramolecular energy transfer since in this copolymer, whenever the excitation is made, direct excitation of indigo is always present (see Fig. 1), due to the broad absorption spectra of indigo. However, the excitation spectrum reveals additional features. Indeed, the difference between the absorption and fluorescence excitation spectra (Fig. S1, ESI†) indicates a more localized nature of the singlet excited state.
In contrast to what was seen in solution, in the solid state (thin films) when excitation was made in the characteristic absorption bands of C12CPDT or indigo, no fluorescence emission was observed for MC and IndC12CPDT. This is a typical signature of fluorescence aggregation-caused quenching (ACQ) in a rigid medium and indicates that for these compounds, in the solid state, the excited state radiative decay becomes a non-competitive deactivation pathway. This behavior, in general, is characteristic of the formation of H-type aggregates in the solid state, i.e., where the molecules are aligned parallel to each other with strong intermolecular interaction and tend to induce the radiationless deactivation process.28
Photophysical data
The fluorescence quantum yields (ϕF) and lifetimes (τF) for the investigated compounds are presented in Table 1. No significant changes are found in the ϕF values between the oligomeric model compound and the copolymer (values in the 0.002–0.004 range) when excited in the absorption band of the indigo moiety (λexc = 552 nm). These values are in good agreement with the ϕF value found for indigo in dimethylformamide solution (ϕF = 0.0023),27 see Table 1, showing that the emission is due to the indigo chromophore.Fluorescence lifetimes for the oligomeric model compound and copolymer were obtained with λexc = 385 nm and collected along the indigo-related emission band using a 3 ps time-resolution TCSPC (see Fig. 3). The global analysis of the fluorescence decays for the MC and IndC12CPDT copolymer in toluene solution reveals that these are well fitted with sums of two and three exponentials, respectively. For the MC, a fast decay component in the 3.7–14 ps range and a longer decay time of 53 ps were found, while for IndC12CPDT three decays times of 3.8 ps, 14 ps and 74 ps were obtained (see Fig. 3). It is possible to observe from Fig. 3 that with the increase in the emission wavelength the absolute value of the negative pre-exponential, which is associated with the short decay time, increases concomitantly with the increase in the pre-exponential value associated with the longer decay component at lower energies (longer wavelengths). This means that on the low energy side of the emission band the longer decay component is being formed during the decay of the shorter components (appearing as a rising component, i.e., a negative pre-exponential value in the analysis, see Fig. 3). In general, this behavior is a typical signature of an excited state process involving two species, with only one present in the ground state and the second being formed during the decay of the first. In the present case, several decay mechanisms can be invoked to explain the decays of the MC and IndC12CPDT, including the occurrence of (i) fast relaxation processes; (ii) excited state proton transfer between the N–H and CO groups of indigo (ESPT in the initially formed excited state keto form to give the enol form7); (iii) energy migration in a ladder-type mechanism (present only in the case of the copolymer). Indeed in previous reports, these relaxation processes have been attributed either to (iii) excitation energy migration along the polymer to lower energy conjugated segments or to (i) conformational relaxation processes in the first singlet excited state.19,29 Since in the MC compound the energy transfer process is inoperative the fast decay component can be attributed to torsional/conformational relaxation (from an instantaneously formed excited state that rapidly relaxes to a lower energy excited state conformation) or (ii) to the occurrence of fast excited state proton transfer within the indigo moiety. In the case of the copolymer all these processes can be operative (energy transfer, torsional/conformational relaxation and ESPT). The contribution of these processes in the excited state of the investigated compounds will be discussed in detail in the following femtosecond transient absorption section.
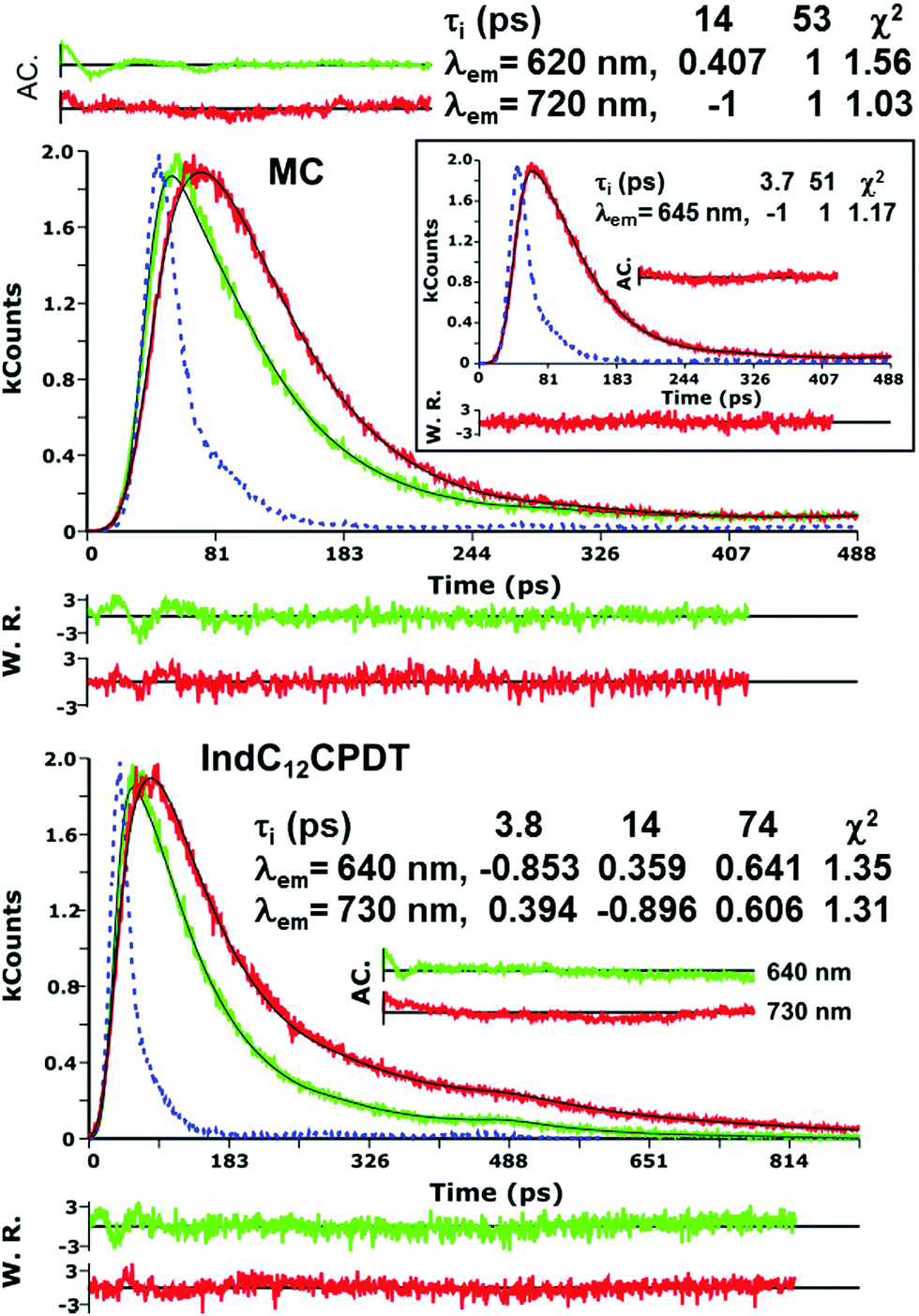 | ||
| Fig. 3 Fluorescence decays for the model compound (MC) and indigo-[didodecyl-cyclopentadithiophene] copolymer, IndC12CPDT, obtained with λexc = 385 nm in toluene solution at 293 K. For a better judgment of the quality of the fits, weighted residuals (W.R.), autocorrelation functions (A.C.) and χ2 values are also presented. The dashed line in the decays is the instrumental response function. | ||
The transient triplet–triplet absorption spectra following laser flash photolysis at 355 nm (nanosecond transient absorption, ns-TA) of degassed toluene solutions were obtained for the model compound and for IndC12CPDT, see Fig. S2 in the ESI.† In addition to ground-state depletion, at shorter wavelengths, broad triplet–triplet absorption bands in the 650–800 nm region were found (Fig. S2, ESI† and Table 1). The transient triplet–triplet absorption spectra and triplet lifetimes (τT = 2.9–6.5 μs) of the MC and IndC12CPDT show, when compared with the data obtained for C12CPDT in methylcyclohexane (transient absorption in the 340–410 nm region and τT = 64 μs) and indigo in benzene solution (transient absorption in the 450–700 nm region and τT = 30 μs),27 electronic delocalization/interactions involving the C12CPDT and indigo units in the triplet state.
Femtosecond transient absorption (fs-TA)
To further investigate the excited state dynamics of IndC12CPDT and the MC, the time-resolved femtosecond transient difference absorption spectra (fs-TA) were recorded in toluene in the 440–830 nm and 800–1600 nm range with excitation at the C12CPDT (370 nm) and indigo (600 nm) characteristic absorption unit bands. The two surfaces were merged (with normalization in the 800–810 nm range) using Surface Xplorer software (see Fig. 4). The fs-TA show that the spectra are dominated by positive broad transient absorption bands (in the 690–1570 nm range for IndC12CPDT and 650–1590 nm for the MC), together with negative transient absorption bands that in agreement with the spectroscopic features presented in Fig. 1 are attributed to the ground state absorption (GSA).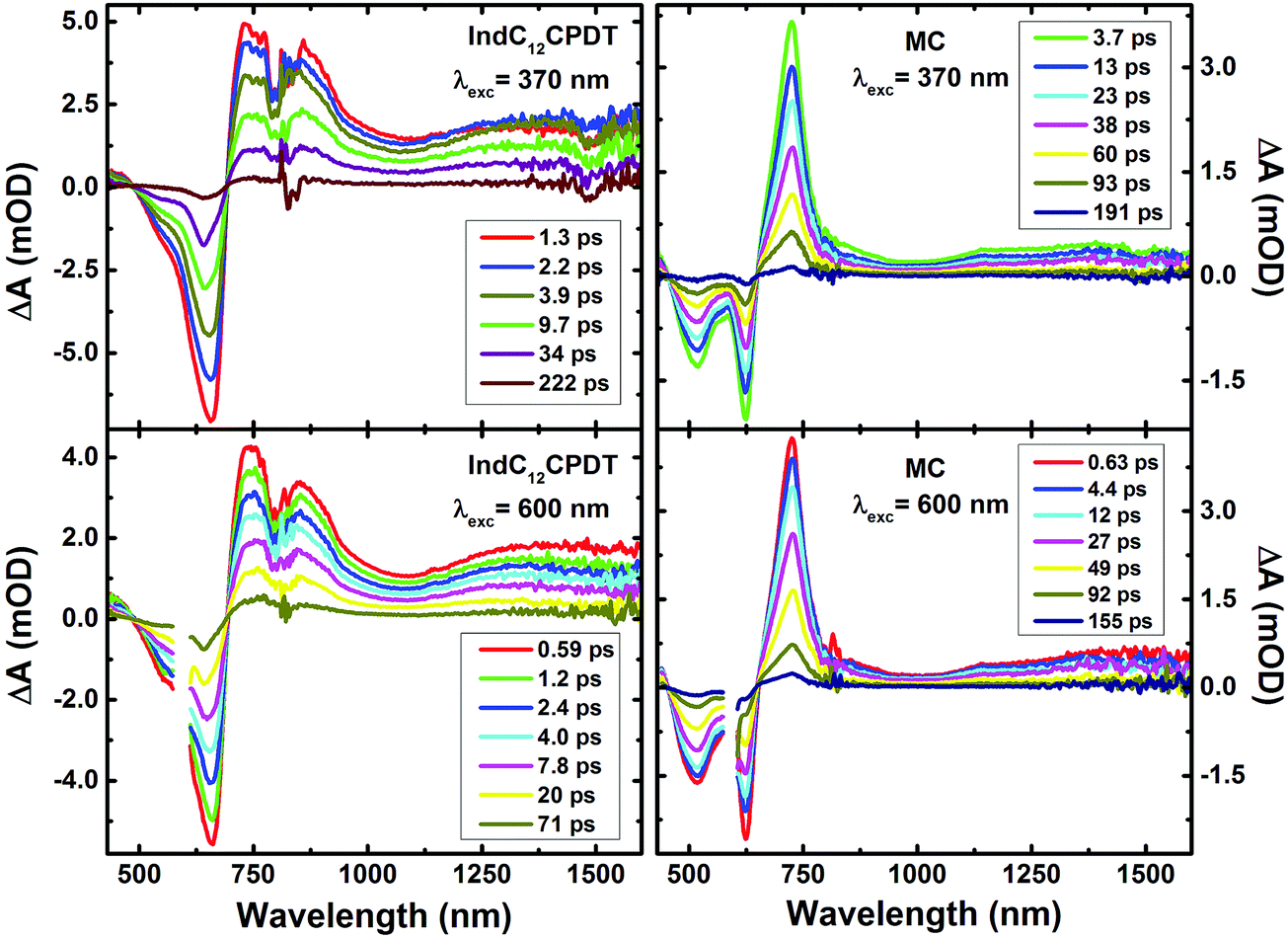 | ||
| Fig. 4 Time-resolved transient absorption data for IndC12CPDT and the MC obtained with excitation at 370 nm and 600 nm in aerated toluene solutions at room temperature. For the data collected with excitation at 600 nm the spectral range 580–610 nm is not shown because it is disturbed by the scattered pump beam. | ||
In addition, for comparison, the transient absorption spectra for the C12CPDT and indigo precursors are presented in the ESI,† see Fig. S3 and S4. The C12CPDT and indigo chromophores present positive transient bands in the 410–700 nm range with maxima at 487 nm and 630 nm, respectively (see Fig. S3 and S4, ESI†), which were attributed to the singlet excited state absorption. Contrary to indigo, the C12CPDT unit also displays an additional positive transient absorption at higher delay times (in the 340–410 nm range) that was assigned to the characteristic transient triplet–triplet absorption with maxima at 385 nm.
Analysis of the fs-TA data for the MC and IndC12CPDT was performed by global fit analysis (using a sequential model) after singular value decomposition (SVD). The best-fit results and representative kinetic traces of the characteristic transient absorption data are presented in Table 2 and Fig. 5.
| Compd | Solvent | λ exc (nm) | Temp. (K) | τ 1 (ps) | τ 2 (ps) | τ 3 (ps) | τ 4 (ps) | τ 5 (μs) |
|---|---|---|---|---|---|---|---|---|
| a This transient lifetime was fixed in the global analysis to the triplet lifetime obtained by ns-TA, see Table 1. b Data from ref. 7. | ||||||||
| Indigob | 2MeTHF | 550 | 293 | 0.09 | 9.4 | — | 99 | — |
| MC | Toluene | 600 | 293 | 0.041 | 6.5 | — | 53 | — |
| Toluene | 370 | 293 | 0.25 | 1.3 | — | 51 | 2.9 | |
| IndC12CPDT | Toluene | 600 | 293 | 0.041 | 2.5 | 18 | 130 | — |
| Toluene | 370 | 293 | 0.734 | 4.4 | 23 | 102 | 6.5 | |
| Toluene | 370 | 280 | 0.831 | 7.4 | 34 | 86 | 6.5 | |
| Toluene | 370 | 269 | 0.017 | 4.7 | 33 | 229 | 6.5 | |
| Toluene | 370 | 256 | 0.018 | 4.3 | 25 | 270 | 6.5 | |
| Toluene | 370 | 234 | 0.445 | 3.32 | 18 | 106 | 6.5 | |
| Toluene | 370 | 213 | 0.649 | 4.8 | 33 | 326 | 6.5 | |
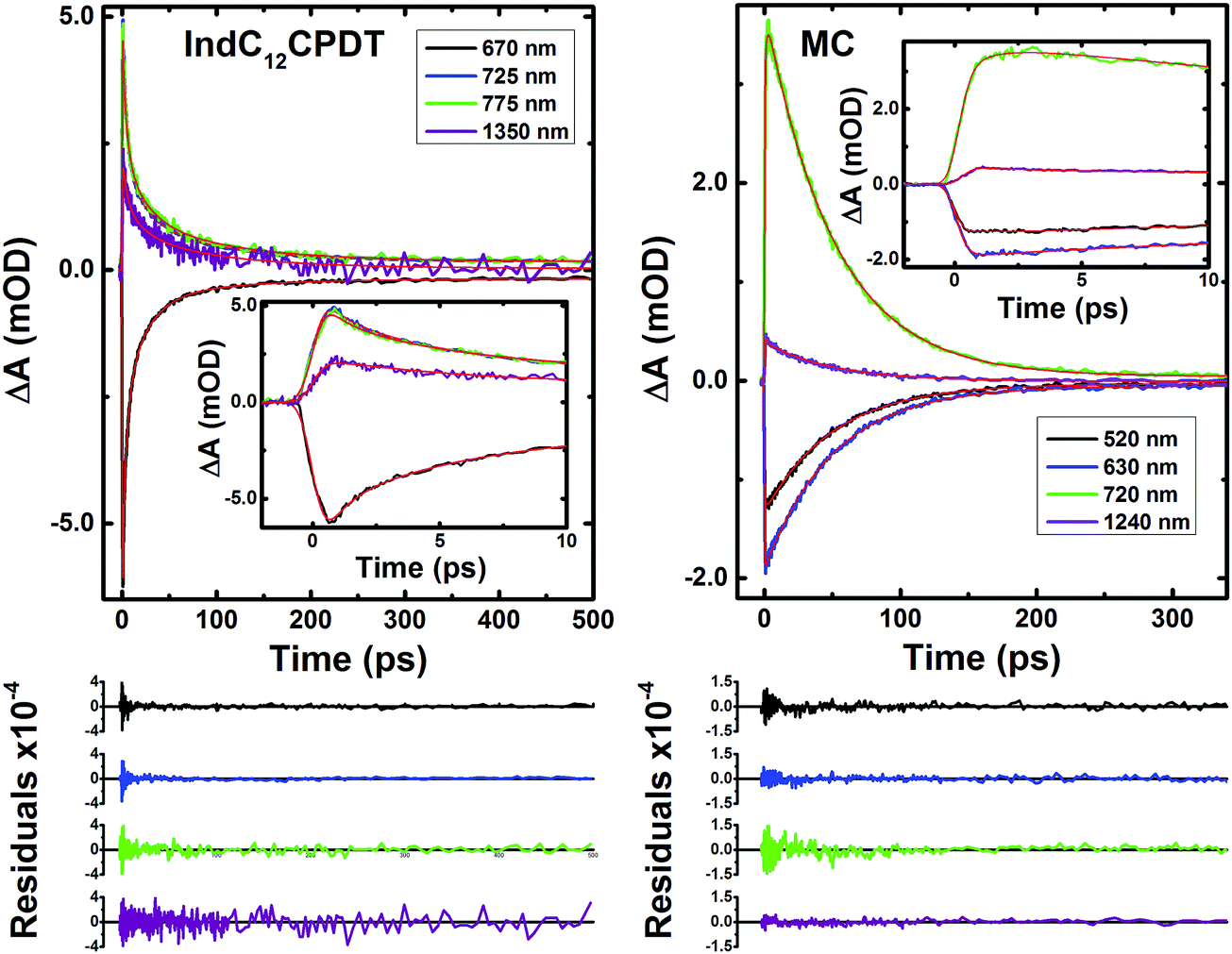 | ||
| Fig. 5 Representative kinetic traces with fits from the global analysis of the transient absorption data for IndC12CPDT and the MC collected in aerated toluene solutions with λexc = 370 nm. Also shown as inset are the decays at shorter times. For a better judgment of the quality of the fits the residuals are also presented. | ||
For these derivatives and depending on whether the excitation was performed in the characteristic absorption bands of indigo (600 nm) or C12CPDT (370 nm), different numbers of decay time constants are needed to properly fit the fs-TA data (in the 440-1450 nm range). Indeed, contrary to what was observed for the MC and IndC12CPDT copolymer with excitation at 370 nm, where a long-lived component (fixed to the triplet state lifetime obtained from laser flash photolysis, 2.9 μs for the MC and 6.5 μs for IndC12CPDT, see Table 1) was necessary to suitably fit the decays (Table 2), when excited at 600 nm the long-lived triplet decay time was not observed. This is observed in Fig. S5 (ESI†) where it can be seen that the kinetic traces completely decay within the monitored time window. Nevertheless, for the investigated compounds, with the exception of the triplet decay time, similar transient lifetimes are observed with excitation at 370 nm or 600 nm, with values ranging from 0.041–0.25 ps, 1.3–6.5 ps and 51–53 ps for the MC and 0.041–0.734 ps, 2.5–4.4 ps, 18–23 ps and 102–130 ps for the IndC12CPDT.
The 51–53 ps and 102–103 ps transient lifetimes found for the model compound and IndC12CPDT, respectively, are in agreement with the longer fluorescence lifetime obtained by TCSPC (53 ps for the MC and 74 ps for IndC12CPDT, see Fig. 3) and thus are assigned to the decay of the relaxed singlet excited state. Concerning the two fastest decay transients these are shorter than the value expected for the solvent relaxation time of toluene, 2.4 ps34 and thus solvent dynamics can be discarded. As previously reported for indigo (Fig. S4, ESI†) the analysis of the fs-TA data revealed the presence of three decay components, 0.090 ps, 9.4 ps and 98.8 ps, which were assigned to vibrational relaxation, formation of the keto form through excited state proton transfer and subsequent decay of the enol form to the ground-state, respectively. Indeed, for both the MC and IndC12CPDT, direct excitation of the indigo chromophore is always possible, thus (i) the sub-picosecond transient lifetimes (0.041–0.25 ps and 0.041–0.734 ps, respectively) are better assigned to vibrational relaxation due to the de-population of a hot vibrational state generated through the 370 nm and 600 nm excitation (since excitation was not performed in the lowest energy 0–0 electronic transition),35 while (ii) the 1.3–6.5 ps and 2.5–4.4 ps decay components, which are in good agreement with the shortest fluorescence lifetimes found for the MC, 3.7 ps (when the analysis is performed in the emission maxima) and IndC12CPDT, 3.8 ps and associated with a rise-time (see Fig. 3), can result from the ESPT within the indigo chromophore.
Noteworthy is the presence of an additional transient decay component for the IndC12CPDT, in the 18–34 ps range, when compared to the MC compound (see the τ3 values in Table 2). This observation is in agreement with the TCSPC data of this copolymer where a similar fluorescence lifetime was found, 14 ps, and was associated with a rise-time (Fig. 3). This time constant has been previously assigned to the occurrence of fast conformational relaxation or to excitation energy migration within the copolymer chromophoric units.36 Since there is an activation energy barrier accompanying the torsional relaxation process the associated rate constant, if this process is present, should be temperature-dependent. When this dependence is absent energy transfer is the dominant deactivation process. Thus, the time-resolved transient absorption behavior of the IndC12CPDT copolymer was also investigated in toluene solution as a function of temperature (see Table 2). From Table 2 and the Arrhenius-type plot in Fig. 6, in general, no significant changes are observed for the first three transient decay component values with the decrease in temperature. In the case of the first decay component this behavior supports the assignment of this sub-picosecond transient to vibrational relaxation. In addition, the Arrhenius-type plot (corrected for the implicit dependence of the solvent refractive index, nD, with temperature) of the reciprocal of the third transient lifetime (Fig. 6) shows that the dynamic process, in the origin of this fast decay, is temperature independent (Ea ≈ 0.01 kJ mol−1) and consequently conformational relaxation in the excited state can be ruled out. Based on these results the transient lifetime in the 18–34 ps range (τ3 in Table 2) should be assigned to the intrachain energy transfer process in IndC12CPDT. Thus, from the overall data obtained for the investigated compounds the average rate constant for the energy transfer process (kET) in the IndC12CPDT copolymer can be obtained by applying eqn (1):37,38
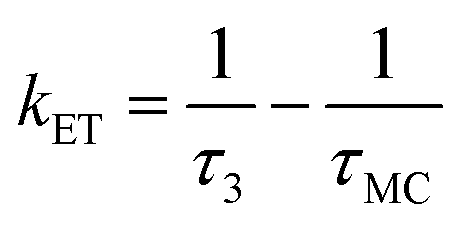 | (1) |
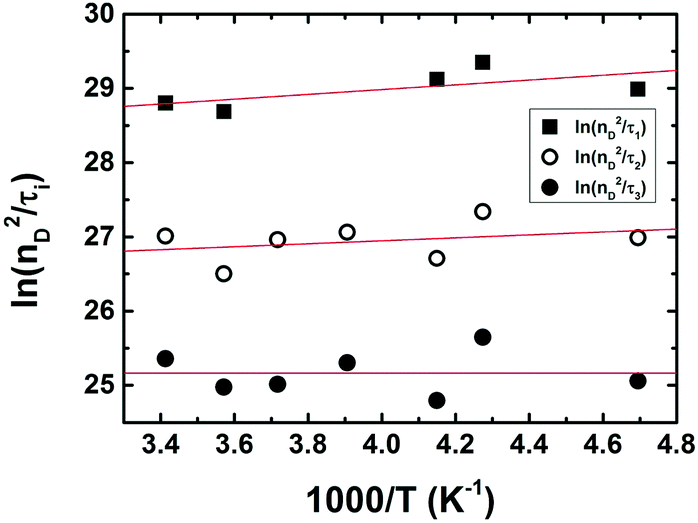 | ||
| Fig. 6 Arrhenius plots accounting for the temperature dependence of the solvent refractive index of the reciprocal of the transient lifetimes (nD2/τi) obtained from the global analysis of the fs-TA data of IndC12CPDT collected as a function of temperature in toluene solution. | ||
In summary in the IndC12CPDT copolymer deactivation of the excited state through conformational relaxation is absent while intra-chain energy transfer and ESPT (in the indigo chromophore) are the dominant deactivation processes.
fs-TA in the solid state (thin films)
The ultrafast dynamics of the MC and IndC12CPDT copolymer were also investigated in the solid state (thin films), Fig. 7. In agreement with what was previously observed in the ground-state absorption spectra the time-resolved femtosecond transient difference absorption spectra in the solid state, obtained with excitation at 370 nm, display similar transient bands in the 440–1600 nm range to those found in solution (see Fig. 5 and 7). In general, the results from the global analysis show that the best fit to the kinetic traces is obtained with the sum of three exponentials in the case of the MC and four exponentials for the IndC12CPDT copolymer (Fig. 8). In agreement with what was found in solution a long lifetime component was needed to properly fit the decays, and was attributed to the transient triplet state (fixed in the global analysis to the triplet lifetime obtained by ns-TA in toluene solution). | ||
| Fig. 7 7 Room temperature time-resolved transient absorption data for IndC12CPDT and the MC in the solid state (thin films) collected with excitation at 370 nm. | ||
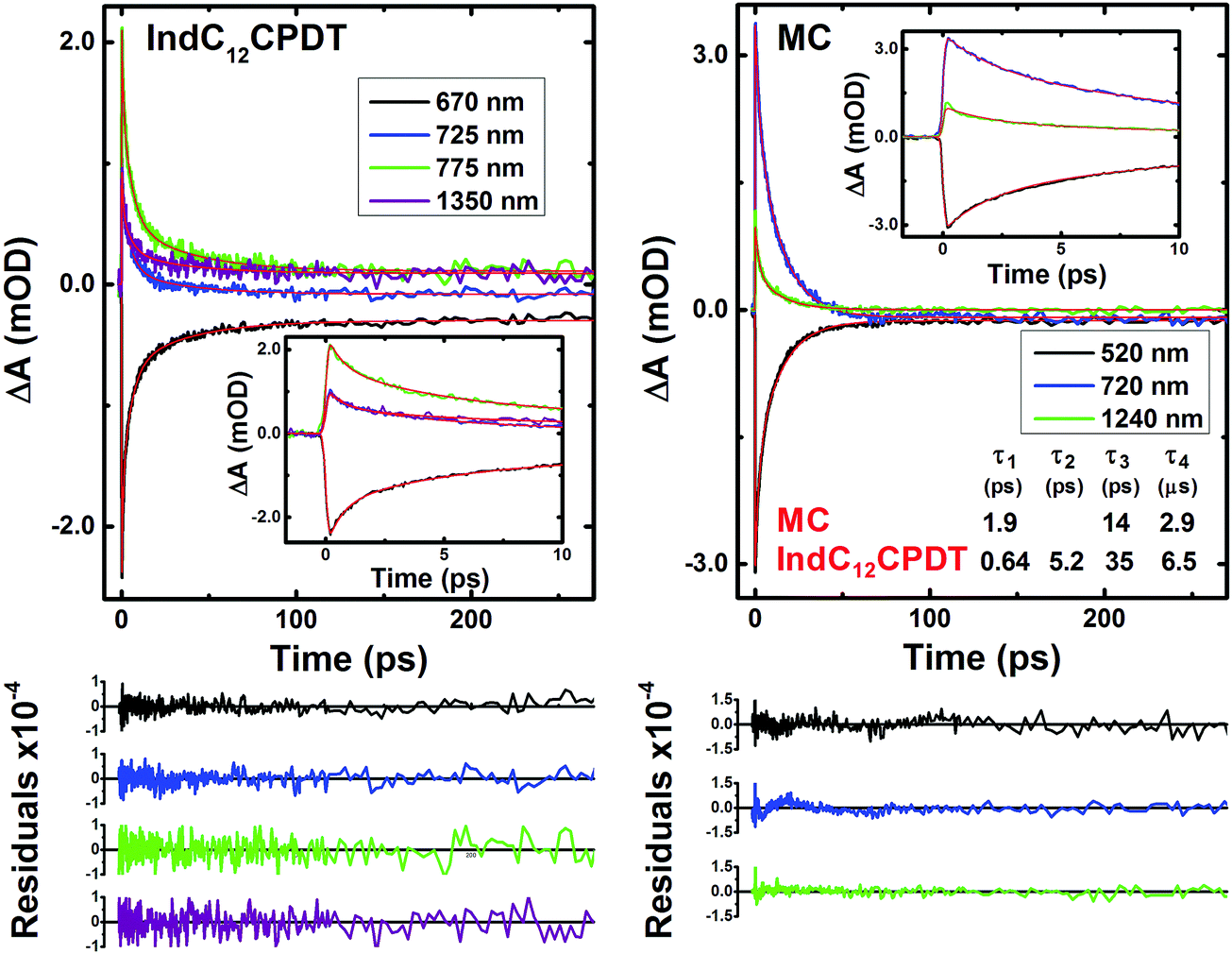 | ||
| Fig. 8 Representative kinetic traces with fits and results (lifetimes, τi) from the global analysis of the transient absorption data for IndC12CPDT and the MC collected in thin films with λexc = 370 nm. Also shown as inset are the decays at shorter times. For a better judgment of the quality of the fits the residuals are also presented. | ||
It is known that ESPT can occur in rigid glasses at very low temperatures and in the solid state.39,40 For the present compounds in the solid state we believe that this process is also active, although, compared to solution we expect a faster ESPT lifetime due to the higher proximity between the N–H and CO groups in the indigo units induced by the increased planarity and restricted movements in the solid state, thus prompting a faster intramolecular ESPT. Noteworthy is that, since in this case (λexc = 370 nm) the occurrence of fast vibrational relaxation cannot be discarded, we expect that the 1.9 ps and 0.64 ps transient lifetimes found for the MC and IndC12CPDT, respectively (Fig. 8), should comprise both the ESPT and vibrational relaxation processes or the ESPT occurs within the instrumental response function lifetime of our fs-TA setup (FWHM ∼ 250 ps).
Moreover, the 14 ps (MC) and 35 ps (IndC12CPDT) decay times are attributed to the non-radiative decay of the relaxed singlet excited state, while the additional lifetime of 5.2 ps (Fig. 8) found for the copolymer should be assigned to the energy transfer process previously identified in solution.
Conclusions
The spectroscopic and photophysical properties of an alternating indigo-[didodecyl-cyclopentadithiophene] copolymer and oligomeric model compound have been evaluated in solution at room and low temperatures. By comparison with indigo and the trimer model compound, it was found that in solution and in the solid state the deactivation of the excited state of the IndC12CPDT copolymer is not made through ESPT and intrachain energy transfer processes with kET = 1.1 × 1010 s−1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding by FEDER (Fundo Europeu de Desenvolvimento Regional) through COMPETE (Programa Operacional Factores de Competitividade). The Coimbra Chemistry Centre is supported by the Fundação para a Ciência e a Tecnologia (FCT), Portuguese Agency for Scientific Research, through the program UID/QUI/UI0313/2013. This work was performed under the project “SunStorage – Harvesting and storage of solar energy”, with reference POCI-01-0145-FEDER-016387, funded by European Regional Development Fund (ERDF), through COMPETE 2020 – Operational Program for Competitiveness and Internationalization (OPCI), and by national funds, through FCT. The FCT is also acknowledged for a post-doctoral grant to J. Pina (ref. SFRH/BPD/108469/2015). The research leading to these results has received funding from Laserlab-Europe (grant agreement no. 284464, EC's Seventh Framework Program).Notes and references
- T. L. Lambert and J. P. Ferraris, J. Chem. Soc., Chem. Commun., 1991, 752–754, 10.1039/C39910000752.
- S. K. Lee, J. H. Seo, N. S. Cho and S. Cho, Thin Solid Films, 2012, 520, 5438–5441 CrossRef CAS.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- H. Huang, L. Yang and B. Sharma, J. Mater. Chem. A, 2017, 5, 11501–11517 CAS.
- J. S. Seixas de Melo, H. D. Burrows and J. Pina, in Photochemistry, ed. E. Fasani and A. Albini, The Royal Society of Chemistry, vol. 43, 2016, pp. 83–102 10.1039/9781782622772-00083.
- J. Pina, J. S. Seixas de Melo, A. Eckert and U. Scherf, J. Mater. Chem. A, 2015, 3, 6373–6382 CAS.
- J. Pina, D. Sarmento, M. Accoto, P. L. Gentili, L. Vaccaro, A. Galvão and J. S. Seixas de Melo, J. Phys. Chem. B, 2017, 121, 2308–2318 CrossRef CAS PubMed.
- J. S. Seixas de Melo, in Photochemistry: Volume 45, ed. E. Fasani, S. Protti and A. Albini, The Royal Society of Chemistry, 2018, vol. 45, pp. 68–100 Search PubMed.
- H. Tanaka, K. Tokuyama, T. Sato and T. Ota, Chem. Lett., 1990, 1813–1816 CrossRef CAS.
- G. Voss, M. Drechsler, S. Eller, M. Gradzielski, D. Gunzelmann, S. Mondal, S. van Smaalen and C. S. Voertler, Helv. Chim. Acta, 2009, 92, 2675–2697 CrossRef CAS.
- R. J. Rondão, J. S. Seixas de Melo, F. B. Schaberle and G. Voss, Phys. Chem. Chem. Phys., 2012, 14, 1778–1783 RSC.
- E. D. Glowacki, D. H. Apaydin, Z. Bozkurt, U. Monkowius, K. Demirak, E. Tordin, M. Himmelsbach, C. Schwarzinger, M. Burian, R. T. Lechner, N. Demitri, G. Voss and N. S. Sariciftci, J. Mater. Chem. C, 2014, 2, 8089–8097 RSC.
- C. Guo, J. Quinn, B. Sun and Y. Li, J. Mater. Chem. C, 2015, 3, 5226–5232 RSC.
- K. J. Fallon, N. Wijeyasinghe, E. F. Manley, S. D. Dimitrov, S. A. Yousaf, R. S. Ashraf, W. Duffy, A. A. Y. Guilbert, D. M. E. Freeman, M. Al-Hashimi, J. Nelson, J. R. Durrant, L. X. Chen, I. McCulloch, T. J. Marks, T. M. Clarke, T. D. Anthopoulos and H. Bronstein, Chem. Mater., 2016, 28, 8366–8378 CrossRef CAS.
- R. S. Becker, J. Seixas de Melo, A. L. Maçanita and F. Elisei, J. Phys. Chem., 1996, 100, 18683–18695 CrossRef CAS.
- D. Magde, J. H. Brannon, T. L. Cremers and J. Olmsted, J. Phys. Chem., 1979, 83, 696–699 CrossRef CAS.
- J. Olmsted, J. Phys. Chem., 1979, 83, 2581–2584 CrossRef CAS.
- M. Montalti, A. Credi, L. Prodi and M. Gandolfi, Handbook of Photochemistry, CRC Presss and Taylor & Francis, Boca Raton, 3rd edn, 2006 Search PubMed.
- J. Pina, J. Seixas de Melo, H. D. Burrows, A. L. Macanita, F. Galbrecht, T. Bunnagel and U. Scherf, Macromolecules, 2009, 42, 1710–1719 CrossRef CAS.
- G. Striker, V. Subramaniam, C. A. M. Seidel and A. Volkmer, J. Phys. Chem. B, 1999, 103, 8612–8617 CrossRef CAS.
- J. Pina, H. D. Burrows, R. S. Becker, F. B. Dias, A. L. Maçanita and J. Seixas de Melo, J. Phys. Chem. B, 2006, 110, 6499–6505 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 22 Search PubMed.
- U. Asawapirom and U. Scherf, Macromol. Rapid Commun., 2001, 22, 746–749 CrossRef CAS.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657–661 CrossRef CAS PubMed.
- S. Ko, R. Mondal, C. Risko, J. K. Lee, S. Hong, M. D. McGehee, J.-L. Brédas and Z. Bao, Macromolecules, 2010, 43, 6685–6698 CrossRef CAS.
- J. Pina, A. Eckert, U. Scherf, A. M. Galvão and J. S. Seixas de Melo, Mater. Chem. Front., 2018 10.1039/C7QM00440K.
- J. Seixas de Melo, A. P. Moura and M. J. Melo, J. Phys. Chem. A, 2004, 108, 6975–6981 CrossRef CAS.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- B. Ferreira, P. F. da Silva, J. S. Seixas de Melo, J. Pina and A. Maçanita, J. Phys. Chem. B, 2012, 116, 2347–2355 CrossRef CAS PubMed.
- J. S. Seixas de Melo, C. Serpa, H. D. Burrows and L. G. Arnaut, Angew. Chem., Int. Ed., 2007, 46, 2094–2096 CrossRef CAS PubMed.
- J. Seixas de Melo, R. Rondão, H. D. Burrows, M. J. Melo, S. Navaratnam, R. Edge and G. Voss, J. Phys. Chem. A, 2006, 110, 13653–13661 CrossRef CAS PubMed.
- J. S. Seixas de Melo, R. Rondão, H. D. Burrows, M. J. Melo, S. Navaratnam, R. Edge and G. Voss, ChemPhysChem, 2006, 7, 2303–2311 CrossRef CAS PubMed.
- T. Okamoto, K. Kudoh, A. Wakamiya and S. Yamaguchi, Chem. – Eur. J., 2007, 13, 548–556 CrossRef CAS PubMed.
- D. S. Larsen, K. Ohta and G. R. Fleming, J. Chem. Phys., 1999, 111, 8970–8979 CrossRef CAS.
- O. Braem, T. J. Penfold, A. Cannizzo and M. Chergui, Phys. Chem. Chem. Phys., 2012, 14, 3513–3519 RSC.
- E. Fron, A. Deres, S. Rocha, G. Zhou, K. Müllen, F. C. De Schryver, M. Sliwa, H. Uji-i, J. Hofkens and T. Vosch, J. Phys. Chem. B, 2010, 114, 1277–1286 CrossRef CAS PubMed.
- F. B. Dias, A. Maçanita, J. Seixas de Melo, H. D. Burrows, R. Güntner, U. Scherf and A. P. Monkman, J. Chem. Phys., 2003, 118, 7119–7126 CrossRef CAS.
- J. Pina, J. S. Seixas de Melo, N. Koenen and U. Scherf, J. Phys. Chem. B, 2013, 117, 7370–7380 CrossRef CAS PubMed.
- B. Dick and N. P. Ernsting, J. Phys. Chem., 1987, 91, 4261–4265 CrossRef CAS.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00439g |
| This journal is © the Partner Organisations 2018 |