
Blackening of aza-BODIPY analogues by simple dimerization: panchromatic absorption of a pyrrolopyrrole aza-BODIPY dimer†
Yuto
Kage
a,
Shigeki
Mori
b,
Marina
Ide
c,
Akinori
Saeki
 *c,
Hiroyuki
Furuta
*ad and
Soji
Shimizu
*ad
*c,
Hiroyuki
Furuta
*ad and
Soji
Shimizu
*ad
aDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, Fukuoka 819-0395, Japan. E-mail: ssoji@cstf.kyushu-u.ac.jp; hfuruta@cstf.kyushu-u.ac.jp
bAdvanced Research Support Center (ADRES), Ehime University, Matsuyama 790-8577, Japan
cDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita 565-0871, Japan. E-mail: saeki@chem.eng.osaka-u.ac.jp
dCenter for Molecular Systems (CMS), Kyushu University, Fukuoka 819-0395, Japan
First published on 17th November 2017
Abstract
Dimerization of the so-called pyrrolopyrrole aza-BODIPY, which is a new class of aza-BODIPY analogues exhibiting intense absorption and emission in the visible (Vis) and near infrared (NIR) region, via a bithienyl linkage led to the creation of a novel black dye with dual emission and panchromatic absorption properties in the Vis/NIR region. The role of the linkage in the dye was unambiguously elucidated by comparison of the optical properties with those of its biphenyl-linked counterpart.
Introduction
Owing to the rapid growth of the research field of optoelectronics, especially of devices such as dye-sensitized solar cells (DSSCs),1 thin-layer organic photovoltaics (OPVs),2 and organic light emitting diodes (OLEDs),3 design and creation of new functional chromophores have been intensively investigated in order to achieve absorption and emission in particular wavelength regions required for respective applications. Among such attempts, broadening of the absorption bands in the visible and near infrared (Vis/NIR) region has been drawing attention due to the potential application in the fields of DSSCs and OPVs with the aim of maximizing solar energy harvesting.4Herein, we describe unique blackening of a boron aza-dipyrromethene (aza-BODIPY) analogue by its simple dimerization through a bithienyl linkage. Recently, we have synthesized an aza-BODIPY analogue referred to as pyrrolopyrrole aza-BODIPY (PPAB) using the Schiff-base formation reaction of diketopyrrolopyrrole and heteroaromatic amines.5 In a previous study, control of the absorption and fluorescence wavelengths of PPAB monomers in the Vis/NIR region was achieved by altering heteroaromatic ring units in the structure of PPAB and aryl groups appended on the pyrrolopyrrole moiety.6 During synthesis research on PPAB monomers, we noticed that a PPAB dimer exhibiting black colour in solution was formed, when a palladium-catalyzed borylation reaction of a PPAB monomer with 2-bromothienyl groups was examined. Although significant effects through a thiophene–thiophene linkage and a thiophene–pyrrolopyrrole linkage have recently been investigated to create fluorescent probes7 and to enhance the two-photon absorption cross section,8 the panchromatic absorption of the PPAB dimer achieved by simple dimerization is still a rare example9–14 and can be suitable for the above-mentioned photo-energy harvesting applications. In order to elucidate this unique broadening mechanism, in which the linkage between each monomer unit is anticipated to play an essential role, we synthesized two kinds of PPAB dimers bearing a bithienyl or biphenyl linkage in this study. The potential application of the black PPAB dimer dye as an OPV material was also attempted for the first time.
Results and discussion
Synthesis and characterization
Bithienyl-linked PPAB dimer 3 was obtained in 22% yield through a palladium-catalyzed borylation reaction of 2-bromothienyl-appended PPAB 2, which was synthesized by bromination of thienyl-substituted PPAB 1 (Scheme 1a). The moderate yield of 3 was partly because of the formation of higher oligomer species. Due to the tendency of 2 towards a homo-coupling reaction and a debromination reaction under palladium-catalyzed borylation reaction conditions, a hetero-coupling reaction of 2 with 9,9-dimethylfluorene-2-boronic acid pinacol ester also provided a dimer 4 bearing dimethylfluorene units at both ends in 7.0% yield, which was comparable to that of the regular coupling product 5 (6.1%, Scheme 1a). In contrast, a borylated product 7 was exclusively obtained from a similar borylation reaction of the PPAB bearing a p-bromophenyl group 6. The biphenyl-linked PPAB dimer was then synthesized in 12% yield via a Suzuki–Miyaura cross coupling reaction of 6 and 7 (Scheme 1b). In addition to these series of dimers, a 9,9-dimethylfluorene-appended PPAB monomer 10 and a PPAB monomer with biphenyl group 11 were also synthesized as reference compounds (Scheme 1c). All these compounds were fully characterized by high-resolution mass spectrometry and 1H NMR spectroscopy (Fig. S1–S13, ESI†). The crystal structures of 2, 5, 10 and 11 were also unambiguously determined by X-ray diffraction analysis (Fig. 1, Fig. S14–S18 and Tables S1–S4, ESI†).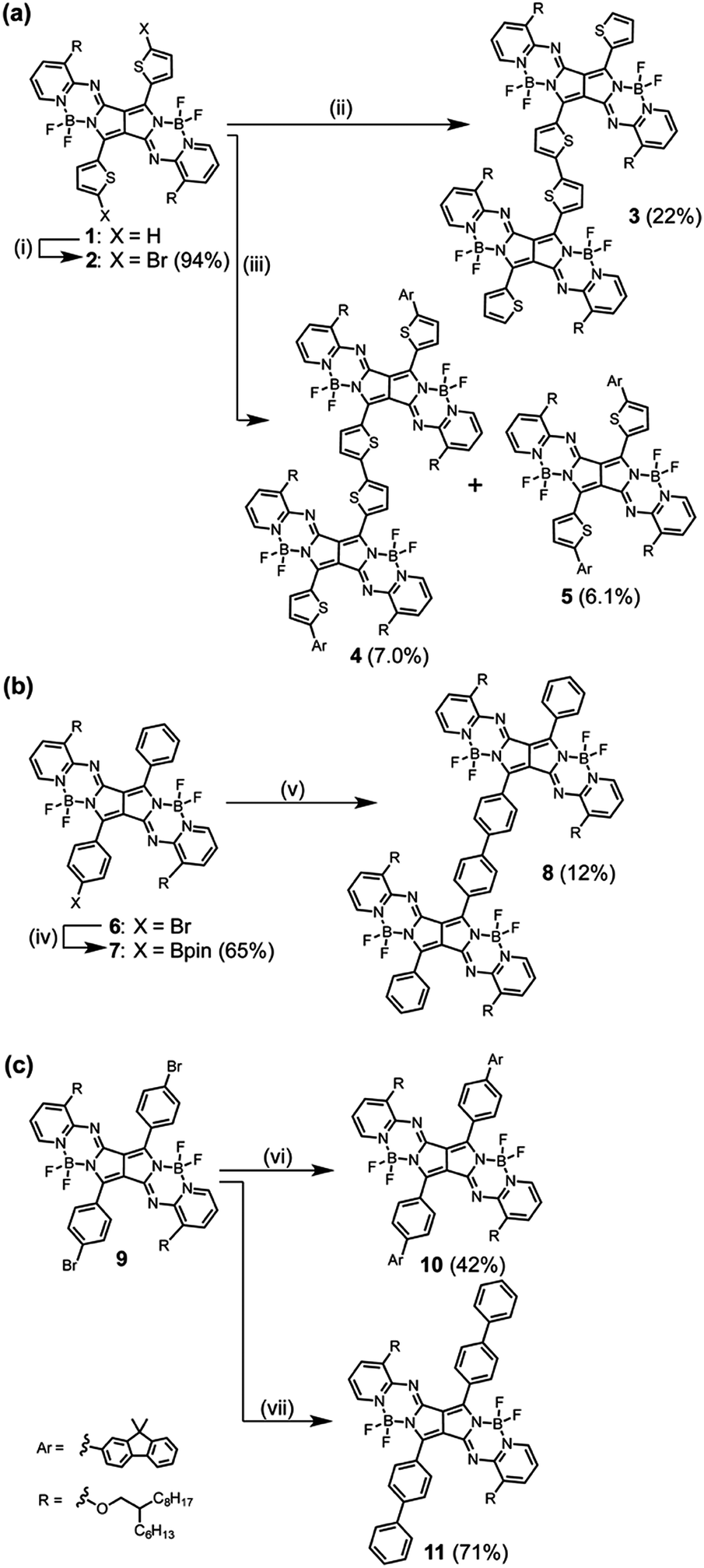 | ||
| Scheme 1 Synthesis of PPAB dimers (3, 4 and 8) and the reference PPAB monomers (5, 10 and 11). Reagents and conditions: (i) NBS, chloroform, r.t.; (ii) (Bpin)2, PdCl2(dppf)·CH2Cl2, KOAc, DMF, 80 °C; (iii) 9,9-dimethylfluorene-2-boronic acid pinacol ester, PdCl2(dppf)·CH2Cl2, KOAc, DMF, 80 °C; (iv) (Bpin)2, PdCl2(PPh3)2, KOAc, 1,4-dioxane, 85 °C; (v) 6, PdCl2(dppf)·CH2Cl2, KOAc, DMF, 80 °C; (vi) 9,9-dimethylfluorene-2-boronic acid pinacol ester, PdCl2(dppf)·CH2Cl2, KOAc, DMF, 80 °C; (vii) phenyl boronic acid, PdCl2(dppf)·CH2Cl2, KOAc, DMF, 80 °C. | ||
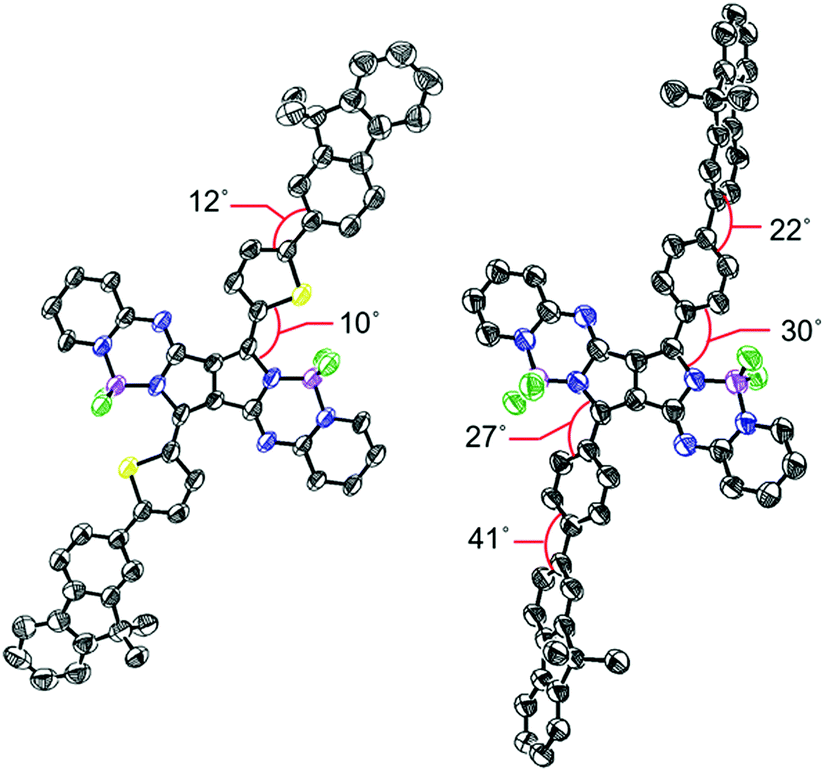 | ||
| Fig. 1 Single crystal X-ray structures of 5 (left) and 10 (right). Hydrogen atoms and alkoxy groups were omitted for clarity. The thermal ellipsoids were scaled to the 50% probability level. | ||
In the crystal structure of 5, the PPAB core structure, thienyl groups, and 9,9-dimethylfluorene groups are arranged in a highly coplanar manner with small dihedral angles. In contrast, significant twists by angles of 22–41° between the substituents and the PPAB core can be observed for 10 and 11.8
Absorption and fluorescence spectra
In chloroform, dimer 3 exhibited panchromatic absorption in the Vis/NIR region with absorption bands at 817, 674 and 565 nm, providing a sharp contrast to the intense absorption band of the corresponding monomer 1 at 693 nm (Fig. 2a). 3 also showed a solvatochromic behaviour. In DMF, an increase of the peak intensity at 673 nm was observed, while a new absorption band at 916 nm appeared in methylcyclohexane at the expense of the band at 673 nm (Fig. S19, ESI†). Compared to 3, 9,9-dimethylfluorene-appended dimer 4 showed a red-shift of the main bands at 817 and 674 nm by ca. 40 nm with only a slight change in the absorption spectral profile (Fig. S20, ESI†). In contrast, the absorption spectrum of 8 in chloroform showed a single sharp band at 671 nm with a similar absorption spectral profile to the PPAB monomers (Fig. 2b). In addition, no such solvatochromic behaviour as 3 was observed for 8 (Fig. S21, ESI†).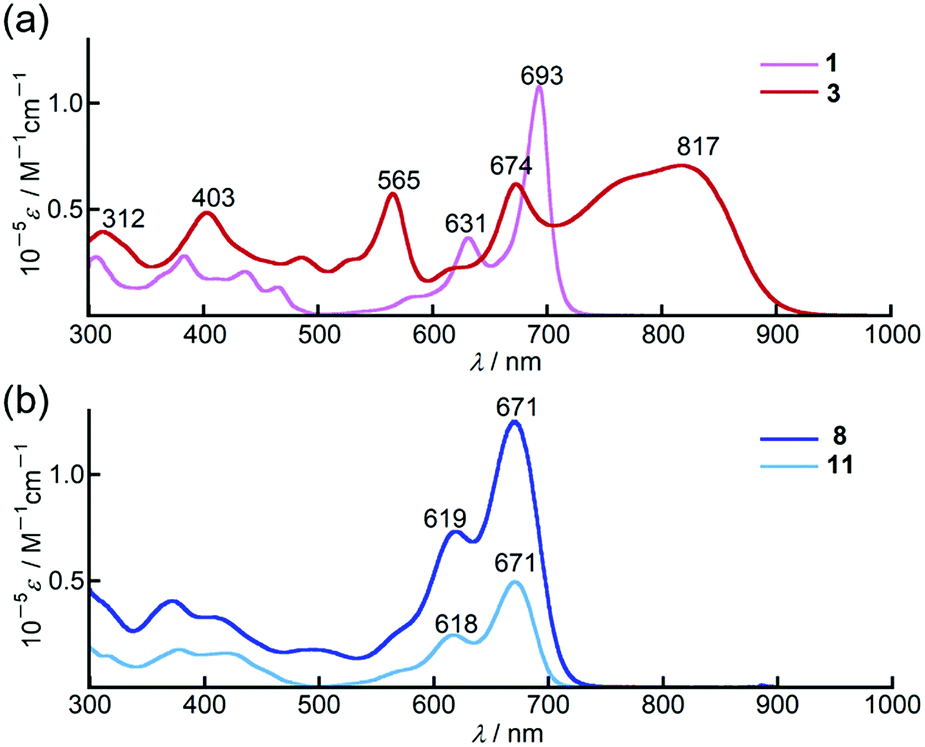 | ||
| Fig. 2 UV/Vis/NIR absorption spectra of (a) 1 (pink) and 3 (red), (b) 8 (blue) and 11 (aqua blue) in chloroform. | ||
Remarkably, 11 exhibited an absorption spectrum almost identical to that of 8, implying that the interaction between two PPAB chromophores in 8 through the biphenyl-linkage is negligibly small due to non-coplanar arrangements of the PPAB moieties and the biphenyl linkage of 8, which can be anticipated from the crystal structures of 10 and 11.
Bithienyl-linked dimers 3 and 4 exhibited unique dual emission centred at 750 and 880 nm and at 770 and 908 nm, respectively, and the relative intensity of each emission band was dependent on the excitation wavelengths (Fig. 3a and Fig. S22, ESI†). On the other hand, the fluorescence spectrum of the biphenyl-linked dimer 8 is rather similar to those of the monomers, showing single intense emission at 702 nm with negligible dependence on the excitation wavelengths (Fig. 3c and Fig. S23–S26, ESI†). It has been reported that a butadiyne-linked porphyrin dimer15 and a meso, meso-linked corrole dimer16 also exhibited similar dual emission properties due to rotational isomerism through the linkage unit. Variable temperature 1H NMR measurements on 3 revealed broadening of the signals at low temperature, which supported free rotational isomerization of 3 at room temperature (Fig. S13, ESI†). In order to gain further insight into the rotational isomerism of 3, the absorption and fluorescence spectra were measured in a viscous solvent consisting of chloroform and paraffin oil (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). In the viscous solution, 3 and 8 showed absorption spectra almost identical to those observed in chloroform. Interestingly, in the fluorescence spectra, 3 exhibited a new emission band at around 685 nm, which was found to be close to the emission band of the corresponding monomer 1 at 704 nm (Fig. 3b and Fig. S23, ESI†). Considering that the emission bands at around 750 and 880 nm were still observed in the viscous solution, this new emission band may have arisen from another rotational isomer, which plausibly changes to other rotational isomers exhibiting emission at 750 or 880 nm in chloroform solution. These results clearly indicate the presence of rotational isomers and isomerization upon excitation. In the case of 8, the fluorescence spectra in the viscous solvent were identical to those in chloroform (Fig. 3d). This is in good agreement with the almost negligible interaction between the PPAB moieties in 8 as discussed above based on the absorption spectra.
:2). In the viscous solution, 3 and 8 showed absorption spectra almost identical to those observed in chloroform. Interestingly, in the fluorescence spectra, 3 exhibited a new emission band at around 685 nm, which was found to be close to the emission band of the corresponding monomer 1 at 704 nm (Fig. 3b and Fig. S23, ESI†). Considering that the emission bands at around 750 and 880 nm were still observed in the viscous solution, this new emission band may have arisen from another rotational isomer, which plausibly changes to other rotational isomers exhibiting emission at 750 or 880 nm in chloroform solution. These results clearly indicate the presence of rotational isomers and isomerization upon excitation. In the case of 8, the fluorescence spectra in the viscous solvent were identical to those in chloroform (Fig. 3d). This is in good agreement with the almost negligible interaction between the PPAB moieties in 8 as discussed above based on the absorption spectra.
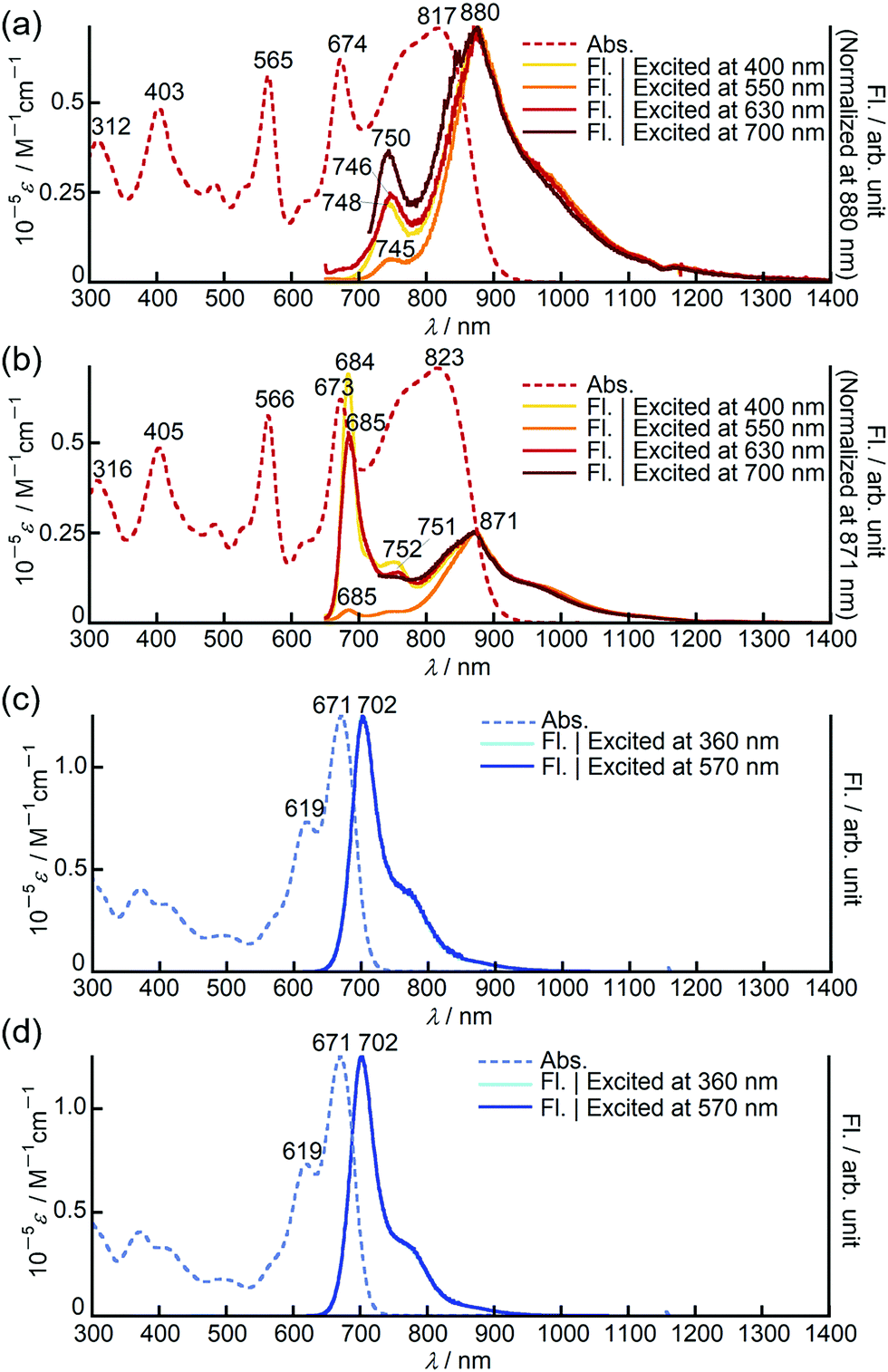 | ||
| Fig. 3 UV/Vis/NIR absorption (dashed lines) and fluorescence (solid lines) spectra of 3 in (a) chloroform and (b) viscous solvents, and 8 in (c) chloroform and (d) viscous solvents. Excitation wavelengths are 400 (yellow), 550 (orange), 630 (red) and 700 nm (dark red) for 3 and 360 (aqua blue) and 570 nm (blue) for 8. A mixture of chloroform and paraffin oil (v/v = 1:2) was used as the viscous solvent. | ||
Theoretical calculation
DFT and TDDFT calculations of 3 were carried out to gain insight into the conformation-dependence of the absorption spectrum. Based on the crystal structure of 2, two model structures with an s-trans (3-a) or s-cis (3-b) bithienyl linkage, respectively, were optimized at the B3LYP/6-31G(d) level (Fig. 4a). In order to estimate the effects of dihedral angles between thiophene rings of the bithienyl linkage and between the pyrrolopyrrole moieties and the bithienyl linkage, model structures 3-c and 3-d with a dihedral angle of 90° were also used for calculations (Fig. 4a). The TDDFT calculations revealed that the broad absorption band at 817 nm in the absorption spectrum of 3 can be attributed to the rotational isomer 3-a or 3-b, whereas the monomer-like absorption bands at 674 and 565 nm can be reproduced satisfactorily by the theoretical absorption of 3-c and 3-d (Fig. 4b and Table 1). Although there could be a number of rotational isomers in solution, high reproducibility of the observed absorption with these model structures indicates that the rotational isomerization not only around the thiophene–thiophene bond, but also around the thiophene–PPAB bond contributes to the panchromatic absorption of 3. The frontier molecular orbital (MO) diagrams also deviate depending on the dihedral angles (Fig. 5 and Fig. S27, ESI†). Compared to the HOMO and LUMO energy levels of 3-c, those of 3-a and 3-b are destabilized and stabilized, respectively, due to the MO interaction of the PPAB monomer through the bithienyl linkage. In the case of 3-d, the LUMO is less stabilized, while the HOMO is destabilized to a similar extent to 3-a and 3-b. For comparison, DFT and TDDFT calculations on PPAB dimers and monomers were also performed at the same level (Fig. S28–S42 and Tables S5–S9, ESI†). The HOMO and LUMO energies and the theoretical absorption of 8 were almost the same as those of the corresponding monomer 11, which again supports the negligible interaction between PPAB moieties via the biphenyl linkage.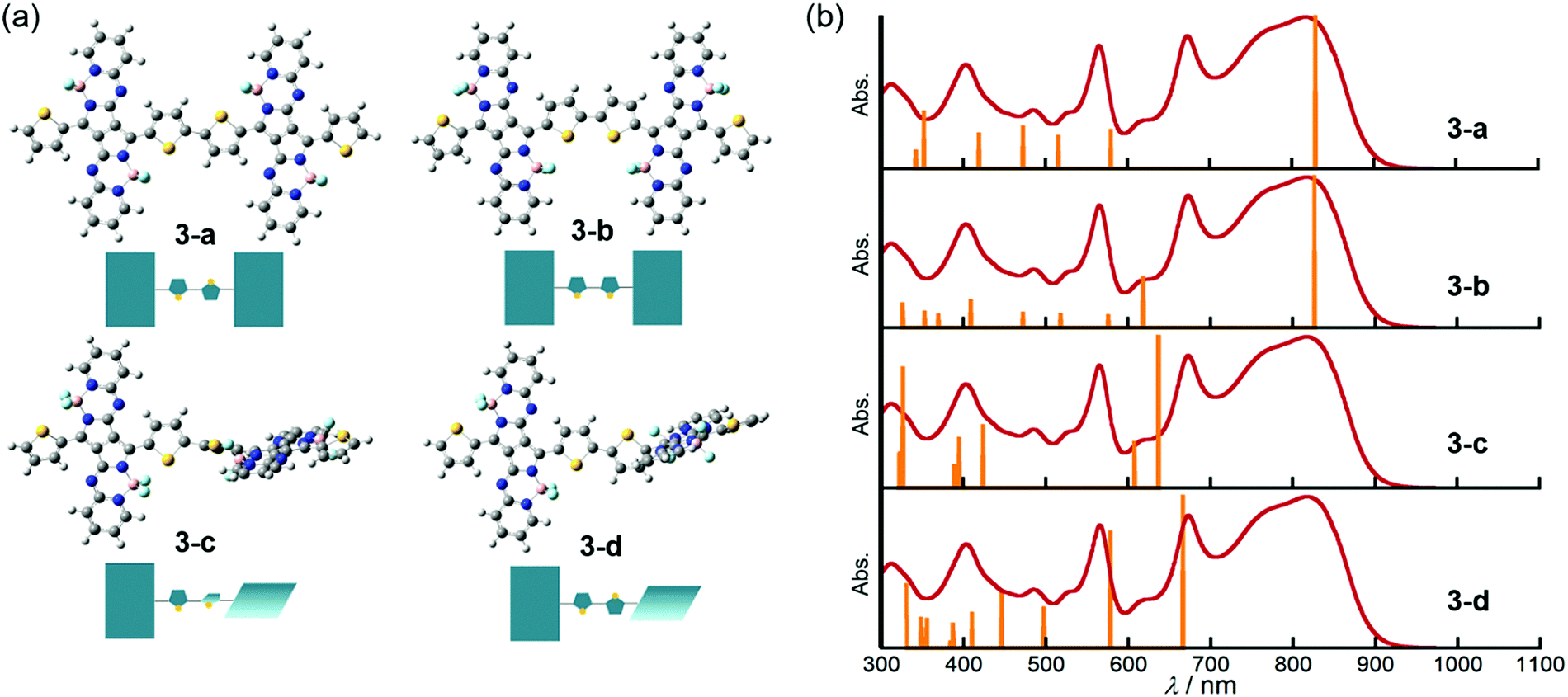 | ||
| Fig. 4 (a) Optimized model structures and schematic representations. (b) Theoretical absorption of each model structure at the B3LYP/6-31G(d) level (orange bars) and the observed absorption spectra of 3 in chloroform (red lines). | ||
| Compound | λ/nm | f | Major contributionsb |
|---|---|---|---|
| a Oscillator strength. b L and H represent the LUMO and HOMO, respectively. | |||
| 3-a | 829 | 1.42 | H → L (100%) |
| 579 | 0.36 | H−1 → L+1 (95%) | |
| 516 | 0.30 | H−2 → L (85%), H → L+2 (10%) | |
| 472 | 0.39 | H → L+2 (86%) | |
| 419 | 0.32 | H−4 → L (15%), H → L+4 (59%) | |
| 3-b | 826 | 1.39 | H → L (100%) |
| 618 | 0.46 | H−1 → L (39%), H → L+1 (61%) | |
| 576 | 0.11 | H−1 → L+1 (95%) | |
| 518 | 0.12 | H−2 → L (87%) | |
| 472 | 0.13 | H → L+2 (88%) | |
| 409 | 0.25 | H−1 → L+2 (30%), H → L+5 (27%) | |
| 3-c | 637 | 1.09 | H−1 → L+1 (59%), H → L (41%) |
| 607 | 0.30 | H−1 → L (49%), H → L+1 (51%) | |
| 423 | 0.41 | H → L+2 (22%), H-2 → L (21%) | |
| 3-d | 666 | 0.84 | H → L+1 (99%) |
| 578 | 0.64 | H−1 → L (96%) | |
| 497 | 0.22 | H−2 → L+1 (88%) | |
| 446 | 0.31 | H → L+3 (83%) | |
| 410 | 0.19 | H → L+4 (25%), H−8 → L+1 (17%) | |
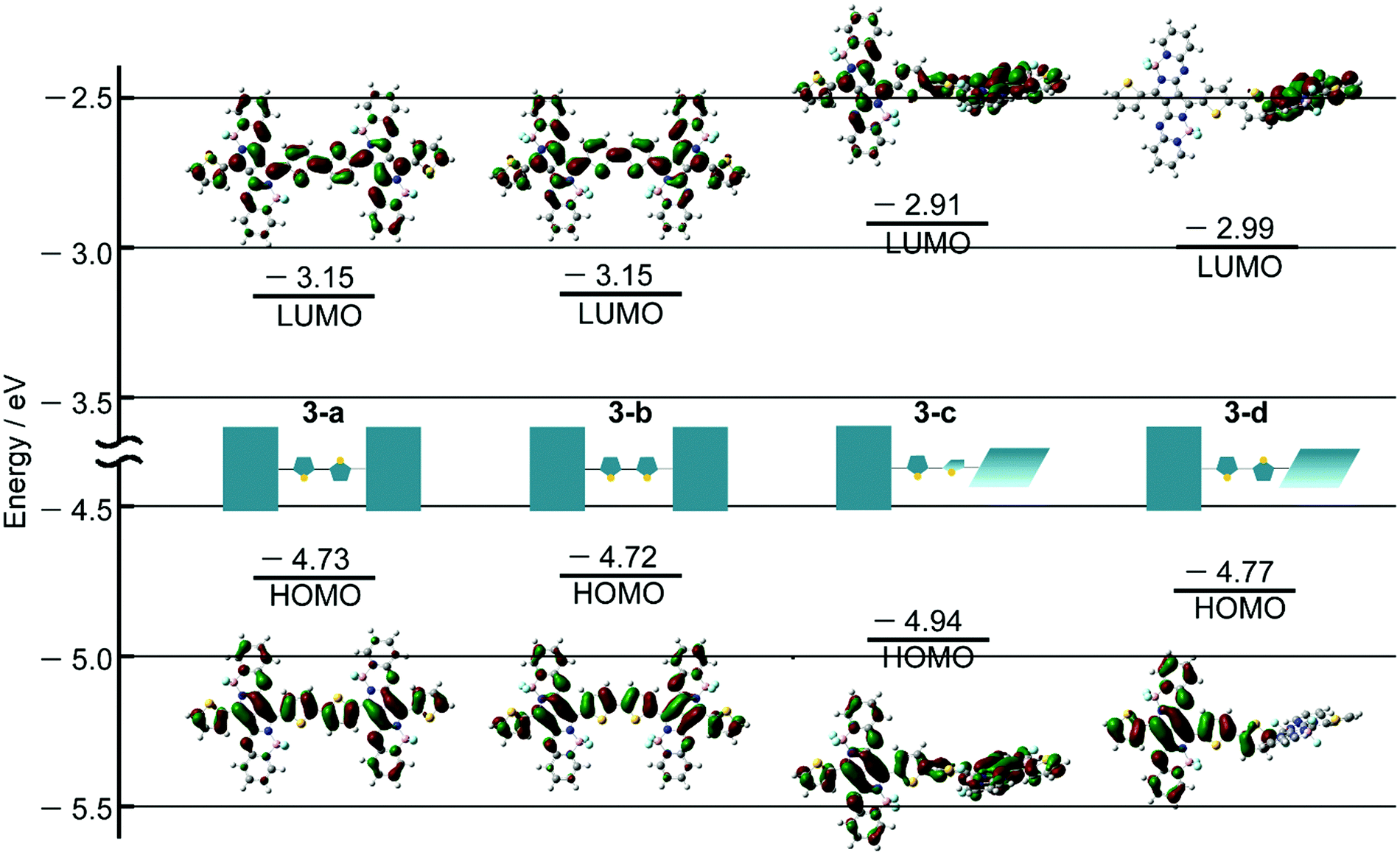 | ||
| Fig. 5 Frontier molecular orbitals of 3-a, 3-b, 3-c and 3-d (B3LYP/6-31G(d)). | ||
Electrochemistry
Cyclic voltammograms of PPAB monomers and dimers were obtained using 0.5 mM sample solutions in o-dichlorobenzene containing 0.1 M tetra-n-butylammonium perchlorate as a supporting electrolyte (Fig. 6). Compared to the first reduction potentials of the PPAB monomers in the current study, which are in the range from −1.21 to −1.24 V (vs. Fc+/Fc), bithienyl-linked dimers 3 and 4 showed a positive shift to −1.12 V. Considering the similar first reduction potential of 8 at −1.25 V to those of the monomers, these positive shifts clearly indicate the interaction between the PPAB moieties through the bithienyl linkage. In addition, a negative shift of the first oxidation potential from 0.27 V (3) to 0.18 V (4) was observed. A similar change was observed for 1 and 5. In contrast, 8 and the corresponding monomers (10 and 11) exhibited similar cyclic voltammograms. This indicates that the types of aryl-substituents have a significant effect on the HOMO energy level, but not on the LUMO energy level in the case of the PPAB monomers and dimers bearing thienyl substituents, whereas no such effect was observed with phenyl substituents. Overall, these electrochemistry results are in good agreement with the observed drastic changes in the absorption spectra between the monomers and dimers with thienyl substituents and no obvious absorption spectral change was observed between the biphenyl-substituted monomers and biphenyl-linked dimers.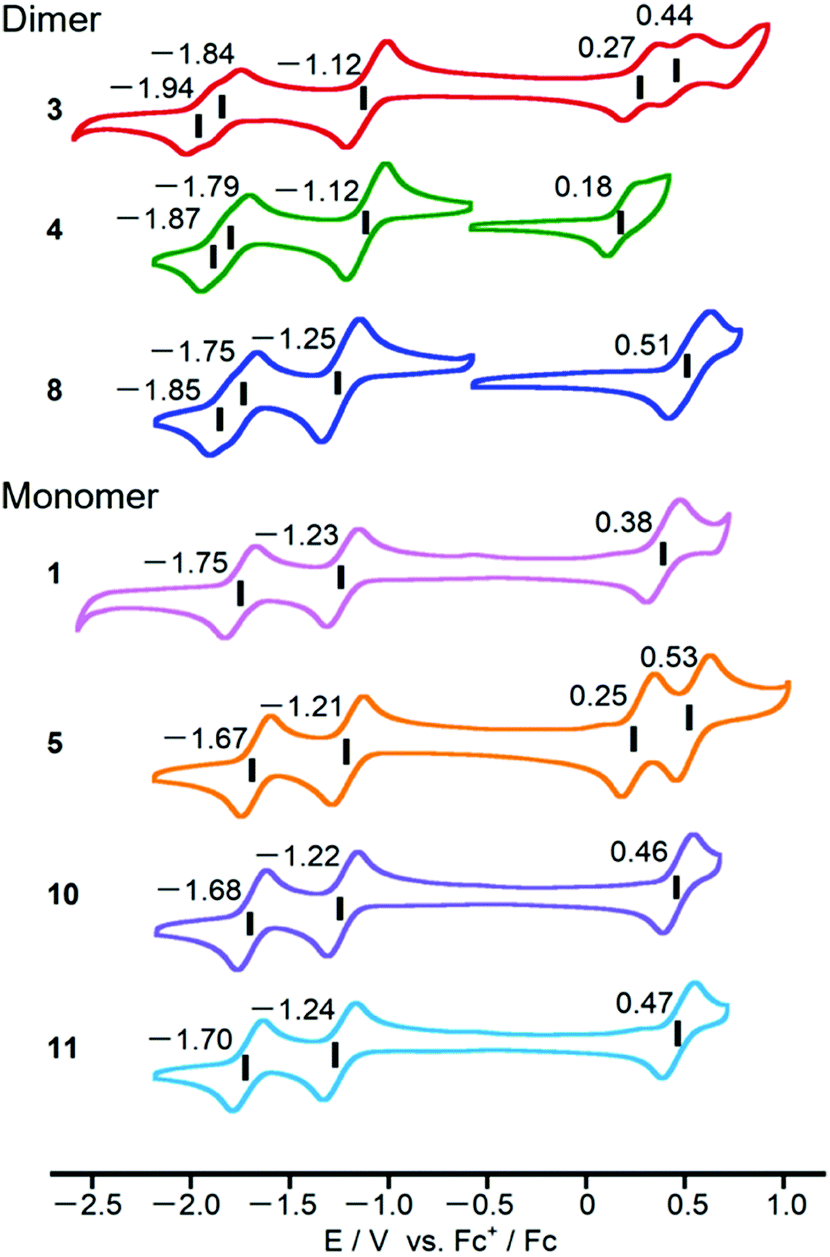 | ||
| Fig. 6 Cyclic voltammograms of PPAB dimers and monomers in o-dichlorobenzene (0.5 mM) containing 0.1 M tetra-n-butylammonium perchlorate as the supporting electrolyte at a scan rate of 100 mV s−1. Values are half-wave potentials. | ||
Photovoltaic application
Owing to the unique panchromatic absorption of 3 in the Vis/NIR region, we examined the potential application of 3 as an OPV material. Based on the first oxidation and reduction potentials of 3 at 0.27 V and −1.12 V (vs. Fc+/Fc, Fig. 6), respectively, the HOMO and LUMO energy levels were estimated to be −5.1 eV and −3.7 eV, respectively, which are almost close to the HOMO of poly(3-hexylthiophene) (P3HT) at −5.1 eV17 and to the LUMO of [6,6]-phenyl C71 butyric acid methyl ester (PC71BM) at −3.9 eV.18 Since these compounds are commonly used as p-type and n-type materials in OPV devices, ambipolar characteristics of 3 were inferred. Prior to device fabrication, we performed time-resolved microwave conductivity (TRMC) measurements19 on the blend films of 3 with P3HT and PC71BM using a white-light pulse from a Xe flash-lamp (Xe-flash)20 as a photoexcitation source. This measurement can provide the value of the maximum transient conductivity, Δσmax, which correlates with the charge separation efficiency and the sum of hole and electron mobilities. In both blends, the Δσmax values were one order of magnitude larger than those of pristine samples (Fig. 7). These results indicated that 3 can actually function as an ambipolar material. From the maximum Δσmax value of the blend film, the best mixing ratios for BHJ (bulk heterojunction)-OPV devices were determined to be 1:2–1:3 (w/w) for 3 and P3HT and 1:1 (w/w) for 3 and PC71BM.
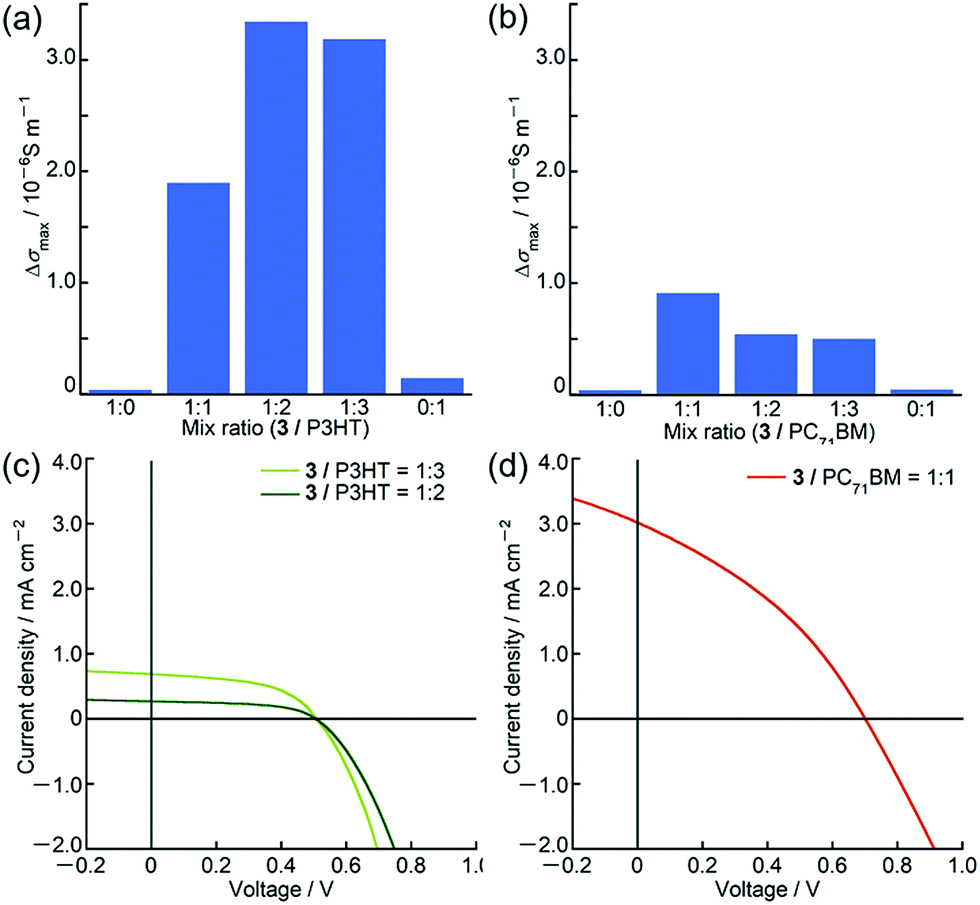 | ||
| Fig. 7 Maximum Xe-flash TRMC signals (Δσmax) of (a) 3/P3HT and (b) 3/PC71BM drop-cast films with various mixing ratios, and J/V curves of (c) 3/P3HT cells (1:3 (w/w), pale green; 1:2 (w/w), green) and (d) 3/PC71BM cells (1:1 (w/w), red). | ||
BHJ-OPV devices with a glass/ITO/ZnO/BHJ/MoO3/Ag inverted cell structure were fabricated (Fig. S43 and Table S10, ESI†). BHJ active layers were spin-coated from chloroform solution of 3 and PC71BM containing 0.5 volume% of solvent additive 1,8-diiodooctane and from o-dichlorobenzene solution of 3 and P3HT. As summarized in Table 2, the best power conversion efficiencies (PCE) of these BHJ-OPV devices were 0.74% with Voc of 0.71 V, Jsc of 3.02 mA cm−2 and FF of 0.35, and 0.18% with Voc of 0.51 V, Jsc of 0.70 mA cm−2 and FF of 0.52 using 3 as donor and acceptor materials, respectively. The low PCEs and small Jsc of these devices implied that suitable bulk heterojunction structures for charge separation were not created probably due to the low solubility of 3 during device fabrication processes. The surface roughness of the active layers observed using an atomic force microscope also supported poor mixing of the donor and acceptor materials (Fig. S44 and S45, ESI†).
Conclusion
In summary, two kinds of PPAB dimers bearing different linkage units were synthesized in order to give an in-depth insight into the unique broadening of absorption. It was found that only bithienyl linked dimers 3 and 4 showed panchromatic absorption and dual emission. Absorption and fluorescence spectra in a viscous solvent and theoretical calculations of 3 revealed that these unique optical properties can be attributed to the presence of rotational isomers. On the other hand, such isomerization has almost a negligible effect in the case of the biphenyl-linked dimer 8 due to the non-coplanar structure and insignificant MO interactions through the biphenyl linkage. Despite the ambipolar properties of 3 as revealed by the CV and TRMC measurements, the OPV performance of 3 was found to be less significant than expected. Since aggregation of 3 during the device fabrication can be considered as the main reason for the low performance, improvement of solubility and optimization of device fabrication conditions is the next step and is actively pursued in our laboratory.Experimental section
A general procedure
Electronic absorption spectra were recorded on a JASCO V-770 spectrophotometer. Fluorescence spectra were recorded on an SPEX Fluorolog-3-NIR spectrometer (HORIBA) with a NIR-PMT R5509 photomultiplier tube (Hamamatsu). Absolute fluorescence quantum yields were measured using a Hamamatsu Photonics A10104-01 calibrated integrating sphere system with self-absorption correction. 1H NMR spectra were recorded on a JEOL JNM-ECX500 spectrometer (operating at 500 MHz for 1H) or a JEOL JNM-AL SERIES FT-NMR spectrometer (operating at 300 MHz for 1H) using a residual solvent as an internal reference for 1H (δ = 7.26 ppm for CDCl3, δ = 5.32 ppm for CD2Cl2 and δ = 2.50 ppm for DMSO-d6). High resolution mass spectrometry was performed on a JEOL LMS-HX-110 spectrometer (FAB mode with 3-nitrobenzyl alcohol (NBA) as the matrix). A viscous solvent was prepared by mixing spectroscopic grade chloroform and liquid paraffin in 1:2 (v/v) ratio. Liquid paraffin was purchased from Ishizu pharmaceutical Co., Ltd. Cyclic voltammograms and differential pulse voltammograms were recorded on a CH Instrument Model 620B (ALS) under an argon atmosphere in o-dichlorobenzene solution with 0.1 M tetra-n-butylammonium perchlorate as the supporting electrolyte. Measurements were made with a glassy carbon electrode, an Ag/AgCl reference electrode, and a Pt-wire counter electrode. The concentration of the solution was fixed at 0.5 mM and the sweep rates were set to 100 mV s−1. The ferrocenium/ferrocene (Fc+/Fc) couple was used as the internal standard. Thin-layer chromatography (TLC) was carried out on aluminum sheets coated with silica gel 60 F254 (MERCK). Preparative separations were performed using silica gel column chromatography (KANTO Silica Gel 60 N, spherical, neutral, 40–50 μm or KANTO Silica Gel 60 N, spherical, neutral, 63–210 μm) and GPC-HPLC (LC-908 recycling preparative HPLC with JAIGEL-2HH and -2.5HH columns (Japan Analytical Industry Co. Ltd.)). All reagents and solvents used for reactions were of commercial reagent grade and were used without further purification unless noted otherwise. All solvents used in optical measurements were of commercial spectroscopic grade.
Crystallographic data collection and structure refinement
Suitable crystals for X-ray analysis were obtained via slow diffusion of methanol into dichloromethane solutions of 2, 5, 10 and 11. Data collection was carried out at 100 K on a Rigaku Saturn 724 CCD diffractometer with MoKα radiation (λ = 0.71069 Å) for 2 and a Rigaku R-AXIS RAPID diffractometer with CuKα radiation (λ = 1.54187 Å) for 5, 10 and 11. The structures were solved by direct methods (Sir-9721 for 2, Sir-201122 for 5, 10 and 11) and refined using a full-matrix least squares technique (SHELXL).23 Yadokari-XG software,24 Olex2,25 and CrystalStructure 4.1.1. were used as GUIs. CCDC 1572423 (2), 1572424 (5), 1572425 (10), and 1572426 (11) contain the supplementary crystallographic data.†Computational methods
The Gaussian 0926 software package was used to carry out DFT and TDDFT calculations using the B3LYP functional with the 6-31G(d) basis set. Structural optimizations were performed on model compounds, in which 2-hexyldecyloxy groups on the pyridyl moieties were replaced with hydrogen atoms for simplicity.Time-resolved microwave conductivity (TRMC)
A resonance cavity was used to obtain a high degree of sensitivity in the conductivity measurement. Resonance frequency and microwave power were set at about 9.1 GHz and 3 mW, respectively, so that the electric field of the microwave was sufficiently small not to disturb the motion of the charge carriers. A microsecond white-light pulse from a Xe flash lamp was used as an excitation source. Photoconductivity, Δσ, was obtained by ΔPr/(APr), in which ΔPr, A and Pr are the transient power change of reflected microwave from a cavity, the sensitivity factor, and the reflected microwave power, respectively. The power of the white-light pulse was 0.3 mJ cm−2 pulse−1. The samples were drop-cast onto a quartz plate from solutions of 3 and P3HT or 3 and PC71BM and dried in a vacuum oven. The TRMC experiments were performed under ambient conditions at room temperature.Organic photovoltaic cells
A ZnO layer was deposited onto a cleaned ITO layer by spin-coating of the ZnO precursor solution (0.1 g mL−1 zinc acetate dihydrate and 0.028 g mL−1 ethanolamine in 2-methoxyethanol). The substrate was annealed on a hot plate at 200 °C for 30 min. An active layer consisting of 3 and P3HT or 3 and PC71BM was cast on top of the ZnO layer in a nitrogen glovebox by spin-coating. The thickness was controlled by changing the rotation speed and was measured using a surface profiler, an ULVAC model Dektak 150. An anode consisting of 10 nm MoO3 and 100 nm Ag layers was sequentially deposited through a shadow mask on top of the active layers by thermal evaporation in a vacuum chamber. The resulting device configuration was ITO (120–160 nm)/ZnO (30 nm)/active layer/MoO3 (10 nm)/Ag (100 nm) with an active area of 7.1 mm2. Current–voltage (J/V) curves were measured using a source-measure unit (ADCMT Corp., 6241 A) under AM1.5G solar illumination at 100 mW cm−2 (1 Sun, monitored using a calibrated standard cell, Bunko Keiki SM-250 KD) from a 300 W solar simulator (SAN-EI Corp., XES-301S). EQE spectra were measured using a Bunko Keiki model BS-520BK equipped with a Keithley model 2401 source meter. The monochromated light power was calibrated using a silicon photovoltaic cell, Bunko Keiki model S1337-1010BQ.Synthesis procedures
1H NMR (500 MHz, CD2Cl2, 295 K): δ [ppm] = 9.94 (d, J = 4.0 Hz, 2H), 9.49 (d, J = 4.0 Hz, 2H), 8.01 (d, J = 5.7 Hz, 2H), 7.96 (d, J = 6.2 Hz, 2H), 7.77 (d, J = 5.1 Hz, 2H), 7.62 (d, J = 4.0 Hz, 2H), 7.37–7.27 (m, 6H), 7.18–7.06 (m, 4H), 4.07 (t, J = 6.8 Hz, 8H), 2.19–1.87 (m, 4H), 1.73–1.04 (m, 96H), 0.95–0.80 (m, 12H), 0.79–0.65 (m, 12H).
HR-MS (FAB) (m/z): 2055.1341 (calcd for C112H154B4F8N12O4S4: 2055.1343 [M+]); 1H NMR (500 MHz, CD2Cl2, 295 K): δ [ppm] = 9.94 (d, J = 4.0 Hz, 2H), 9.49 (d, J = 4.0 Hz, 2H), 8.01 (d, J = 5.7 Hz, 2H), 7.96 (d, J = 6.2 Hz, 2H), 7.77 (d, J = 5.1 Hz, 2H), 7.62 (d, J = 4.0 Hz, 2H), 7.37–7.27 (m, 6H), 7.18–7.06 (m, 4H), 4.07 (t, J = 6.8 Hz, 8H), 2.19–1.87 (m, 4H), 1.73–1.04 (m, 96H), 0.95–0.80 (m, 12H), 0.79–0.65 (m, 12H); UV/Vis/NIR (CHCl3): λmax [nm] (ε/M−1 cm−1) = 403 (48600), 565 (57400), 674 (61900), 817 (70700).
:2 (v/v)) and recrystallized from dichloromethane and methanol. The products were obtained as dark red solids (4: 11 mg, 7.0%, 5: 11 mg, 6.1%).
4: HRMS (FAB) (m/z): 2439.3228 (calcd for C142H178B4F8N12O4S4: 2439.3221 [M+]); 1H NMR (500 MHz, CDCl3, 308 K): δ [ppm] = 10.03 (d, J = 4.5 Hz, 2H), 9.91 (d, J = 4.0 Hz, 2H), 8.03 (d, J = 6.0 Hz, 2H), 7.98 (d, J = 6.0 Hz, 2H), 7.82 (s, 2H), 7.80–7.75 (m, 6H), 7.60 (d, J = 4.0 Hz, 2H), 7.58 (d, J = 4.0 Hz, 2H), 7.48–7.47 (m, 2H), 7.39–7.35 (m, 4H), 7.08–7.03 (m, 4H), 4.05 (m, 8H), 2.10 (m, 4H), 1.62–0.75 (m, 144H); UV/Vis/NIR (CHCl3): λmax [nm] (ε/M−1 cm−1) = 332 (58800), 430 (55900), 538 (47800), 701 (56400), 861 (83800).
5: HRMS (FAB) (m/z): 1412.7620 (calcd for C86H102B2F4N6O2S2: 1412.76 [M+]); 1H NMR (500 MHz, CD2Cl2, 292 K): δ [ppm] = 9.95 (d, J = 4.5 Hz, 2H), 7.98 (d, J = 6.2 Hz, 2H), 7.83 (s, 2H), 7.75–7.79 (m, 6H), 7.59 (d, J = 4.0 Hz, 2H), 7.47–7.48 (m, 2H), 7.34–7.37 (m, 4H), 7.32 (d, J = 7.9 Hz, 2H), 7.11 (t, J = 7.1 Hz, 2H), 4.05 (d, J = 5.7 Hz, 4H), 2.06–2.10 (m, 2H), 1.05–1.60 (m, 68H), 0.76–0.85 (m, 12H); UV/Vis/NIR (CHCl3): λmax [nm] (ε/M−1 cm−1) = 332 (48400), 426 (40500), 518 (35100), 683 (42800), 751 (122000); ΦF = 0.24 (CHCl3).
:85 (v/v)) to give 7 as blue solids (66 mg, 65%).
1H NMR (500 MHz, CD2Cl2, 295 K): δ [ppm] = 8.37–8.32 (m, 4H), 7.92 (d, J = 8.3 Hz, 2H), 7.82 (d, J = 6.1 Hz, 2H), 7.56–7.49 (m, 3H), 7.26 (d, J = 8.3 Hz, 2H), 7.08–7.02 (m, 2H), 3.91 (t, J = 5.3 Hz, 4H), 1.97–1.86 (m, 2H), 1.45–1.21 (m, 50H), 0.89–0.82 (m, 12H).
:2 (v/v)) and recrystallized from dichloromethane and methanol to give 8 as blue green solids (12 mg, 12%).
HR-MS (FAB) (m/z): 2032.3152 (calcd for C120H163B4F8N12O4: 2032.3218 [M+ + H]); 1H NMR (500 MHz, CD2Cl2, 295 K): δ [ppm] = 8.71 (d, J = 8.3 Hz, 4H), 8.38 (m, 4H), 7.94 (d, J = 8.3 Hz, 4H), 7.89 (d, J = 6.1 Hz, 2H), 7.85 (d, J = 6.1 Hz, 2H), 7.57–7.53 (m, 6H), 7.29 (d, J = 6.8 Hz, 4H), 7.11–7.06 (m, 4H), 3.97 (d, J = 6.1 Hz, 4H), 3.95 (d, J = 5.3 Hz, 4H), 2.04–1.86 (m, 4H), 1.49–1.07 (m, 76H), 0.88 (m, 12H), 0.76 (t, J = 6.8 Hz, 12H); UV/Vis/NIR (CHCl3): λmax [nm] (ε/M−1 cm−1) = 372 (41000), 404 (33000), 488 (18000), 619 (73000), 671 (125000); ΦF = 0.18 (CHCl3).
:2 (v/v)) and recrystallized from dichloromethane and methanol. 10 was obtained as green solids (25 mg, 42%).
HRMS (FAB) (m/z): 1400.8496 (calcd for C90H106B2F4N6O2: 1400.85 [M+]); 1H NMR (500 MHz, CDCl3, 292 K): δ [ppm] = 8.65 (d, J = 8.5 Hz, 4H), 7.91 (d, J = 6.0 Hz, 2H), 7.87 (d, J = 9.0 Hz, 4H), 7.82–7.76 (m, 6H), 7.71 (dd, J = 8.0, 2.0 Hz, 2H), 7.48 (d, J = 9.0 Hz, 2H), 7.40–7.33 (m, 4H), 7.23 (d, J = 7.0 Hz, 2H), 7.05–7.01 (m, 2H), 3.93 (d, J = 5.5 Hz, 4H), 1.96 (m, 2H), 1.59–1.16 (m, 60H), 0.84–0.79 (m, 12H); UV/Vis/NIR (CHCl3): λmax [nm] (ε/M−1 cm−1) = 313 (67200), 385 (26900), 451 (30500), 622 (35600), 676 (71100); ΦF = 0.51 (CHCl3).
:2 (v/v)). 11 was obtained as green solids (34 mg, 71%).
HRMS (FAB) (m/z): 1168.7250 (calcd for C72H90B2F4N6O2: 1168.7248 [M+]); 1H NMR (500 MHz, CDCl3, 292 K): δ [ppm] = 8.61 (d, J = 8.5 Hz, 4H), 7.89 (d, J = 6.2 Hz, 2H), 7.79 (d, J = 8.5 Hz, 4H), 7.71 (d, J = 7.4 Hz, 4H), 7.47 (t, J = 7.7 Hz, 4H), 7.39 (t, J = 7.4 Hz, 2H), 7.22 (d, J = 7.9 Hz, 2H), 7.01–7.05 (m, 2H), 3.91 (d, J = 5.7 Hz, 4H), 1.91–1.96 (m, 2H), 1.09–1.36 (m, 48H), 0.81–0.89 (m, 12H); UV/Vis/NIR (CHCl3): λmax [nm] (ε/M−1 cm−1) = 378 (17900), 419 (16000), 618 (24600), 671 (49700); ΦF = 0.56 (CHCl3).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid for Young Scientists A (JSPS KAKENHI Grant Number JP26708003), Scientific Research on Innovative Areas, “New Polymeric Materials Based on Element-Blocks29 (No. 2401)” (JSPS KAKENHI Grant Number JP15H00756) and “π-System Figuration: Control of Electron and Structural Dynamism for Innovative Functions (No. 2601)” (JSPS KAKENHI Grant Number JP17H05160).References
- (a) B. Hardin, H. Snaith and M. McGehee, Nat. Photonics, 2012, 6, 162 CrossRef CAS; (b) A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed; (c) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- (a) J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821 CrossRef CAS PubMed; (b) L. Lu, T. Zheng, Q. Wu, A. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666 CrossRef CAS PubMed; (c) S. Collins, N. Ran, M. Heiber and T. Nguyen, Adv. Energy Mater., 2017, 7, 1602242 CrossRef.
- (a) M. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed; (b) L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926 CrossRef CAS PubMed.
- (a) N. Robertson, Angew. Chem., Int. Ed., 2008, 47, 1012 CrossRef CAS PubMed; (b) B. He, D. Zherebetskyy, H. Wang, M. Kolaczkowski, L. Klivansky, T. Tan, L. Wang and Y. Liu, Chem. Sci., 2016, 7, 3857 RSC; (c) S. Erten-Ela, D. Yilmaz, B. Icli, Y. Dede, S. Icli and E. Akkaya, Org. Lett., 2008, 10, 3299 CrossRef CAS PubMed; (d) S. Banala, T. Rühl, P. Sintic, K. Wurst and B. Kräutler, Angew. Chem., Int. Ed., 2009, 48, 599 CrossRef CAS PubMed.
- S. Shimizu, T. Iino, Y. Araki and N. Kobayashi, Chem. Commun., 2013, 49, 1621 RSC.
- S. Shimizu, T. Iino, A. Saeki, S. Seki and N. Kobayashi, Chem. – Eur. J., 2015, 21, 2893 CrossRef CAS PubMed.
- (a) A. Fin, A. V. Jentzsch, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2012, 51, 12736 CrossRef CAS PubMed; (b) M. D. Molin and S. Matile, Org. Biomol. Chem., 2013, 11, 1952 RSC; (c) D. A. Doval and S. Matile, Org. Biomol. Chem., 2013, 11, 7467 RSC; (d) D. A. Doval, M. D. Molin, S. Ward, A. Fin, N. Sakai and S. Matile, Chem. Sci., 2014, 5, 2819 RSC.
- M. Grzybowski, E. Glodkowska-Mrowka, V. Hugues, W. Brutkowski, M. Blanchard-Desce and D. T. Gryko, Chem. – Eur. J., 2015, 21, 9101 CrossRef CAS PubMed.
- (a) A. Osuka and H. Shimidzu, Angew. Chem., Int. Ed. Engl., 1997, 36, 135 CrossRef CAS; (b) S. Ooi, T. Tanaka and A. Osuka, Inorg. Chem., 2016, 55, 8920 CrossRef CAS PubMed.
- F. Odobel, S. Suresh, E. Blart, Y. Nicolas, J. Quintard, P. Janvier, J. Questel, B. Illien, D. Rondeau, P. Richomme, T. Häupl, S. Wallin and L. Hammarström, Chem. – Eur. J., 2002, 8, 3027 CrossRef CAS.
- J. Mei, K. Graham, R. Stalder and J. Reynolds, Org. Lett., 2010, 12, 660 CrossRef CAS PubMed.
- S. Hayashi, Y. Inokuma and A. Osuka, Org. Lett., 2010, 12, 4148 CrossRef CAS PubMed.
- (a) M. Bröring, R. Krüger, S. Link, C. Kleeberg, S. Köhler, X. Xie, B. Ventura and L. Flamigni, Chem. – Eur. J., 2008, 14, 2976 CrossRef PubMed; (b) A. Nepomnyashchii, M. Bröring, J. Ahrens and A. Bard, J. Am. Chem. Soc., 2011, 133, 19498 CrossRef CAS PubMed.
- Y. Hayashi, S. Yamaguchi, W. Cha, D. Kim and H. Shinokubo, Org. Lett., 2011, 13, 2992 CrossRef CAS PubMed.
- (a) M. Winters, J. Kärnbratt, M. Eng, C. Wilson, H. Anderson and B. Albinsson, J. Phys. Chem. C, 2007, 111, 7192 CrossRef CAS; (b) H. Doan, S. Raut, D. Yale, M. Balaz, S. Dzyuba and Z. Gryczynski, Chem. Commun., 2016, 52, 9510 RSC.
- K. Park, S. Ooi, T. Kim, T. Tanaka, A. Osuka and D. Kim, Phys. Chem. Chem. Phys., 2016, 18, 23374 RSC.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789 CrossRef CAS.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970 RSC.
- (a) F. Grozema and L. Siebbeles, J. Phys. Chem. Lett., 2011, 2, 2951 CrossRef CAS; (b) A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193 CrossRef CAS PubMed; (c) Y. Shimata and A. Saeki, J. Phys. Chem. C, 2017, 121, 18351 CrossRef CAS.
- A. Saeki, S. Yoshikawa, M. Tsuji, Y. Koizumi, M. Ide, C. Vijayakumar and S. Seki, J. Am. Chem. Soc., 2012, 134, 19035 CrossRef CAS PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, M. Mallamo, A. Mazzone, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2012, 45, 357 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3 CrossRef PubMed.
- Yadokari-XG, Software for Crystal Structure Analyses, K. Wakita, (2001); Release of Software (Yadokari-XG 2009) for Crystal Structure Analyses, C. Kabuto, S. Akine, T. Nemoto and E. Kwon, J. Crystallogr. Soc. Jpn., 2009, 51, 218 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- G. Zhang, L. Song, S. Bi, Y. Wu, J. Yu and L. Wang, Dyes Pigm., 2014, 102, 100 CrossRef CAS.
- S. Hitosugi, D. Tanimoto, W. Nakanishi and H. Isobe, Chem. Lett., 2012, 41, 972 CrossRef CAS.
- Y. Chujo and K. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1572423 (2), 1572424 (5), 1572425 (10), and 1572426 (11). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00438a |
| This journal is © the Partner Organisations 2018 |